
Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Patients and Method
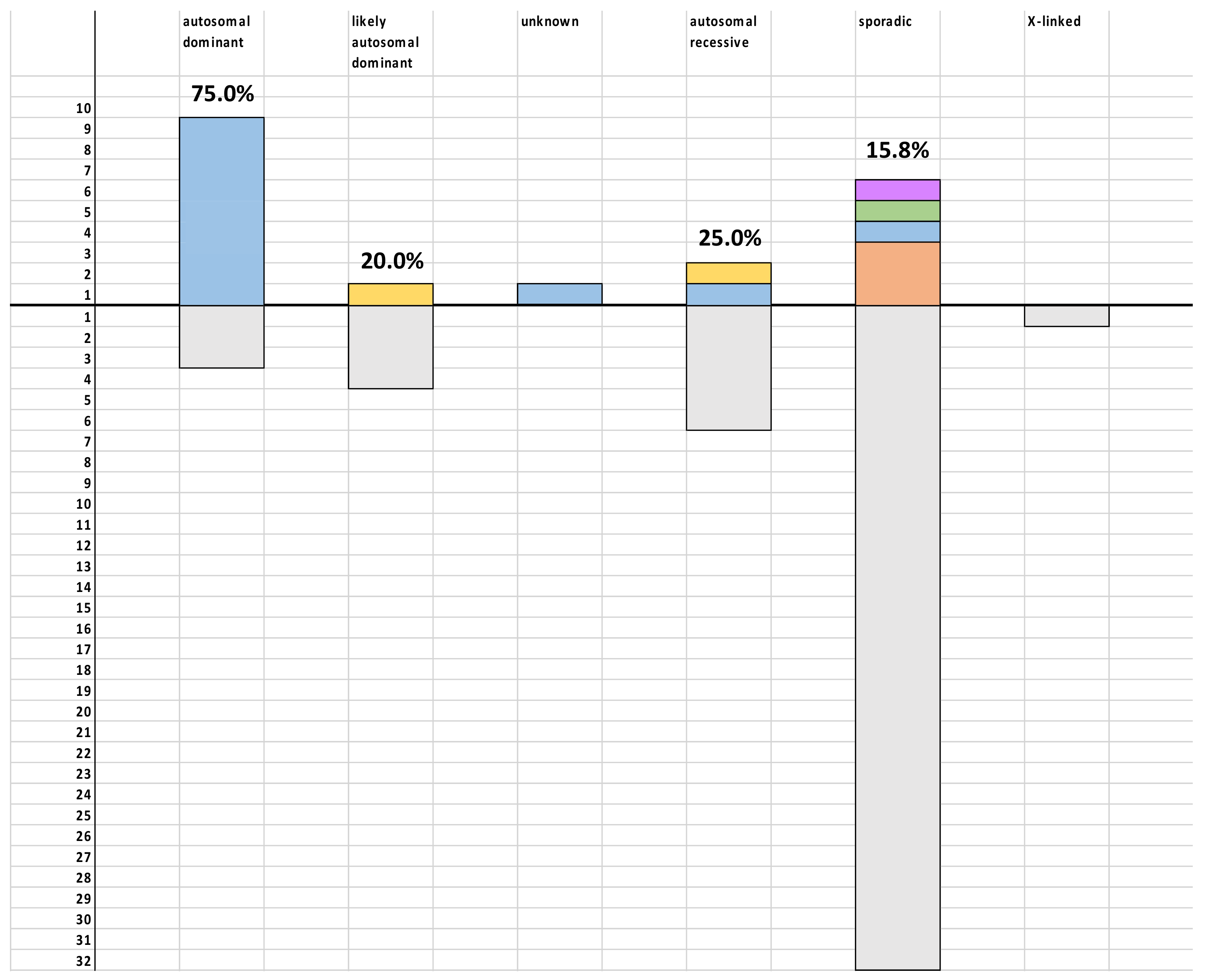
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef]
- Agosta, F.; Scarlato, M.; Spinelli, E.G.; Canu, E.; Benedetti, S.; Bassi, M.T.; Casali, C.; Sessa, M.; Copetti, M.; Pagani, E.; et al. Hereditary Spastic Paraplegia: Beyond Clinical Phenotypes toward a Unified Pattern of Central Nervous System Damage. Radiology 2015, 276, 207–218. [Google Scholar] [CrossRef]
- Meyyazhagan, A.; Kuchi Bhotla, H.; Pappuswamy, M.; Orlacchio, A. The Puzzle of Hereditary Spastic Paraplegia: From Epidemiology to Treatment. Int. J. Mol. Sci. 2022, 23, 7665. [Google Scholar] [CrossRef]
- Darios, F.; Coarelli, G.; Durr, A. Genetics in hereditary spastic paraplegias: Essential but not enough. Curr. Opin. Neurobiol. 2022, 72, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Rydning, S.L.; Wedding, I.M.; Koht, J.; Pihlstrøm, L.; Rengmark, A.H.; Henriksen, S.P.; Tallaksen, C.M.; Toft, M. Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. PLoS ONE 2017, 12, e0174667. [Google Scholar] [CrossRef] [PubMed]
- Méreaux, J.L.; Banneau, G.; Papin, M.; Coarelli, G.; Valter, R.; Raymond, L.; Kol, B.; Ariste, O.; Parodi, L.; Tissier, L.; et al. Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain 2022, 145, 1029–1037. [Google Scholar] [CrossRef]
- Arnoldi, A.; Tonelli, A.; Crippa, F.; Villani, G.; Pacelli, C.; Sironi, M.; Pozzoli, U.; D’Angelo, M.G.; Meola, G.; Martinuzzi, A.; et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum. Mutat. 2008, 29, 522–531. [Google Scholar] [CrossRef]
- Koh, K.; Ishiura, H.; Tsuji, S.; Takiyama, Y. JASPAC: Japan Spastic Paraplegia Research Consortium. Brain Sci. 2018, 8, 153. [Google Scholar] [CrossRef]
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 2014, 261, 518–539. [Google Scholar] [CrossRef]
- Lan, M.Y.; Chang, Y.Y.; Yeh, T.H.; Lai, S.C.; Liou, C.W.; Kuo, H.C.; Wu, Y.R.; Lyu, R.K.; Hung, J.W.; Chang, Y.C.; et al. High frequency of SPG4 in Taiwanese families with autosomal dominant hereditary spastic paraplegia. BMC Neurol. 2014, 14, 216. [Google Scholar] [CrossRef]
- Dong, E.L.; Wang, C.; Wu, S.; Lu, Y.Q.; Lin, X.H.; Su, H.Z.; Zhao, M.; He, J.; Ma, L.X.; Wang, N.; et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 2018, 13, 36. [Google Scholar] [CrossRef]
- Hazan, J.; Fonknechten, N.; Mavel, D.; Paternotte, C.; Samson, D.; Artiguenave, F.; Davoine, C.S.; Cruaud, C.; Dürr, A.; Wincker, P.; et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999, 23, 296–303. [Google Scholar] [CrossRef] [PubMed]
- McDermott, C.J.; Burness, C.E.; Kirby, J.; Cox, L.E.; Rao, D.G.; Hewamadduma, C.; Sharrack, B.; Hadjivassiliou, M.; Chinnery, P.F.; Dalton, A. UK and Irish HSP Consortium: Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology 2006, 67, 45–51. [Google Scholar] [CrossRef]
- Orlacchio, A.; Patrono, C.; Gaudiello, F.; Rocchi, C.; Moschella, V.; Floris, R.; Bernardi, G.; Kawarai, T. Silver syndrome variant of hereditary spastic paraplegia: A locus to 4p and allelism with SPG4. Neurology 2008, 70, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- De Souza, P.V.; Bortholin, T.; Naylor, F.G.; de Rezende Pinto, W.B.; Oliveira, A.S. Infantile-onset ascending spastic paraplegia phenotype associated with SPAST mutation. J. Neurol. Sci. 2016, 371, 34–35. [Google Scholar] [CrossRef]
- Klebe, S.; Stevanin, G.; Depienne, C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015, 171, 505–530. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Johnsen, B.; Koefoed, P.; Scheuer, K.H.; Grønbech-Jensen, M.; Law, I.; Krabbe, K.; Nørremølle, A.; Eiberg, H.; Søndergård, H.; et al. Hereditary spastic paraplegia with cerebellar ataxia: A complex phenotype associated with a new SPG4 gene mutation. Eur. J. Neurol. 2004, 11, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Kawarai, T.; Totaro, A.; Errico, A.; St George-Hyslop, P.H.; Rugarli, E.I.; Bernardi, G. Hereditary spastic paraplegia: Clinical genetic study of 15 families. Arch. Neurol. 2004, 61, 849–855. [Google Scholar] [CrossRef]
- Ribaï, P.; Depienne, C.; Fedirko, E.; Jothy, A.C.; Viveweger, C.; Hahn-Barma, V.; Brice, A.; Durr, A. Mental deficiency in three families with SPG4 spastic paraplegia. Eur. J. Hum. Genet. 2008, 16, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, L.; Fenu, S.; Stevanin, G.; Durr, A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev. Neurol. 2017, 173, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Lindig, T.; Bender, B.; Hauser, T.K.; Mang, S.; Schweikardt, D.; Klose, U.; Karle, K.N.; Schüle, R.; Schöls, L.; Rattay, T.W. Gray and white matter alterations in hereditary spastic paraplegia type SPG4 and clinical correlations. J. Neurol. 2015, 262, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016, 139 Pt 7, 1904–1918. [Google Scholar] [CrossRef]
- Rubegni, A.; Storti, E.; Tessa, A.; Federico, A.; Santorelli, F.M. Hereditary spastic paraplegia type 11 with a very late onset. J. Neurol. 2015, 262, 1987–1989. [Google Scholar] [CrossRef]
- Kawarai, T.; Miyamoto, R.; Mori, A.; Oki, R.; Tsukamoto-Miyashiro, A.; Matsui, N.; Miyazaki, Y.; Orlacchio, A.; Izumi, Y.; Nishida, Y.; et al. Late-onset spastic paraplegia: Aberrant SPG11 transcripts generated by a novel splice site donor mutation. J. Neurol. Sci. 2015, 359, 250–255. [Google Scholar] [CrossRef]
- Doleckova, K.; Roth, J.; Stellmachova, J.; Gescheidt, T.; Sigut, V.; Houska, P.; Jech, R.; Zech, M.; Vyhnalek, M.; Vyhnalkova, E.; et al. SPG11: Clinical and genetic features of seven Czech patients and literature review. Neurol. Res. 2022, 44, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Di Fonzo, A.; Ghezzi, S.; Locatelli, F.; Stevanin, G.; Costa, A.; Corti, S.; Bresolin, N.; Comi, G.P. SPG11: A consistent clinical phenotype in a family with homozygous spatacsin truncating mutation. Neurogenetics 2007, 8, 301–305. [Google Scholar] [CrossRef]
- Pippucci, T.; Panza, E.; Pompilii, E.; Donadio, V.; Borreca, A.; Babalini, C.; Patrono, C.; Zuntini, R.; Kawarai, T.; Bernardi, G.; et al. Autosomal recessive hereditary spastic paraplegia with thin corpus callosum: A novel mutation in the SPG11 gene and further evidence for genetic heterogeneity. Eur. J. Neurol. 2009, 16, 121–126. [Google Scholar] [CrossRef]
- Chrestian, N.; Dupré, N.; Gan-Or, Z.; Szuto, A.; Chen, S.; Venkitachalam, A.; Brisson, J.D.; Warman-Chardon, J.; Ahmed, S.; Ashtiani, S.; et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 2016, 3, e122. [Google Scholar] [CrossRef]
- Wagner, F.; Titelbaum, D.S.; Engisch, R.; Coskun, E.K.; Waugh, J.L. Subtle Imaging Findings Aid the Diagnosis of Adolescent Hereditary Spastic Paraplegia and Ataxia. Clin. Neuroradiol. 2019, 29, 215–221. [Google Scholar] [CrossRef]
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; Rosa, A.L.; Lerer, I.; Hamri, A.; Alegria, P.; et al. SPATAX consortium. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008, 131 Pt 3, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135 Pt 10, 2980–2993. [Google Scholar] [CrossRef]
- Yahikozawa, H.; Yoshida, K.; Sato, S.; Hanyu, N.; Doi, H.; Miyatake, S.; Matsumoto, N. Predominant cerebellar phenotype in spastic paraplegia 7 (SPG7). Hum. Genome Var. 2015, 2, 15012. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, K.L.; van der Heijden, C.D.; de Bot, S.T.; den Dunnen, W.F.; van den Berg, L.H.; Verschuuren-Bemelmans, C.C.; Kremer, H.P.; Veldink, J.H.; Kamsteeg, E.J.; Scheffer, H.; et al. Genotype-phenotype correlations in spastic paraplegia type 7: A study in a large Dutch cohort. Brain 2012, 135 Pt 10, 2994–3004. [Google Scholar] [CrossRef]
- Sánchez-Ferrero, E.; Coto, E.; Beetz, C.; Gámez, J.; Corao, A.I.; Díaz, M.; Esteban, J.; del Castillo, E.; Moris, G.; Infante, J.; et al. SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V. Clin. Genet. 2013, 83, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Petrucci, L.; Ienco, E.C.; Chico, L.; Simi, P.; Fogli, A.; Baldinotti, F.; Simoncini, C.; LoGerfo, A.; Carlesi, C.; et al. Hereditary spastic paraparesis in adults. A clinical and genetic perspective from Tuscany. Clin. Neurol. Neurosurg. 2014, 120, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137 Pt 5, 1323–1336. [Google Scholar] [CrossRef]
- Goizet, C.; Boukhris, A.; Mundwiller, E.; Tallaksen, C.; Forlani, S.; Toutain, A.; Carriere, N.; Paquis, V.; Depienne, C.; Durr, A.; et al. Complicated forms of autosomal dominant hereditary spastic paraplegia are frequent in SPG10. Hum. Mutat. 2009, 30, E376–E385. [Google Scholar] [CrossRef]
- Crimella, C.; Baschirotto, C.; Arnoldi, A.; Tonelli, A.; Tenderini, E.; Airoldi, G.; Martinuzzi, A.; Trabacca, A.; Losito, L.; Scarlato, M.; et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 2012, 82, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P. Hereditary Spastic Paraplegia Overview. 2000 August 15 [Updated 2021 Feb 11]. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallance, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1509/ (accessed on 6 September 2022).


{kind=link}
| Family ID | Patient ID | Presumed Inheritance | Disease Subtype | Gene Name | Transcript | Variant | Zygosity | HGMD Identifier # | gnomAD Frequency | ACMG Pathogenicity Class | ACMG Pathogenicity Evidence |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HSP24 | AD | SPG4 | SPAST | NM_014946.3 | c.1245+1delG | het | - | 0 | Likely pathogenic (novel) | PM2, PM5, PP1, PP3, PP4 |
| HSP94 | AD | SPG4 | SPAST | NM_014946.3 | c.1245+1delG | het | |||||
| 2 | HSP32 | AD | SPG4 | SPAST | NM_014946.3 | deletion of exon 15 and 16 | het | CG1415382 | - | Pathogenic (recurrent) | |
| 3 | HSP33 | AD | SPG4 | SPAST | NM_014946.3 | c.425delA (p.Lys142Argfs*19) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 4 | HSP42 | AD | SPG4 | SPAST | NM_014946.3 | c.1069_1070insA (p.I357Nfs*10) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 5 | HSP4 | AR | SPG4 | SPAST | NM_014946.3 | c.1351A>G (p.R451G) | het | - | 0 | Likely pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 6 | HSP68 | sporadic | SPG4 | SPAST | NM_014946.3 | deletion exons 8–17 and further downstream | het | CG1415377 | - | Pathogenic (recurrent) | |
| 7 | HSP52 | AD | SPG4 | SPAST | NM_014946.3 | c.820_827del (p.M274Wfs*14) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 8 | HSP80 | AD | SPG4 | SPAST | NM_014946.3 | c.1494G>T (p.R498S) | het | CM1512265 | 0 | Pathogenic (recurrent) | |
| HSP81 | AD | SPG4 | SPAST | NM_014946.3 | c.1494G>T (p.R498S) | het | |||||
| 9 | HSP114 | unknown (adopted) | SPG4 | SPAST | NM_014946.3 | c.308C>A (p.S103*) | het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1 |
| 10 | HSP9 | AD | SPG4 | SPAST | NM_014946.3 | c.1495C>T (p.R499C) | het | CM992676 | 0 | Pathogenic (recurrent) | |
| HSP10 | AD | SPG4 | SPAST | NM_014946.3 | c.1495C>T (p.R499C) | het | |||||
| 11 | HSP11 | AD | SPG4 | SPAST | NM_014946.3 | c.1672_1673del (p.L558Gfs*18) | het | CD021858 | 0 | Pathogenic (recurrent) | |
| HSP12 | AD | SPG4 | SPAST | NM_014946.3 | c.1672_1673del (p.L558Gfs*18) | het | |||||
| 12 | HSP112 | AD | SPG4 | SPAST | NM_014946.3 | deletion of 5’UTR-ex1 | het | CG052756 | - | Pathogenic (recurrent) | |
| 13 | HSP118 | AR | SPG7 | SPG7 | NM_003119.3 | c.233T>A (p.L78*) | hom | CM081826 | 0 | Pathogenic (recurrent) | |
| 14 | HSP119 | likely AD | SPG7 | SPG7 | NM_003119.3 | c.233T>A (p.L78*) | hom | CM081826 | 0 | Pathogenic (recurrent) | |
| 15 | HSP113 | sporadic | SPG11 | SPG11 | NM_025137.4 | c.5381T>C (p.L1794P) | hom | CM166061 | 0 | Pathogenic (recurrent) | |
| 16 | HSP128 | sporadic | SPG11 | SPG11 | NM_025137.4 | c.5381T>C (p.L1794P) | comp het | CM166061 | 0 | Pathogenic (recurrent) | |
| duplication spanning intron27-ex29 | CN166911 | 0 | Pathogenic (recurrent) | ||||||||
| 17 | HSP35 | sporadic | SPG11 | SPG11 | NM_025137.4 | duplication spanning intron27-ex29 | hom | CN166911 | - | Pathogenic (recurrent) | |
| 18 | HSP34_HSP69 | sporadic | SPG10 | KIF5A | NM_004984.4 | c.746T>C (p.Leu249Pro) | het | - | 0 | Likely pathogenic (novel) | PM2, PM5, PP1, PP3, PP4 |
| 19 | DIST690 | sporadic | SPG15 | ZFYVE26 | NM_015346.4 | c.2114dupC (p.E706*) | comp het | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1, PP3, PP4, PP5 |
| c.2357delC (p.P786Hfs*10) | - | 0 | Pathogenic (novel) | PVS1, PM2, PM4, PP1, PP3, PP4 |
| Features | Number of Patients |
|---|---|
| % females | 50% |
| Age at onset | <1–6.2% 6–10–18.8% 11–20–6.2% 21–40–31.2% 41–60–31.2% >60–6.2% |
| Age | 42.9 ± 11. |
| Mobility | normal—6.2% abnormal, but no aids—93.8% |
| Lower limbs | hyperreflexia—31.2% hyperreflexia, weakness—56.2% hyperreflexia, weakness, distal muscle atrophy—12.5% |
| Upper limbs | normal—18.8% hyperreflexia—75.0% hyperreflexia, distal muscle weakness—6.2% |
| Sphincters | normal—50.0% bladder impairment—12.5% bladder and bowel impairment—25.0% |
| Sensibility | normal—56.2% vibration impaired in lower limbs—37.5% vibration and touch impaired in lower limbs—6.2% |
| Additional features | 56.2% |
| Patient ID | HSP113 | HSP128 | HSP35 |
|---|---|---|---|
| Gender | female | male | male |
| Age at onset | 41–60 | 41–60 | 21–40 |
| Age | 52 | 53 | 25 |
| Mobility | abnormal, but no aids | abnormal, but no aids | abnormal, but no aids |
| LL | hyperreflexia, weakness | asymmetric hyperreflexia | hyperreflexia |
| UL | normal | normal | hyperreflexia |
| Sphincter dysfunction | no | no | no |
| Sensibility impairment | normal | normal | normal |
| Additional features | foot dystonia | no | mild mental retardation (IQ 78), mild dysarthria, mild postural tremor |
| NCS | normal | bilateral carpal tunnel syndrome | normal |
| SSEP | not done | not done | abnormal |
| Brain MRI | normal | temporal arachnoid cyst | thin corpus callosum, mild periventricular WMHLs |
| Spine MRI | normal | normal | normal |
| Serbian p.L78* Patients | Italian p.L78* Patients | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HSP118 | HSP119 | P1-EM9-06 | P2-EM18-08 | ||||||
| Marker | Position (Mb) | ||||||||
| D16S3123 | 87.6500 | 99 | 101 | 101 | 109 | 101 | 109 | 101 | 109 |
| rs8191483 | 88.8770 | C | C | C | C | C | C | C | C |
| D16S3026 | 89.4930 | 202 | 202 | 202 | 202 | 202 | 202 | 202 | 202 |
| D16S3121 | 89.4985 | 73 | 73 | 73 | 73 | 73 | 73 | 73 | 73 |
| p.L78* | 89.5769 | A | A | A | A | A | A | A | A |
| rs12960 | 89.6203 | G | G | G | G | G | G | G | G |
| rs174035 | 89.6277 | C | C | C | C | C | C | C | C |
| rs455527 | 89.6440 | T | T | T | T | T | T | T | T |
| rs352935 | 89.6486 | C | C | C | C | C | C | C | C |
| rs2162943 | 89.7607 | C | C | C | C | C | C | C | C |
| Patient ID | HSP118 | HSP119 |
|---|---|---|
| Gender | male | male |
| Age at onset | 41–60 | 41–60 |
| Age | 54 | 57 |
| Mobility | unilateral support | abnormal, but no aids |
| LL | hyperreflexia, weakness | hyperreflexia, weakness |
| UL | hyperreflexia | hyperreflexia |
| Sphincter dysfunction | no | bladder |
| Sensibility impairment | no | vibration in LL |
| Additional features | postural tremor | torticollis |
| NCS | not done | normal |
| SSEP | not done | not done |
| Brain MRI | one small supratentorial WMHLs | severe cortical and cerebellar atrophy |
| Spine MRI | normal | normal |
| Patient ID | HSP34 | DIST690 |
|---|---|---|
| HSP type | SPG10 | SPG15 |
| Gender | female | female |
| Age at onset | 6–10 | 11–20 |
| Age | 20 | 23 |
| Mobility | abnormal, but no aids | abnormal, but no aids |
| LL | hyperreflexia, weakness | hyperreflexia, weakness |
| UL | hyperreflexia | normal |
| Sphincter dysfunction | bladder | no |
| Sensibility impairment | vibration and touch in LL | normal |
| Additional features | foot deformities, mild static hand tremor | foot dystonia, mild dysarthria, mild limb ataxia and tremor |
| NCS | predominantly motor, axonal polyneuropathy | normal |
| SSEP | abnormal | normal |
| Brain MRI | normal | normal |
| Spine MRI | normal | normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perić, S.; Marković, V.; Candayan, A.; De Vriendt, E.; Momčilović, N.; Savić, A.; Dragašević-Mišković, N.; Svetel, M.; Stević, Z.; Božović, I.; et al. Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia. Cells 2022, 11, 2804. https://doi.org/10.3390/cells11182804
Perić S, Marković V, Candayan A, De Vriendt E, Momčilović N, Savić A, Dragašević-Mišković N, Svetel M, Stević Z, Božović I, et al. Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia. Cells. 2022; 11(18):2804. https://doi.org/10.3390/cells11182804
Chicago/Turabian StylePerić, Stojan, Vladana Marković, Ayşe Candayan, Els De Vriendt, Nikola Momčilović, Andrija Savić, Nataša Dragašević-Mišković, Marina Svetel, Zorica Stević, Ivo Božović, and et al. 2022. "Phenotypic and Genetic Heterogeneity of Adult Patients with Hereditary Spastic Paraplegia from Serbia" Cells 11, no. 18: 2804. https://doi.org/10.3390/cells11182804