
Oxidative Glucose Metabolism Promotes Senescence in Vascular Endothelial Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Antibodies
2.3. Cell Culture
2.4. Cell Lysis and Western Blot
2.5. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
2.6. Glucose Consumption
2.7. Lactate Production
2.8. ATP Assay
2.9. Seahorse Real-Time Cell Metabolic Analyses
2.10. Glucose Flux Measurement
2.11. Mitochondrial ROS Measurement
2.12. Detection of Mitochondrial Mass
2.13. Statistical Analysis
3. Results
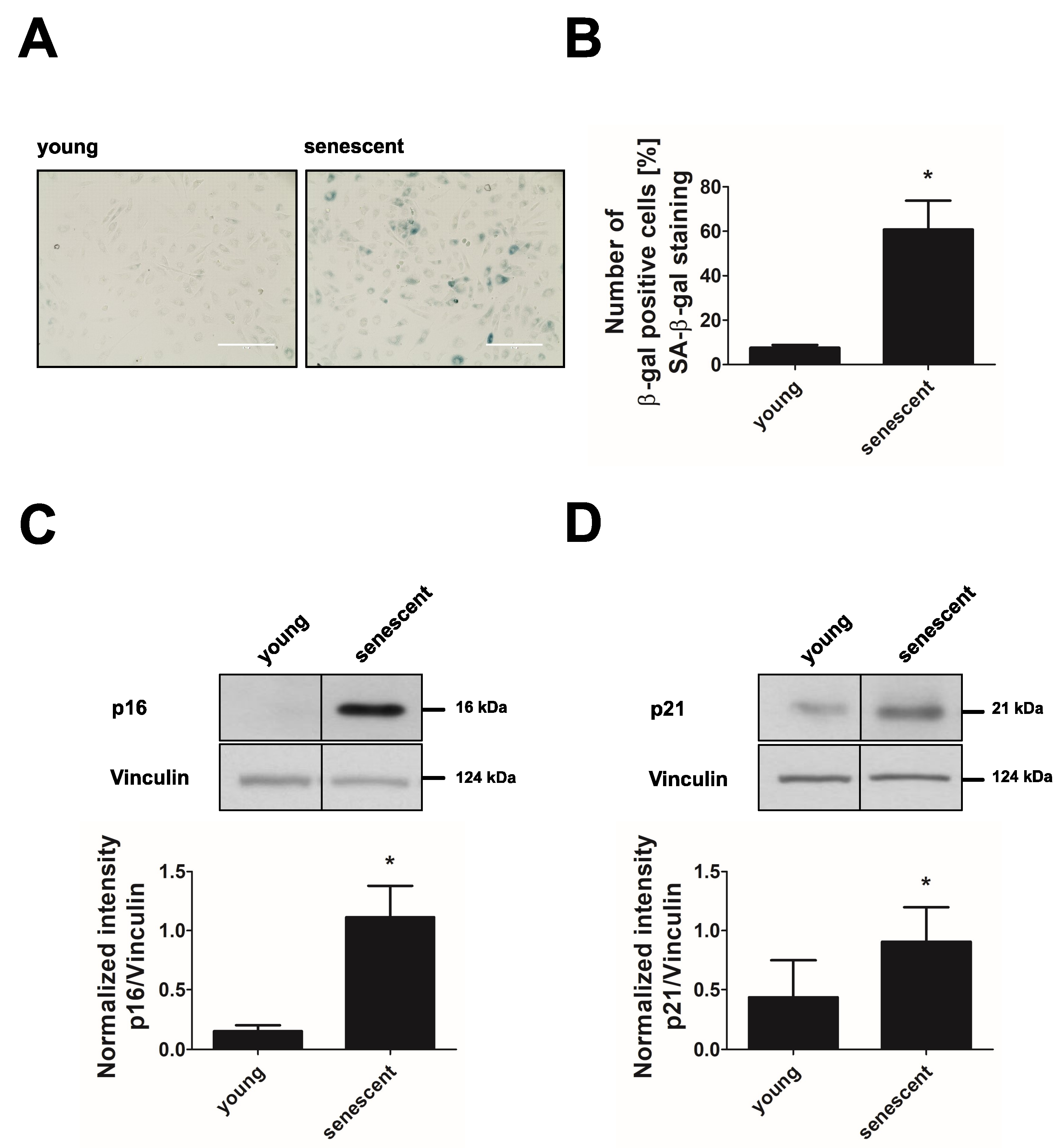
3.1. Long-Term Culture Leads to Replicative Senescence of Endothelial Cells
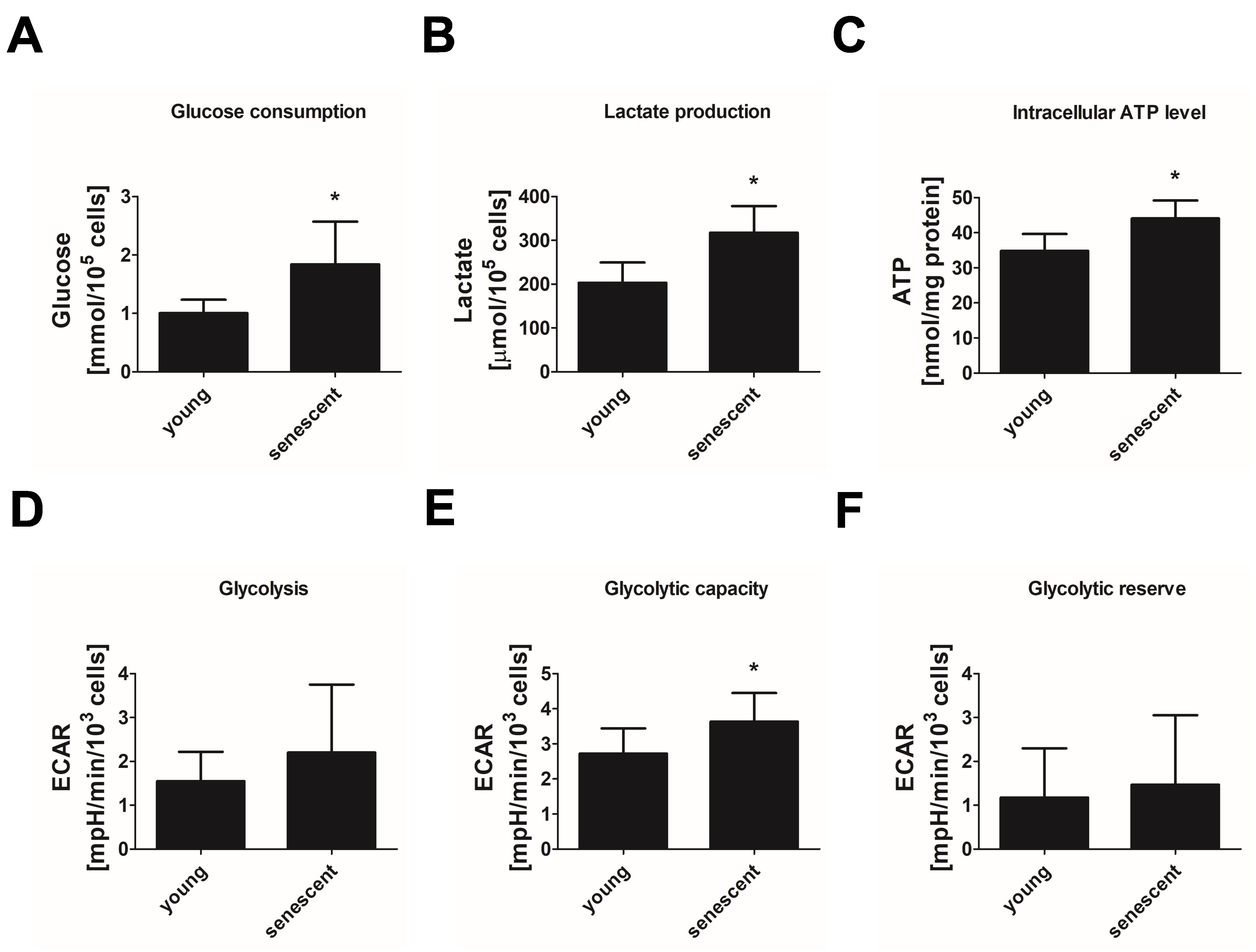
3.2. Replicative Senescent Endothelial Cells Exhibit Increased Glycolytic Activity
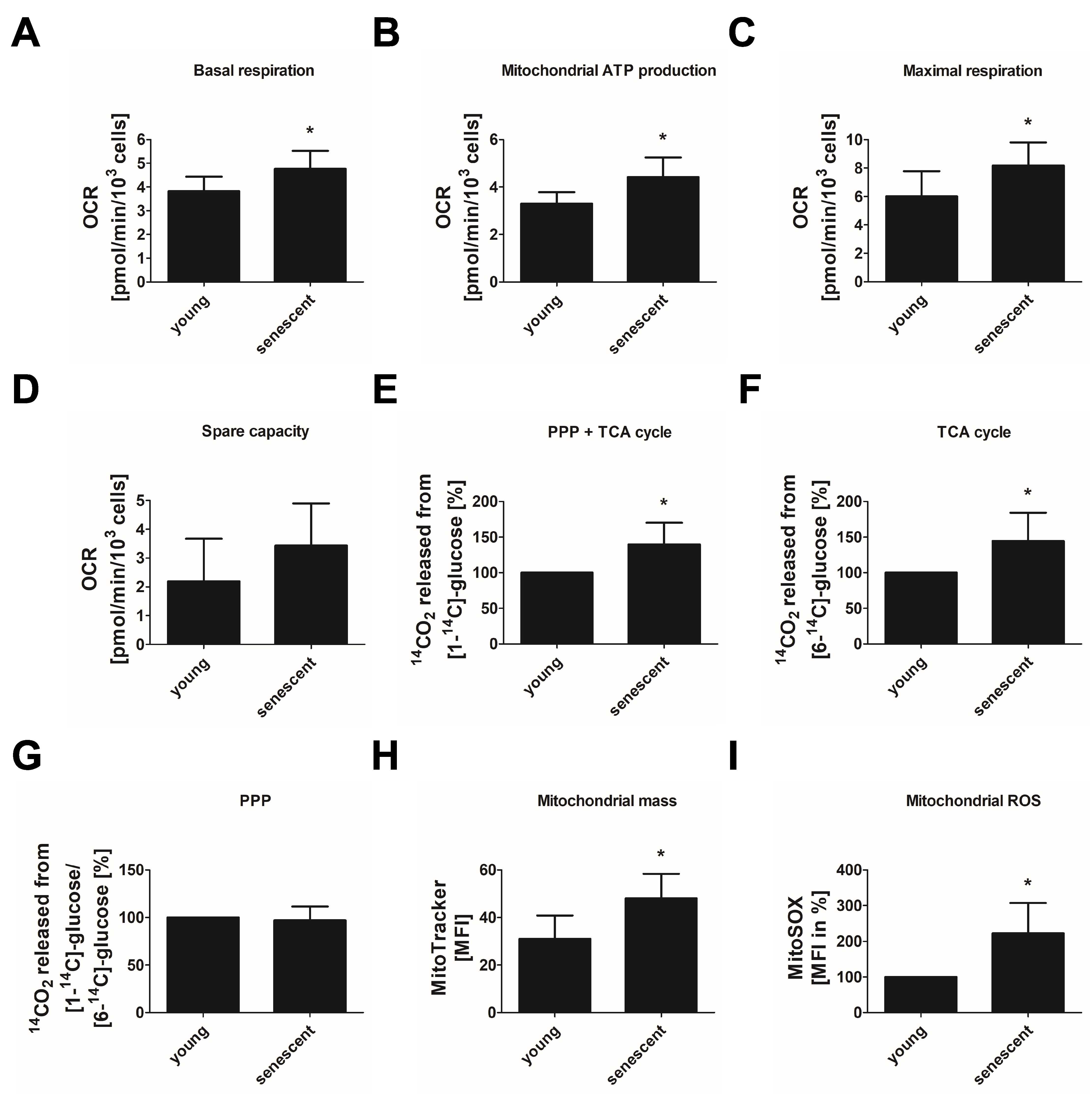
3.3. Replicative Senescent Endothelial Cells Show Increased Oxidative Phosphorylation
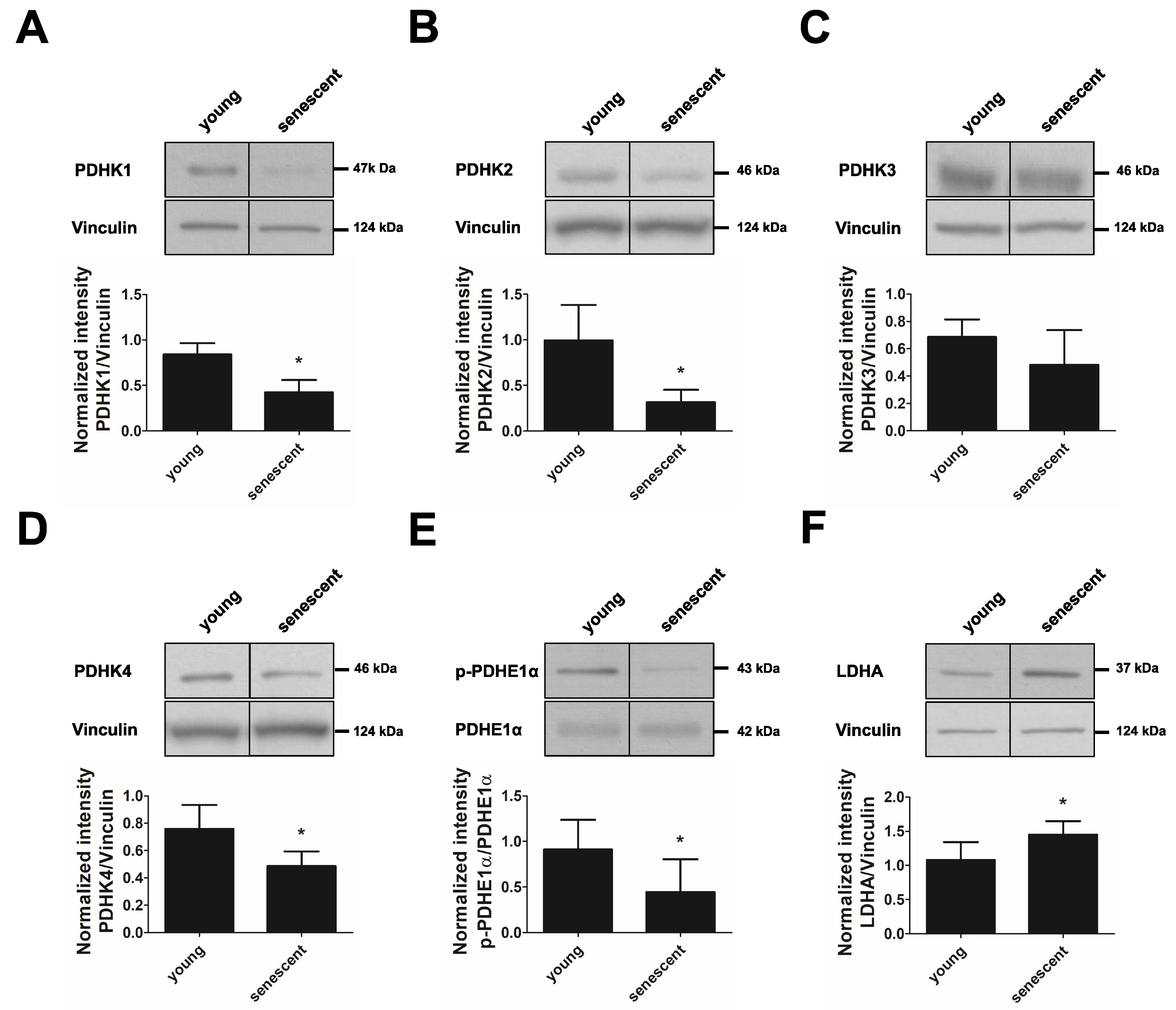
3.4. PDHK Isoforms 1–4 Are Downregulated in Replicative Senescent Endothelial Cells
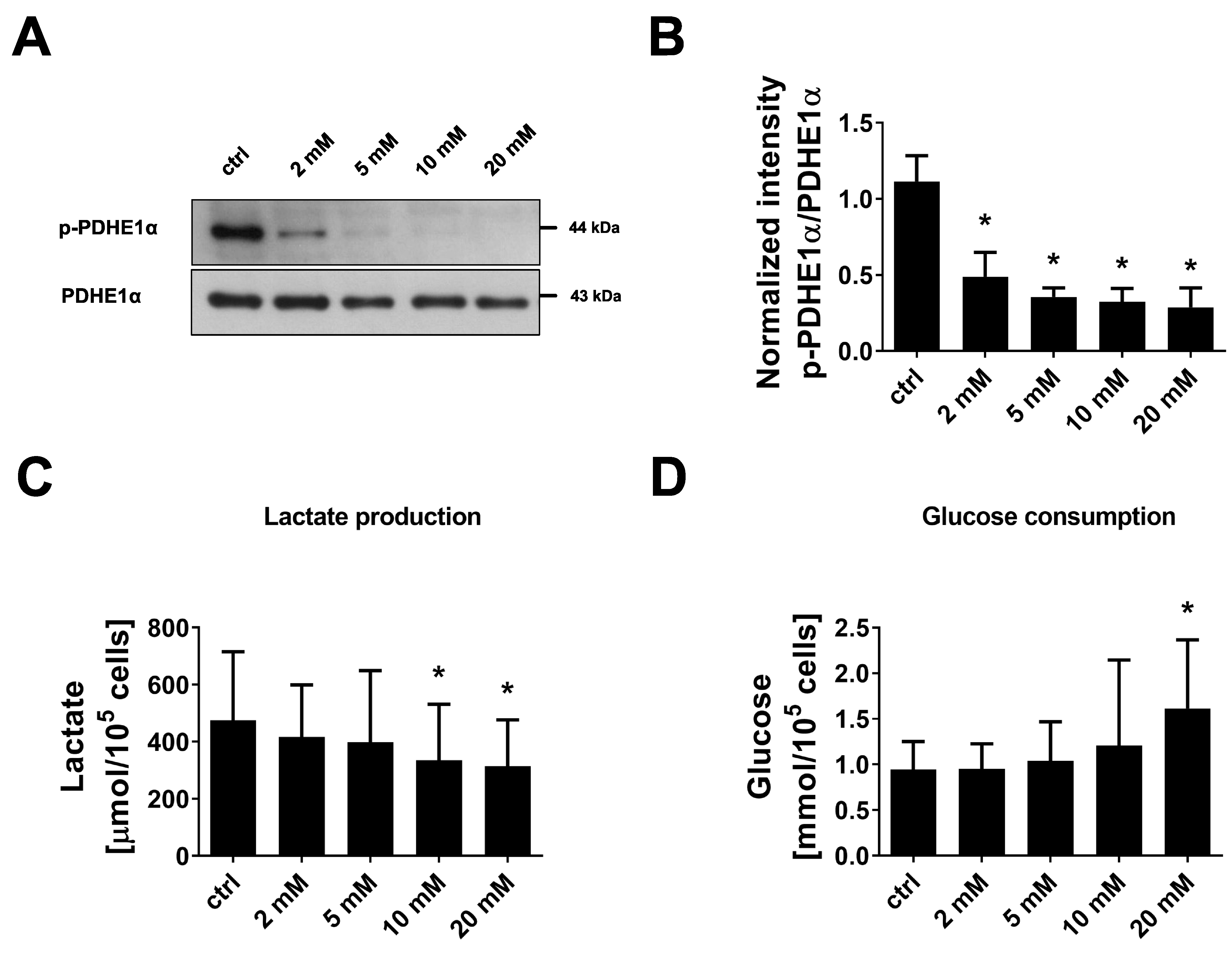
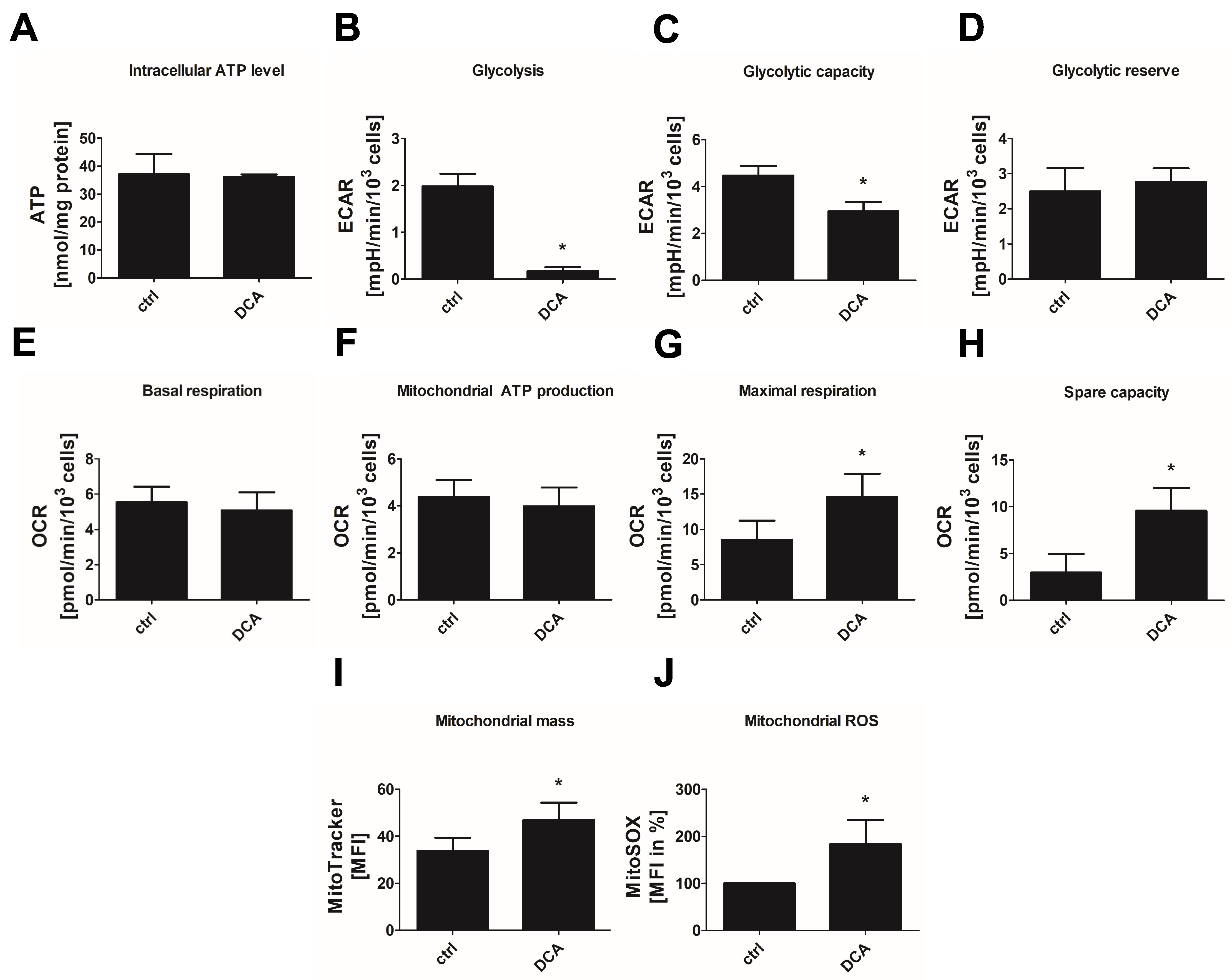
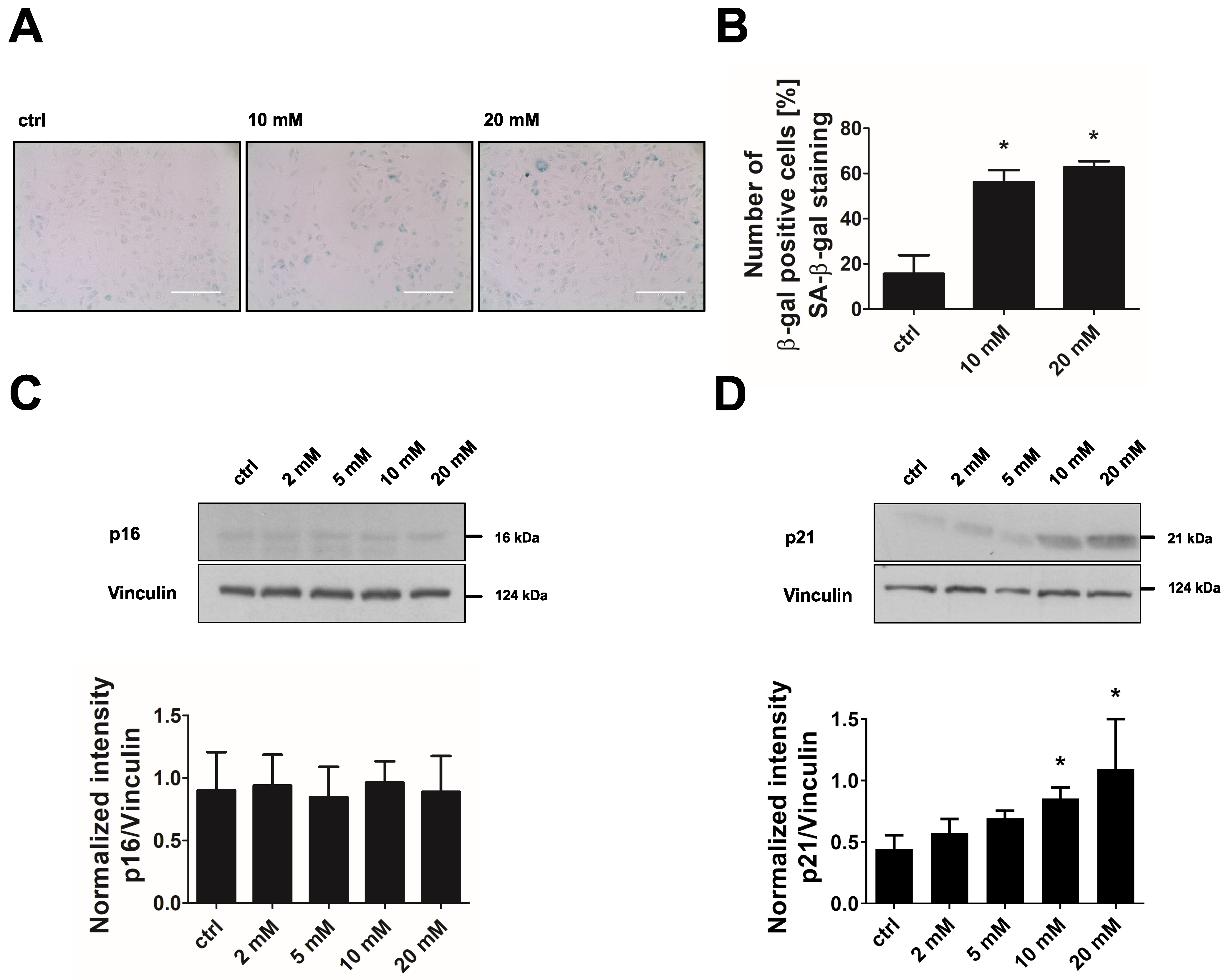
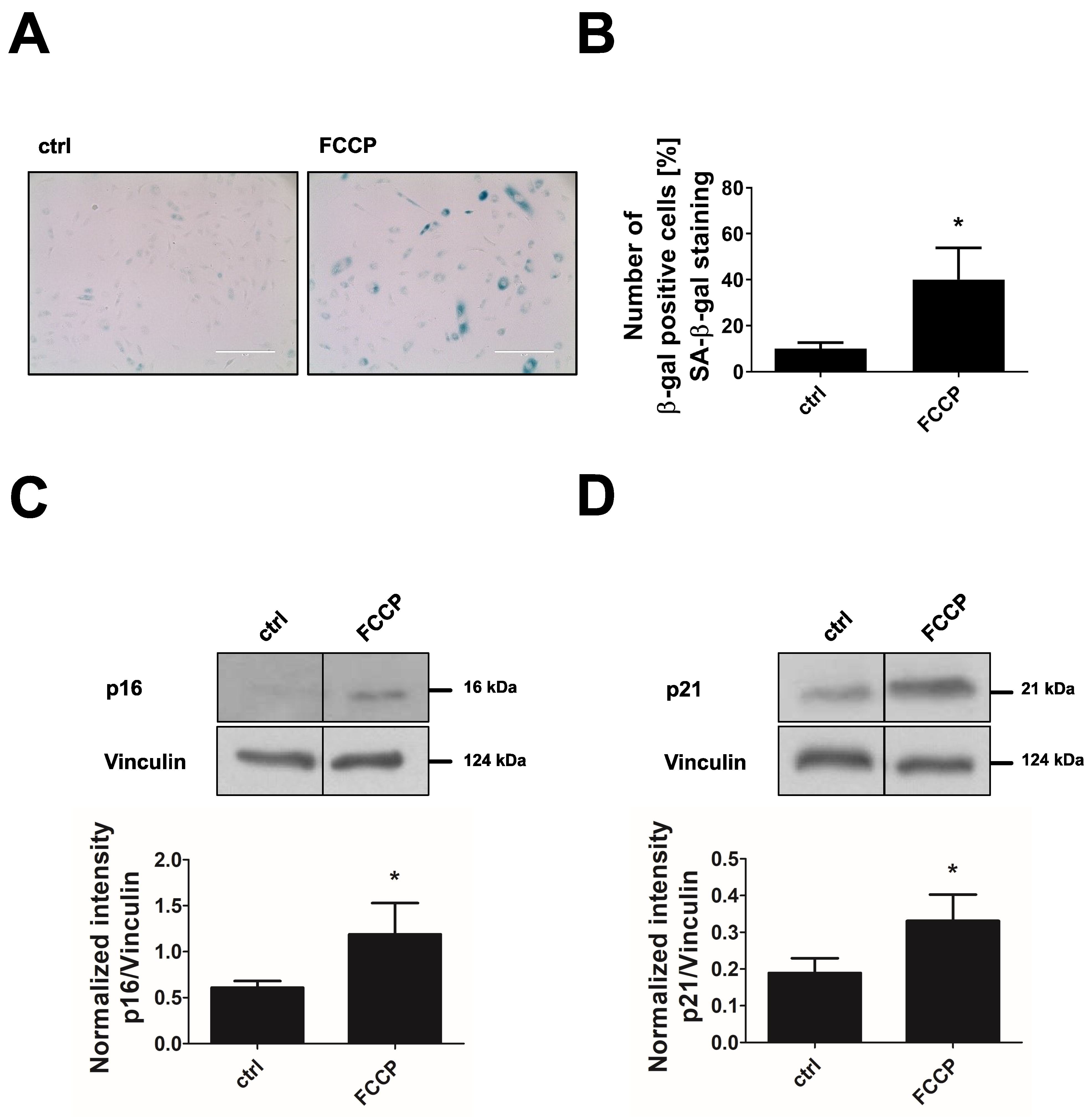
3.5. Inhibition of PDHKs by DCA Enhances Oxidative Glucose Metabolism and Induces Premature Senescence
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Regina, C.; Panatta, E.; Candi, E.; Melino, G.; Amelio, I.; Balistreri, C.R.; Annicchiarico-Petruzzelli, M.; Di Daniele, N.; Ruvolo, G. Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech. Ageing Dev. 2016, 159, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Marchand, A.; Atassi, F.; Gaaya, A.; Leprince, P.; Le Feuvre, C.; Soubrier, F.; Lompre, A.M.; Nadaud, S. The Wnt/beta-catenin pathway is activated during advanced arterial aging in humans. Aging Cell 2011, 10, 220–232. [Google Scholar] [CrossRef]
- Morgan, R.G.; Ives, S.J.; Lesniewski, L.A.; Cawthon, R.M.; Andtbacka, R.H.; Noyes, R.D.; Richardson, R.S.; Donato, A.J. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am. J. Physiol Heart Circ. Physiol 2013, 305, H251–H258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossman, M.J.; Kaplon, R.E.; Hill, S.D.; McNamara, M.N.; Santos-Parker, J.R.; Pierce, G.L.; Seals, D.R.; Donato, A.J. Endothelial cell senescence with aging in healthy humans: Prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H890–H895. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.G.; Ives, S.J.; Walker, A.E.; Cawthon, R.M.; Andtbacka, R.H.; Noyes, D.; Lesniewski, L.A.; Richardson, R.S.; Donato, A.J. Role of arterial telomere dysfunction in hypertension: Relative contributions of telomere shortening and telomere uncapping. J. Hypertens 2014, 32, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Park, J.Y.; Lee, H.; Song, E.S.; Kuk, M.U.; Joo, J.; Oh, S.; Kwon, H.W.; Park, J.T.; Park, S.C. Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence. Cells 2021, 10, 3003. [Google Scholar] [CrossRef]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Shen, X.; Yan, Y.; Li, H. Pyruvate dehydrogenase kinases (PDKs): An overview toward clinical applications. Biosci. Rep. 2021, 4, BSR20204402. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [Green Version]
- Coutelle, O.; Hornig-Do, H.T.; Witt, A.; Andree, M.; Schiffmann, L.M.; Piekarek, M.; Brinkmann, K.; Seeger, J.M.; Liwschitz, M.; Miwa, S.; et al. Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol. Med. 2014, 6, 624–639. [Google Scholar] [CrossRef]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuosmanen, S.M.; Sihvola, V.; Kansanen, E.; Kaikkonen, M.U.; Levonen, A.L. MicroRNAs mediate the senescence-associated decline of NRF2 in endothelial cells. Redox Biol. 2018, 18, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Unterluggauer, H.; Mazurek, S.; Lener, B.; Hutter, E.; Eigenbrodt, E.; Zwerschke, W.; Jansen-Durr, P. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 2008, 9, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, S.; Cooper, R.H.; Randle, P.J. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 1974, 141, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Spengler, K.; Kryeziu, N.; Grosse, S.; Mosig, A.S.; Heller, R. VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKalpha1. Cells 2020, 9, 687. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Chen, S.-H.; Kou, G.H.; Kuo, C.-M. An Enzymatic Microassay for Lactate Concentration in Blood and Hemolymph. Acta Zool. Taiwanica 1999, 10, 91–101. [Google Scholar]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Ballantyne, S.R.; Robson, A.L.; Moerman, E.J. Energy metabolism in cultured human fibroblasts during aging in vitro. J. Cell Physiol. 1982, 112, 419–424. [Google Scholar] [CrossRef]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, K.S.; Mueller, M.M.; Tanneur, V.; Wallisch, S.; Fedorenko, O.; Palmada, M.; Lang, F.; Broer, S.; Heilig, C.W.; Schleicher, E.; et al. Regulation of cytosolic pH and lactic acid release in mesangial cells overexpressing GLUT1. Kidney Int. 2003, 64, 1338–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Dabritz, J.H.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Yin, P.H.; Chi, C.W.; Wei, Y.H. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J. Biomed. Sci. 2002, 9, 517–526. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBiomed. Icine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kato, M.; Chuang, D.T. Pivotal role of the C-terminal DW-motif in mediating inhibition of pyruvate dehydrogenase kinase 2 by dichloroacetate. J. Biol. Chem. 2009, 284, 34458–34467. [Google Scholar] [CrossRef] [Green Version]
- Knoechel, T.R.; Tucker, A.D.; Robinson, C.M.; Phillips, C.; Taylor, W.; Bungay, P.J.; Kasten, S.A.; Roche, T.E.; Brown, D.G. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 2006, 45, 402–415. [Google Scholar] [CrossRef]
- Waleke, J.P.; Diamond, M.P.; Saed, G.M. Dichloroacetate inhibition of angiogenesis caused by hypoxia treatment of normal peritoneal and adhesion fibroblasts in human umbilical vein endothelial cells. Fertil. Steril. 2004, 82, S17–S18. [Google Scholar] [CrossRef]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Agnoletto, C.; Melloni, E.; Casciano, F.; Rigolin, G.M.; Rimondi, E.; Celeghini, C.; Brunelli, L.; Cuneo, A.; Secchiero, P.; Zauli, G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget 2014, 5, 4347–4360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoonjans, C.A.; Mathieu, B.; Joudiou, N.; Zampieri, L.X.; Brusa, D.; Sonveaux, P.; Feron, O.; Gallez, B. Targeting Endothelial Cell Metabolism by Inhibition of Pyruvate Dehydrogenase Kinase and Glutaminase-1. J. Clin. Med. 2020, 9, 3308. [Google Scholar] [CrossRef] [PubMed]
- Harting, T.P.; Stubbendorff, M.; Hammer, S.C.; Schadzek, P.; Ngezahayo, A.; Murua Escobar, H.; Nolte, I. Dichloroacetate affects proliferation but not apoptosis in canine mammary cell lines. PLoS ONE 2017, 12, e0178744. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stabenow, L.K.; Zibrova, D.; Ender, C.; Helbing, D.L.; Spengler, K.; Marx, C.; Wang, Z.-Q.; Heller, R. Oxidative Glucose Metabolism Promotes Senescence in Vascular Endothelial Cells. Cells 2022, 11, 2213. https://doi.org/10.3390/cells11142213
Stabenow LK, Zibrova D, Ender C, Helbing DL, Spengler K, Marx C, Wang Z-Q, Heller R. Oxidative Glucose Metabolism Promotes Senescence in Vascular Endothelial Cells. Cells. 2022; 11(14):2213. https://doi.org/10.3390/cells11142213
Chicago/Turabian StyleStabenow, Leonie K., Darya Zibrova, Claudia Ender, Dario L. Helbing, Katrin Spengler, Christian Marx, Zhao-Qi Wang, and Regine Heller. 2022. "Oxidative Glucose Metabolism Promotes Senescence in Vascular Endothelial Cells" Cells 11, no. 14: 2213. https://doi.org/10.3390/cells11142213