
CKII Control of Axonal Plasticity Is Mediated by Mitochondrial Ca2+ via Mitochondrial NCLX
 ,
,
Abstract
:1. Introduction
2. Results
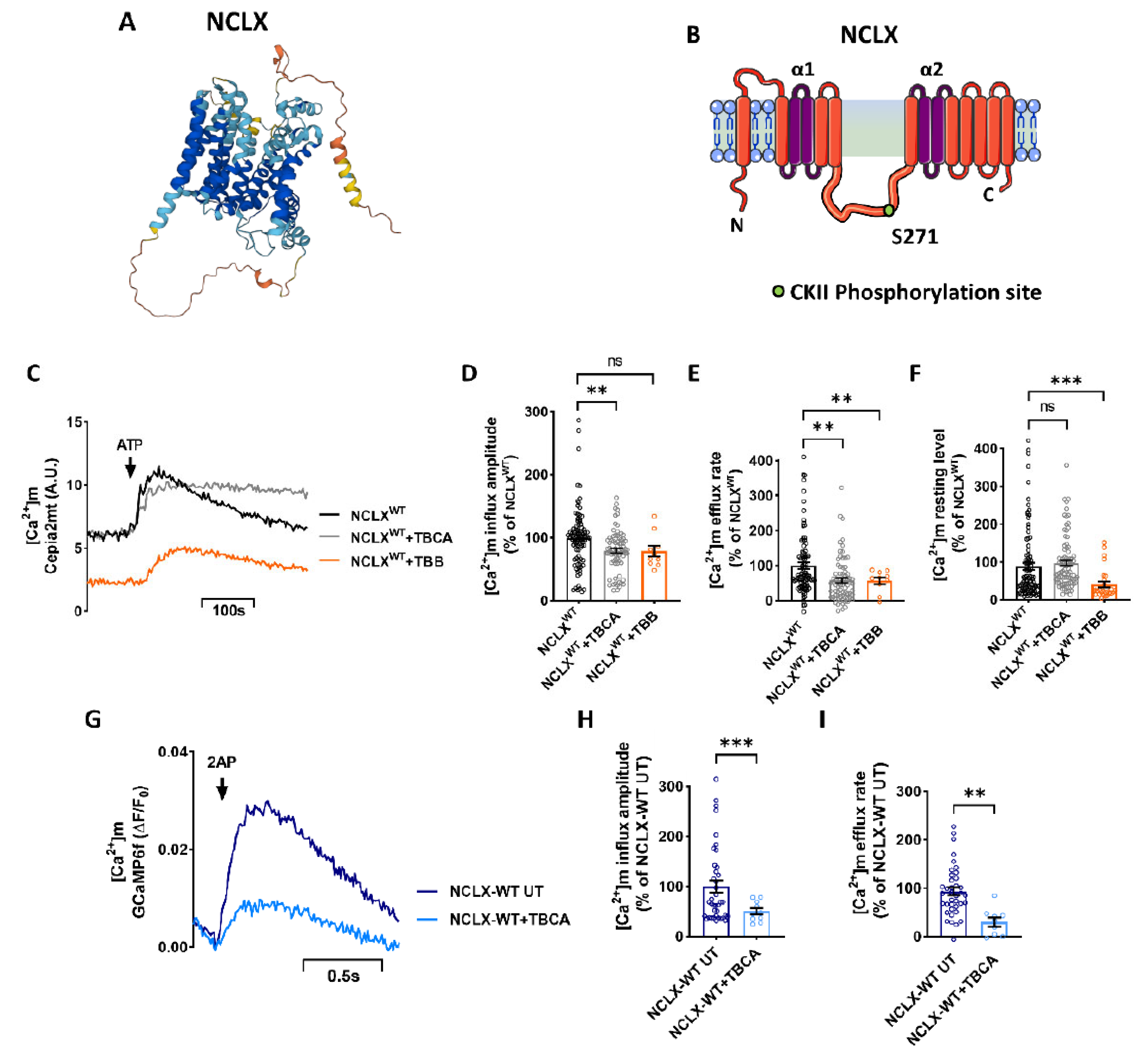
2.1. Inhibition of CKII by TBB and TBCA Modulates the [Ca2+]m Transient
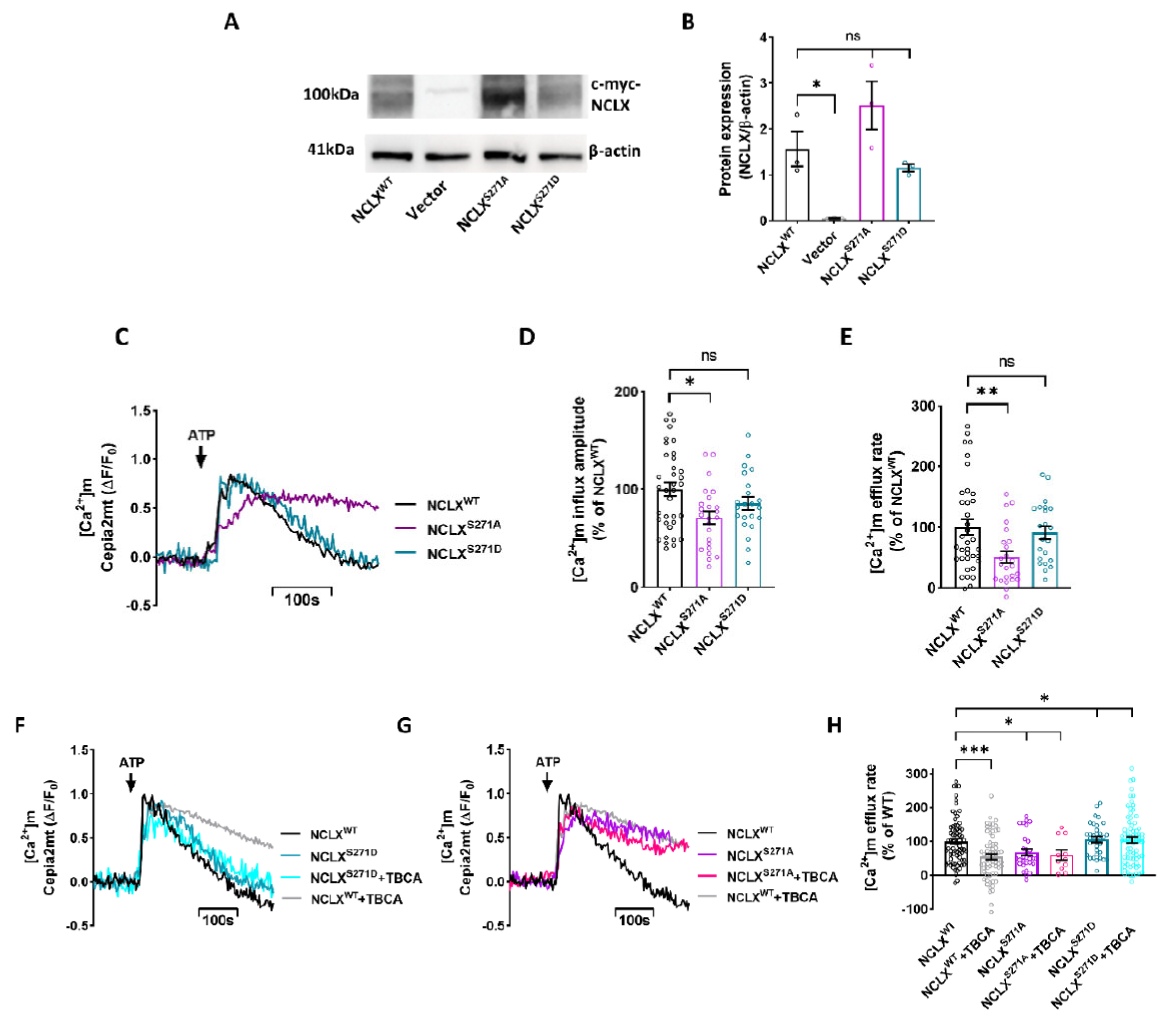
2.2. NCLX S271 Mutants Mimic a Phosphorylated State of the CKII Site
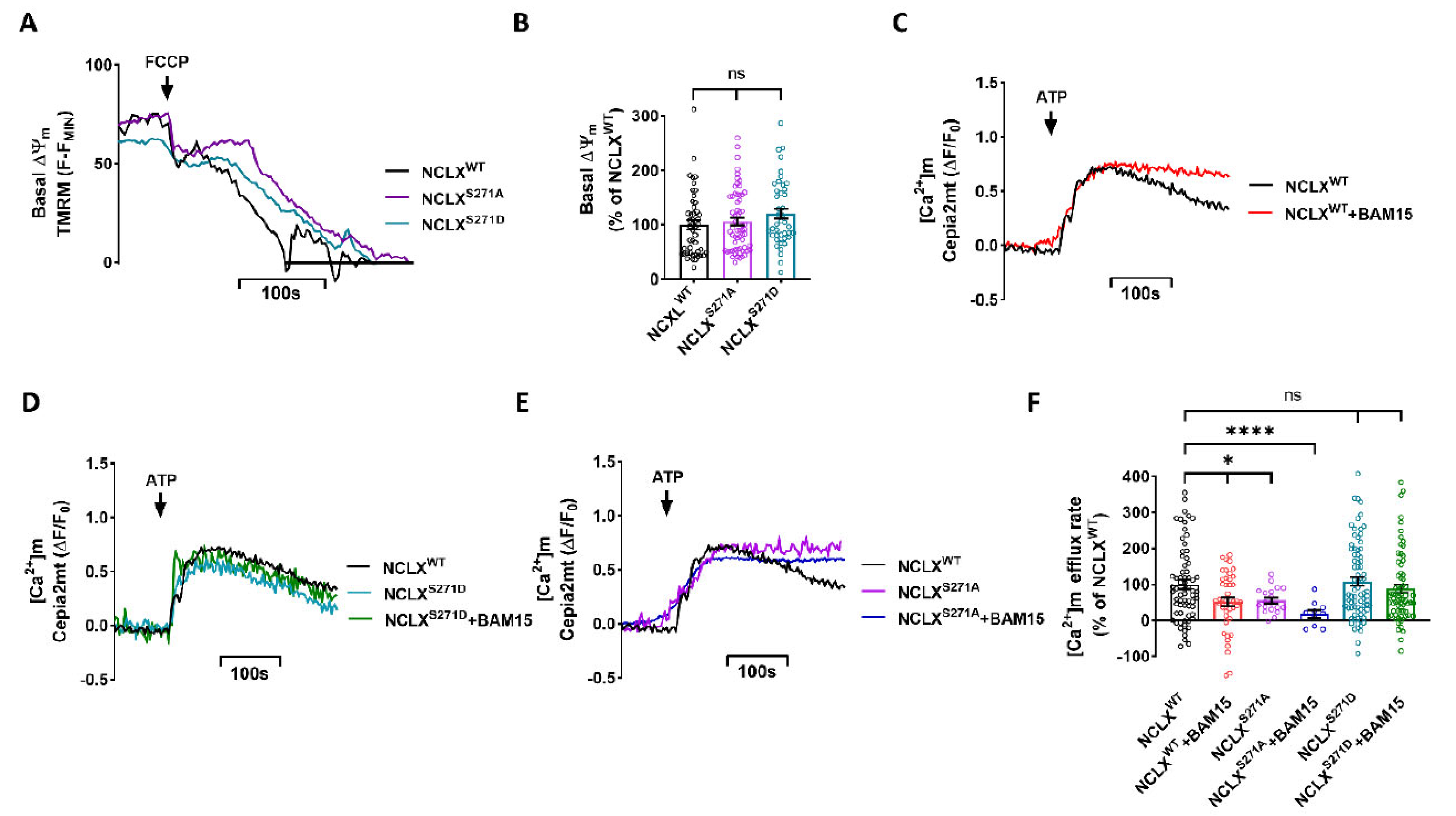
2.3. Role of NCLX CKII Regulatory Site in Controlling the Allosteric Regulation of NCLX by ΔΨm
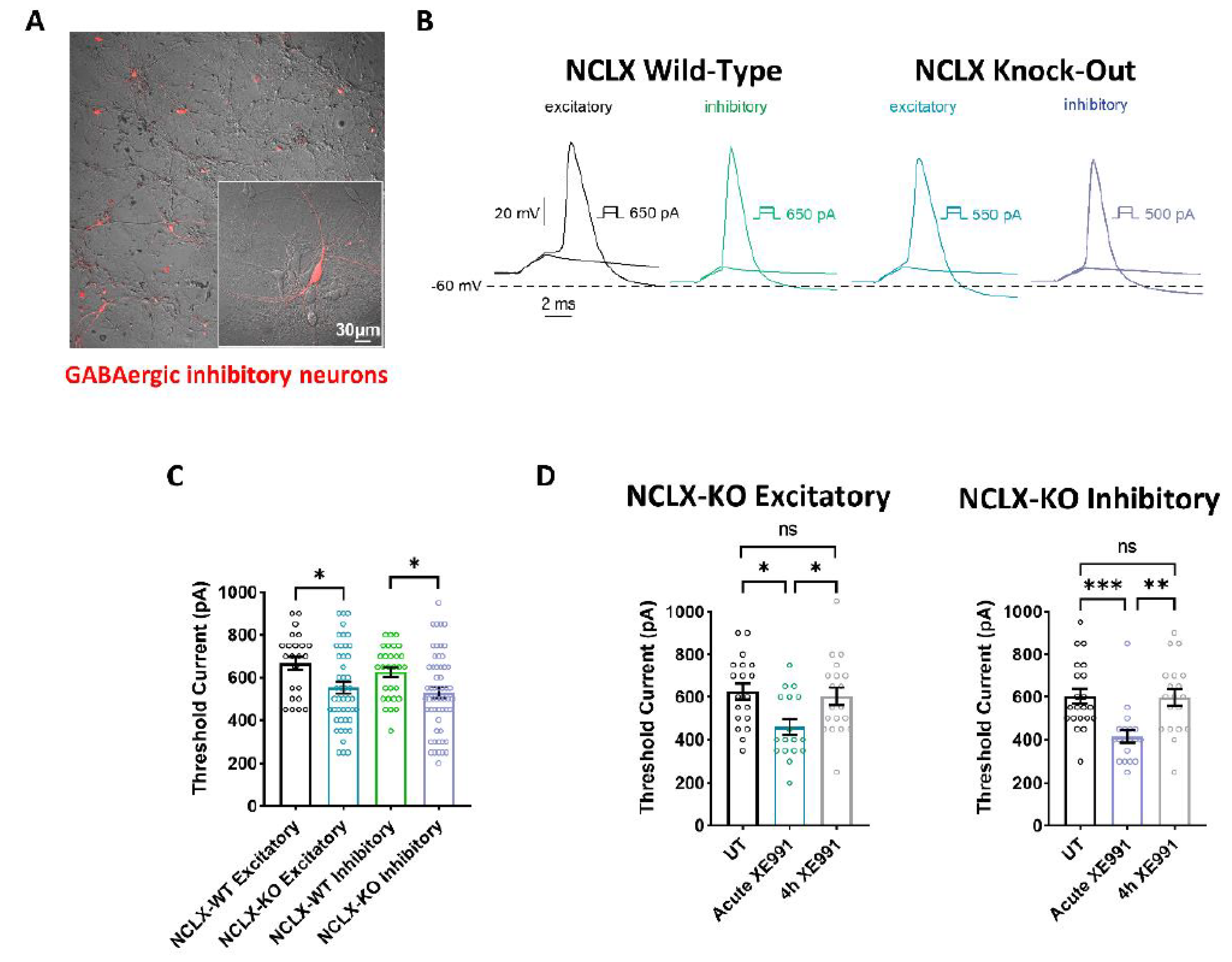
2.4. Intrinsic Excitability Properties of Hippocampal Neurons Derived from WT and NCLX-KO Mice
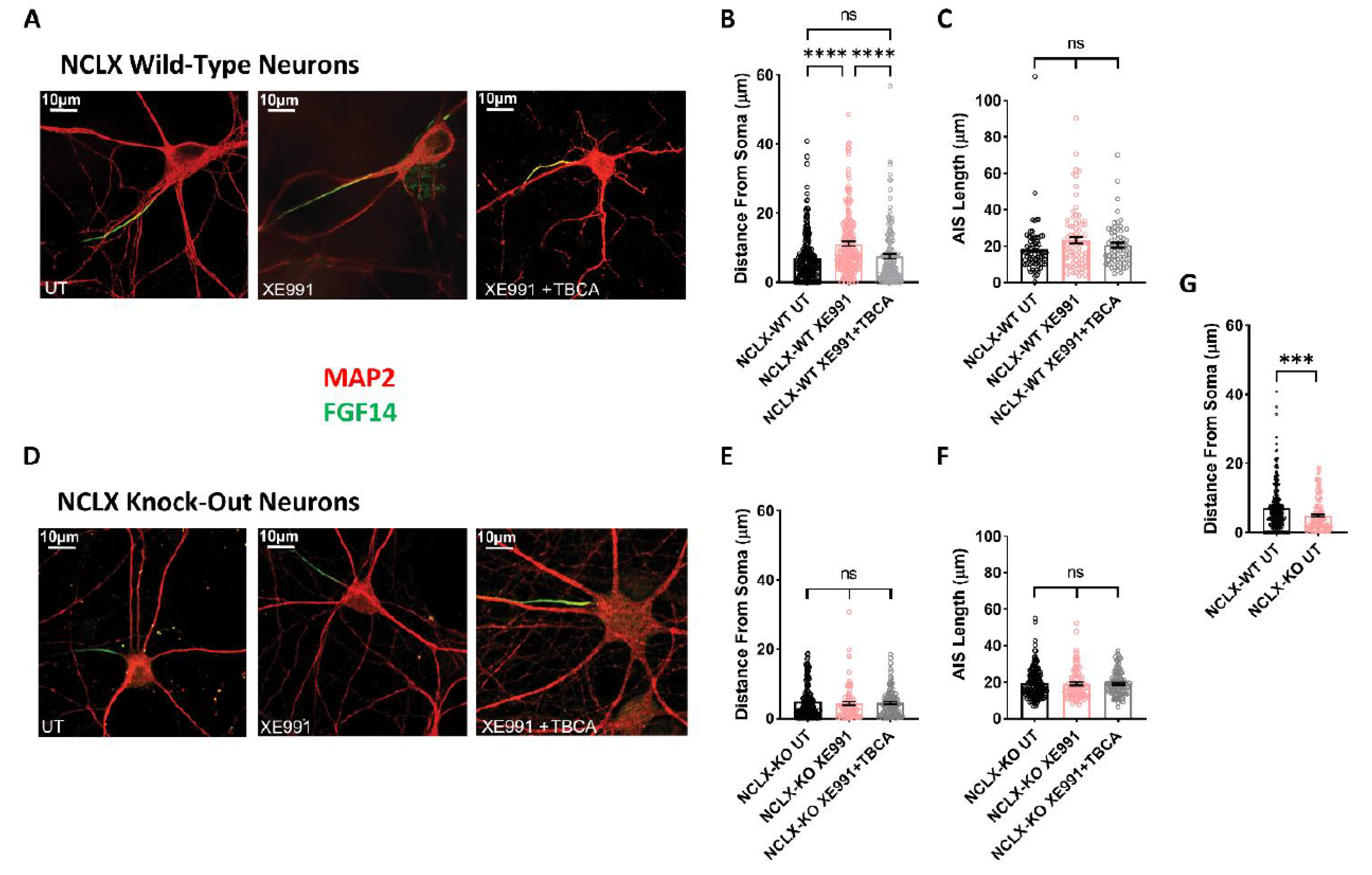
2.5. Plasticity of the Axon Initial Segment in Hippocampal Neurons Derived from WT and NCLX-KO Mice
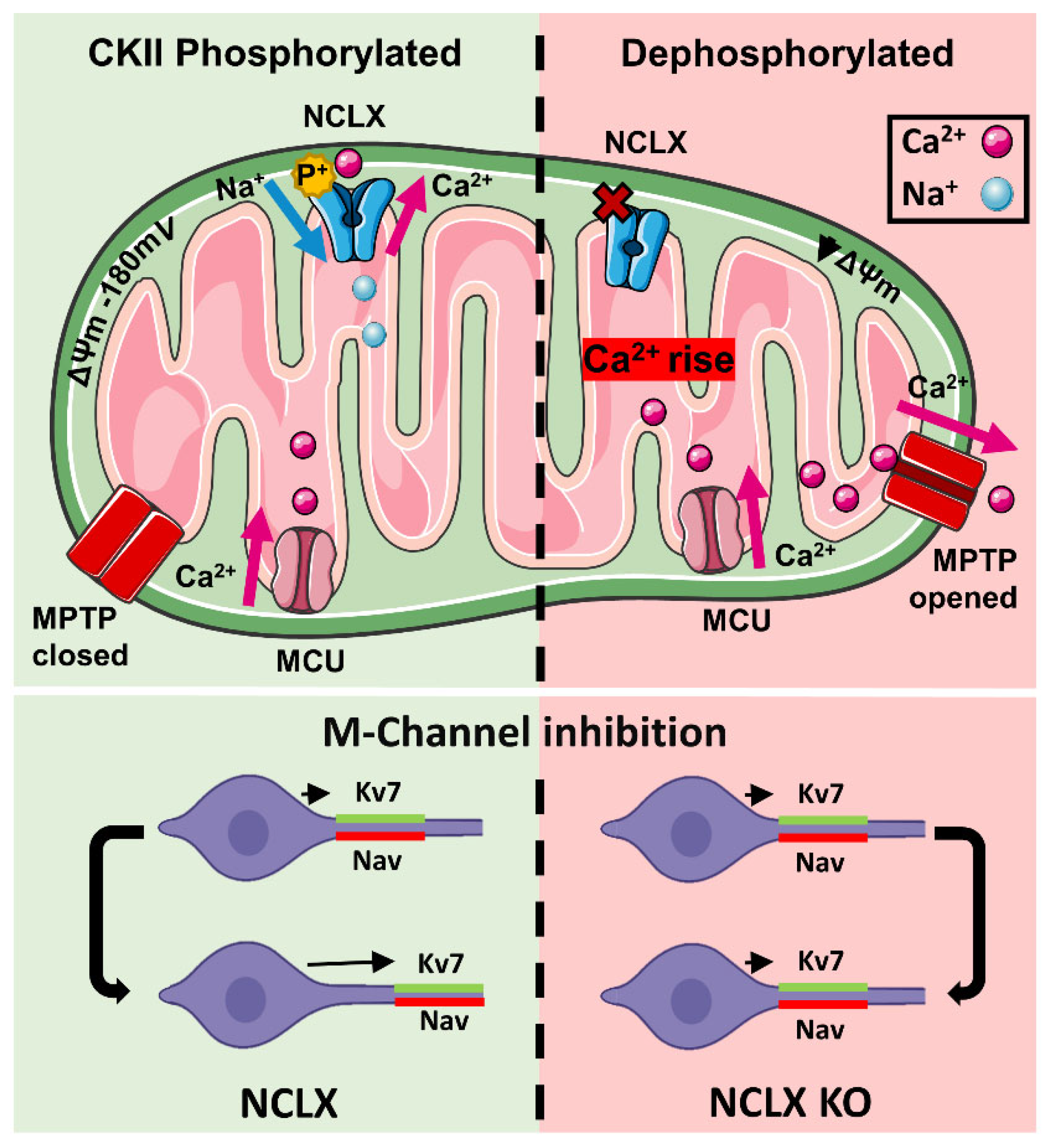
3. Discussion
4. Materials and Methods
4.1. Primary Neuronal Culture and Infection
4.2. Cell Cultures and Transfection
4.3. Live Fluorescence Imaging
4.4. Western Blotting
4.5. Plasmids
- NCLXS271A forward: GTTACTCCAGAGATCCTCGCAGACTCCGAG;
- NCLXS271A reverse: CTCGGAGTCTGCGAGGATCTCTGGAGTAAC;
- NCLXS271D forward: CTCCAGAGATCCTCGATGACTCCGAGGAGGAC;
- NCLXS271D reverse: GTCCTCCTCGGAGTCATCGAGGATCTCTGGAG.
4.6. [Ca2+]m Measurements
4.7. Cytosolic Ca2+Measurements
4.8. ΔΨm Measurements
4.9. Drugs
4.10. Primary Cultures of Hippocampal Neurons
4.11. Recombinant AAV-Dlx-mCherry Plasmid and Infection
4.12. Patch-Clamp Electrophysiology
4.13. Immunostaining
4.14. Antibodies
4.15. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixit, D.; Sharma, V.; Ghosh, S.; Mehta, V.S.; Sen, E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012, 3, e271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogosawa, S.; Yoshida, K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci. 2018, 109, 3376–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng Liang, L.; Shih Shun, C. Activation of Casein Kinase II by Gallic Acid Induces BIK–BAX/BAK-Mediated ER Ca++-ROS-Dependent Apoptosis of Human Oral Cancer Cells. Front. Physiol. 2017, 8, 761. [Google Scholar] [CrossRef] [Green Version]
- Bettegazzi, B.; Sebastian Monasor, L.; Bellani, S.; Codazzi, F.; Restelli, L.M.; Colombo, A.V.; Deigendesch, N.; Frank, S.; Saito, T.; Saido, T.C.; et al. Casein Kinase 2 dependent phosphorylation of eIF4B regulates BACE1 expression in Alzheimer’s disease. Cell Death Dis. 2021, 12, 769. [Google Scholar] [CrossRef]
- Blanquet, P.R. Casein kinase 2 as a potentially important enzyme in the nervous system. Prog. Neurobiol. 2000, 60, 211–246. [Google Scholar] [CrossRef]
- Qaiser, F.; Trembley, J.H.; Kren, B.T.; Wu, J.-J.; Naveed, A.K.; Ahmed, K. Protein Kinase CK2 Inhibition Induces Cell Death via Early Impact on Mitochondrial Function. J. Cell. Biochem. 2014, 115, 2103–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanki, T.; Kurihara, Y.; Jin, X.; Goda, T.; Ono, Y.; Aihara, M.; Hirota, Y.; Saigusa, T.; Aoki, Y.; Uchiumi, T.; et al. Casein kinase 2 is essential for mitophagy. EMBO Rep. 2013, 14, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.W.; Wang, J.K.; Wang, L.C.; Guo, Q.; Liu, T.T.; Wang, F.J.; Feng, N.; Zhang, X.W.; Liao, L.X.; Zhao, M.M.; et al. Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct. Target. Ther. 2021, 6, 71. [Google Scholar] [CrossRef]
- Kostic, M.; Ludtmann, M.H.R.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Parnis, J.; Montana, V.; Delgado-Martinez, I.; Matyash, V.; Parpura, V.; Kettenmann, H.; Sekler, I.; Nolte, C. Mitochondrial Exchanger NCLX Plays a Major Role in the Intracellular Ca2+ Signaling, Gliotransmission, and Proliferation of Astrocytes. J. Neurosci. 2013, 33, 7206–7219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, M.; Katoshevski, T.; Sekler, I. Allosteric Regulation of NCLX by Mitochondrial Membrane Potential Links the Metabolic State and Ca2+ Signaling in Mitochondria. Cell Rep. 2018, 25, 3465–3475.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, J.G.; Denton, R.M. Intracellular calcium ions and intramitochondrial Ca2+ in the regulation of energy metabolism in mammalian tissues. Proc. Nutr. Soc. 1990, 49, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A. Ca2+ binding to citrate cycle dehydrogenases. Int. J. Biochem. 1990, 22, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Zenebe, W.J.; Nazarewicz, R.R.; Parihar, M.S.; Ghafourifar, P. Hypoxia/Reoxygenation of isolated rat heart mitochondria causes cytochrome c release and oxidative stress; evidence for involvement of mitochondrial nitric oxide synthase. J. Mol. Cell. Cardiol. 2007, 43, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Fluegge, D.; Moeller, L.M.; Cichy, A.; Gorin, M.; Weth, A.; Veitinger, S.; Cainarca, S.; Lohmer, S.; Corazza, S.; Neuhaus, E.M.; et al. Mitochondrial Ca2+ mobilization is a key element in olfactory signaling. Nat. Neurosci. 2012, 15, 754–762. [Google Scholar] [CrossRef]
- Chang, D.T.W.; Reynolds, I.J. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Neurobiol. 2006, 80, 241–268. [Google Scholar] [CrossRef]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef]
- Katoshevski, T.; Ben-Kasus Nissim, T.; Sekler, I. Recent studies on NCLX in health and diseases. Cell Calcium 2021, 94, 102345. [Google Scholar] [CrossRef]
- Opalinska, M.; Meisinger, C. Mitochondrial protein import under kinase surveillance. Microb. Cell 2014, 1, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Lezmy, J.; Gelman, H.; Katsenelson, M.; Styr, B.; Tikochinsky, E.; Lipinsky, M.; Peretz, A.; Slutsky, I.; Attali, B. M-Current Inhibition in Hippocampal Excitatory Neurons Triggers Intrinsic and Synaptic Homeostatic Responses at Different Temporal Scales. J. Neurosci. 2020, 40, 3694–3706. [Google Scholar] [CrossRef] [PubMed]
- Grubb, M.S.; Shu, Y.; Kuba, H.; Rasband, M.N.; Wimmer, V.C.; Bender, K.J. Short- and Long-Term Plasticity at the Axon Initial Segment. J. Neurosci. 2011, 31, 16049–16055. [Google Scholar] [CrossRef] [Green Version]
- Lezmy, J.; Lipinsky, M.; Khrapunsky, Y.; Patrich, E.; Shalom, L.; Peretz, A.; Fleidervish, I.A.; Attali, B. M-current inhibition rapidly induces a unique CK2-dependent plasticity of the axon initial segment. Proc. Natl. Acad. Sci. USA 2017, 114, E10234–E10243. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.A.; Poletto, G.; Di Maira, G.; Cozza, G.; Ruzzene, M.; Sarno, S.; Bain, J.; Elliott, M.; Moro, S.; Zagotto, G.; et al. Tetrabromocinnamic Acid (TBCA) and Related Compounds Represent a New Class of Specific Protein Kinase CK2 Inhibitors. ChemBioChem 2007, 8, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of Protein Kinase CK2 Inhibitors Derived from 4,5,6,7-Tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyman, L.; Williams, G.S.B.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Stavsky, A.; Stoler, O.; Kostic, M.; Katoshevsky, T.; Assali, E.A.; Savic, I.; Amitai, Y.; Prokisch, H.; Leiz, S.; Daumer-Haas, C.; et al. Aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol. 2021, 4, 666. [Google Scholar] [CrossRef]
- Vais, H.; Mallilankaraman, K.; Mak, D.-O.D.; Hoff, H.; Payne, R.; Tanis, J.E.; Foskett, J.K. EMRE Is a Matrix Ca2+ Sensor that Governs Gatekeeping of the Mitochondrial Ca2+ Uniporter. Cell Rep. 2016, 14, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Moreau, B.; Parekh, A.B. Ca2+-Dependent Inactivation of the Mitochondrial Ca2+ Uniporter Involves Proton Flux through the ATP Synthase. Curr. Biol. 2008, 18, 855–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-Calcium Exchange Is Mediated by a Distinct Potassium-independent Sodium-Calcium Exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef] [Green Version]
- Plotegher, N.; Filadi, R.; Pizzo, P.; Duchen, M.R. Excitotoxicity Revisited: Mitochondria on the Verge of a Nervous Breakdown. Trends Neurosci. 2021, 44, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.; Wills, Z.; Chu, C.T. Excitatory Dendritic Mitochondrial Calcium Toxicity: Implications for Parkinson’s and Other Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 523. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [Green Version]
- Styr, B.; Gonen, N.; Zarhin, D.; Ruggiero, A.; Atsmon, R.; Gazit, N.; Braun, G.; Frere, S.; Vertkin, I.; Shapira, I.; et al. Mitochondrial Regulation of the Hippocampal Firing Rate Set Point and Seizure Susceptibility. Neuron 2019, 102, 1009–1024.e8. [Google Scholar] [CrossRef]
- Dimidschstein, J.; Chen, Q.; Tremblay, R.; Rogers, S.L.; Saldi, G.-A.; Guo, L.; Xu, Q.; Liu, R.; Lu, C.; Chu, J.; et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci. 2016, 19, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Pablo, J.L.; Pitt, G.S. FGF14 is a regulator of KCNQ2/3 channels. Proc. Natl. Acad. Sci. USA 2017, 114, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 1992, 267, 8834–8839. [Google Scholar] [CrossRef]
- Kim, B.; Matsuoka, S. Cytoplasmic Na + -dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J. Physiol. 2008, 586, 1683–1697. [Google Scholar] [CrossRef]
- Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria control store-operated Ca 2+ entry through Na + and redox signals. EMBO J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cell Dev. Biol. 2019, 94, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hernansanz-Agustín, P.; Enríquez, J.A. Sodium in Mitochondrial Redox Signaling. Antioxid. Redox Signal. 2022, 37, 290–300. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Lu, T.; Meng, L.; Luo, Y.; Fu, X.; Hou, Y. Mitochondrial Ca2+ Overload Leads to Mitochondrial Oxidative Stress and Delayed Meiotic Resumption in Mouse Oocytes. Front. Cell Dev. Biol. 2020, 8, 580876. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.C.; Lee, Y.M.; Kim, H.D. Mitochondrial disease and epilepsy. Brain Dev. 2013, 35, 757–761. [Google Scholar] [CrossRef]
- Khurana, D.S.; Valencia, I.; Goldenthal, M.J.; Legido, A. Mitochondrial Dysfunction in Epilepsy. Semin. Pediatr. Neurol. 2013, 20, 176–187. [Google Scholar] [CrossRef]
- Hadley, S.H.; Bahia, P.K.; Taylor-Clark, T.E. Sensory Nerve Terminal Mitochondrial Dysfunction Induces Hyperexcitability in Airway Nociceptors via Protein Kinase C. Mol. Pharmacol. 2014, 85, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, M.; Nedergaard, S. Effect of acute mitochondrial dysfunction on hyperexcitable network activity in rat hippocampus in vitro. Brain Res. 2021, 1751, 147193. [Google Scholar] [CrossRef]
- Duprat, F.; Guillemare, E.; Romey, G.; Fink, M.; Lesage, F.; Lazdunski, M.; Honore, E. Susceptibility of cloned K+ channels to reactive oxygen species. Proc. Natl. Acad. Sci. USA 1995, 92, 11796–11800. [Google Scholar] [CrossRef] [Green Version]
- Müller, W.; Bittner, K. Differential Oxidative Modulation of Voltage-Dependent K + Currents in Rat Hippocampal Neurons. J. Neurophysiol. 2002, 87, 2990–2995. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef] [PubMed]
- De La Rossa, A.; Laporte, M.H.; Astori, S.; Marissal, T.; Montessuit, S.; Sheshadri, P.; Ramos-Fernández, E.; Mendez, P.; Khani, A.; Quairiaux, C.; et al. Paradoxical neuronal hyperexcitability in a mouse model of mitochondrial pyruvate import deficiency. eLife 2022, 11, e72595. [Google Scholar] [CrossRef] [PubMed]
- Grubb, M.S.; Burrone, J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 2010, 465, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita, I.I.; Hershfinkel, M.; Fishman, D.; Ozeri, E.; Rutter, G.; Sensi, S.L.; Khananshvili, D.; Lewis, E.C.; Sekler, I. The Mitochondrial Na+/Ca2+ Exchanger Upregulates Glucose Dependent Ca2+ Signalling Linked to Insulin Secretion. PLoS ONE 2012, 7, e46649. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Manufacturer | Catalog Number |
|---|---|---|
| Mouse monoclonal anti-Myc tag | Abcam | Cat# AB18185 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat#A2228; RRID:AB_476697 |
| Goat anti-mouse IgG | Jackson | Cat# 115-035-146; RRID:AB_2307392 |
| Mouse a-FGF14 | NeuroMab | Cat#75 096 |
| Rabbit a-MAP2 | Millipore | Cat#AB5622 |
| Donkey a-mouse Alexa488 | Jackson | Cat#715- 545-150 |
| Donkey a-rabbit Cy3 | Jackson | Cat#711-165-152 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katoshevski, T.; Bar, L.; Tikochinsky, E.; Harel, S.; Ben-Kasus Nissim, T.; Bogeski, I.; Hershfinkel, M.; Attali, B.; Sekler, I. CKII Control of Axonal Plasticity Is Mediated by Mitochondrial Ca2+ via Mitochondrial NCLX. Cells 2022, 11, 3990. https://doi.org/10.3390/cells11243990
Katoshevski T, Bar L, Tikochinsky E, Harel S, Ben-Kasus Nissim T, Bogeski I, Hershfinkel M, Attali B, Sekler I. CKII Control of Axonal Plasticity Is Mediated by Mitochondrial Ca2+ via Mitochondrial NCLX. Cells. 2022; 11(24):3990. https://doi.org/10.3390/cells11243990
Chicago/Turabian StyleKatoshevski, Tomer, Lior Bar, Eliav Tikochinsky, Shimon Harel, Tsipi Ben-Kasus Nissim, Ivan Bogeski, Michal Hershfinkel, Bernard Attali, and Israel Sekler. 2022. "CKII Control of Axonal Plasticity Is Mediated by Mitochondrial Ca2+ via Mitochondrial NCLX" Cells 11, no. 24: 3990. https://doi.org/10.3390/cells11243990