

Paclitaxel Inhibits KCNQ Channels in Primary Sensory Neurons to Initiate the Development of Painful Peripheral Neuropathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Strains and Genotyping
2.2. Administration of Paclitaxel
2.3. Behavioral Tests
2.4. DRG Neuron Dissociation and Culturing
2.5. Chinese Hamster Ovary (CHO) Cell Transfection
2.6. Electrophysiological Recordings of DRG Neurons and CHO Cells
2.7. Intraepidermal Nerve Fiber Staining and Quantification
2.8. Statistics
3. Results
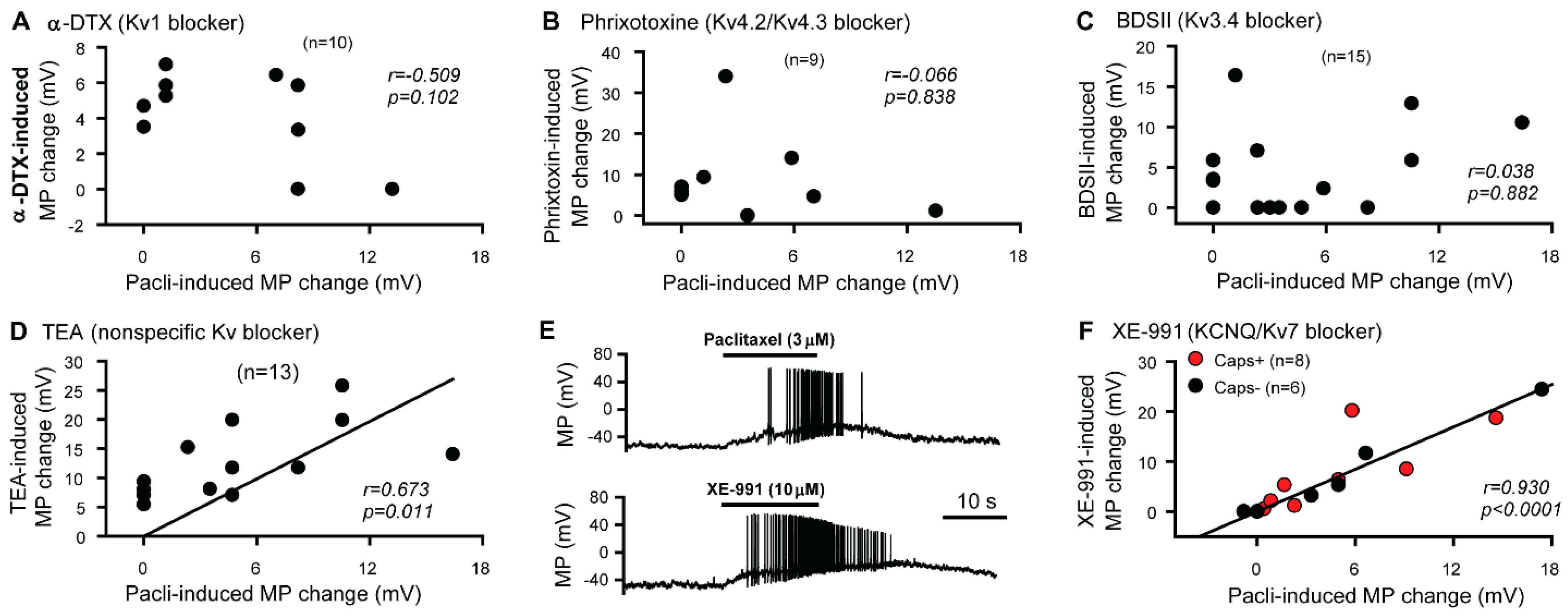
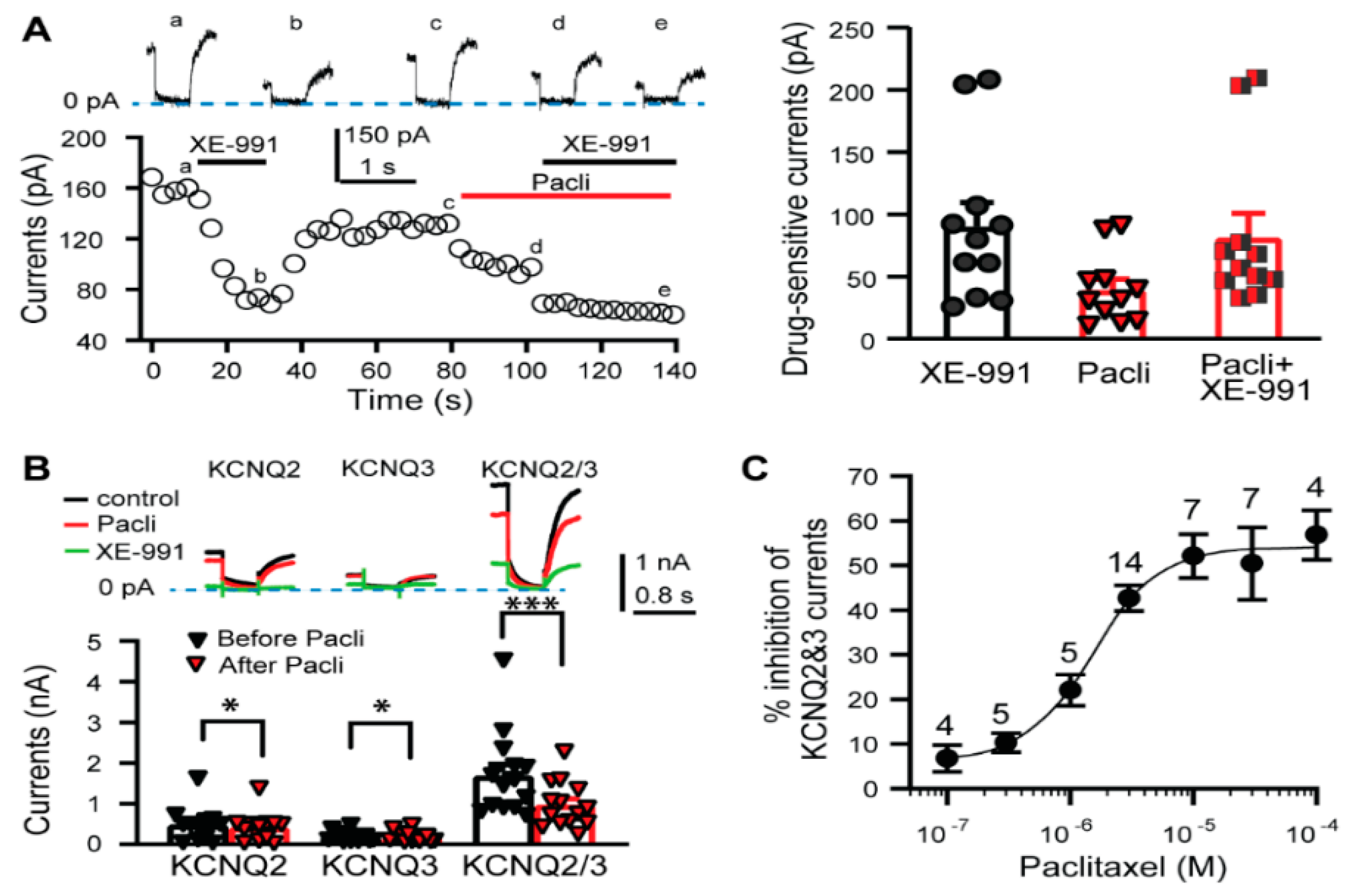
3.1. KCNQ Channels Are Involved in the Paclitaxel-Induced Excitation/Depolarization of DRG Neurons
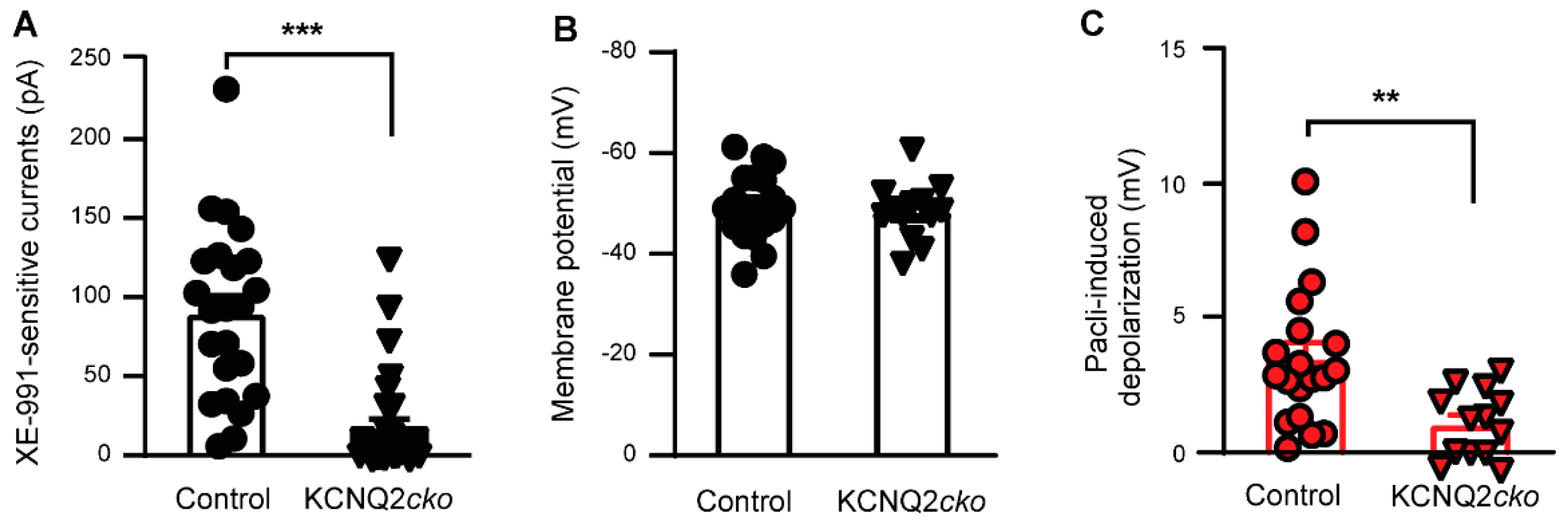
3.2. Paclitaxel Fails to Induce Acute Neuronal Excitation in KCNQ2 Conditional Knockout Mice
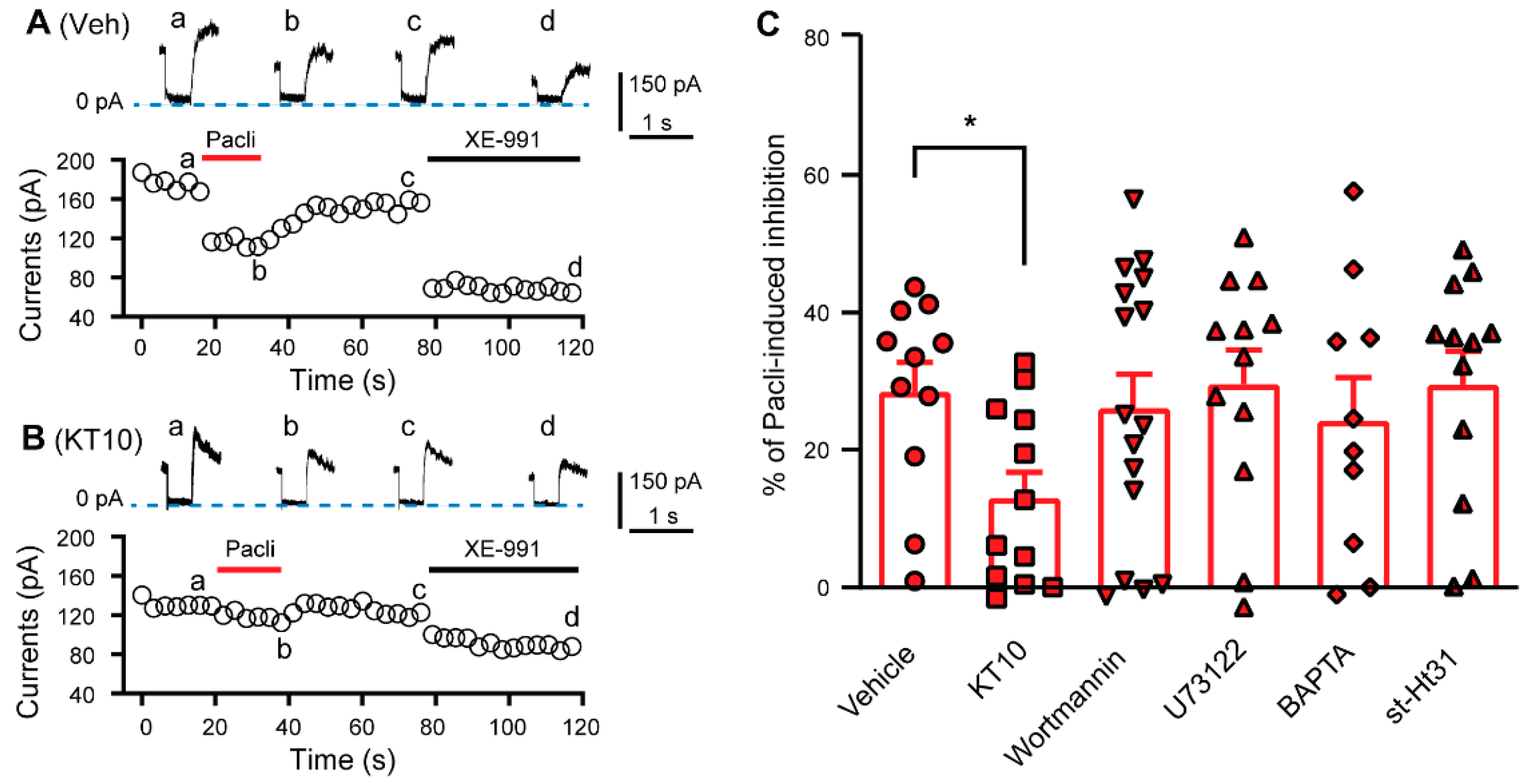
3.3. PIP2 Is Involved in the Paclitaxel-Induced Inhibition of KCNQ Channels
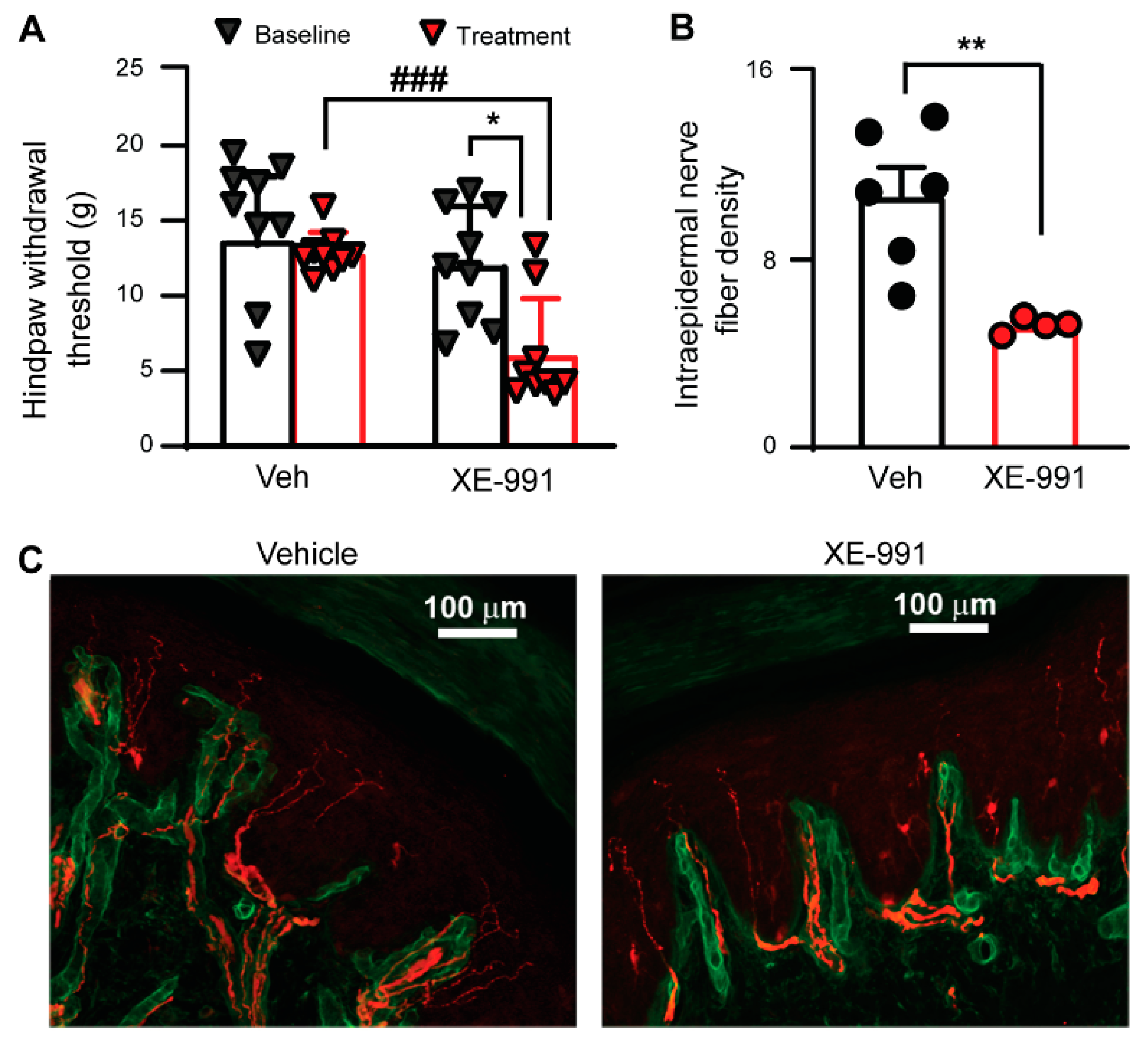
3.4. Inhibiting KCNQ Channels In Vivo Induces PIPN-like Pathological Alterations
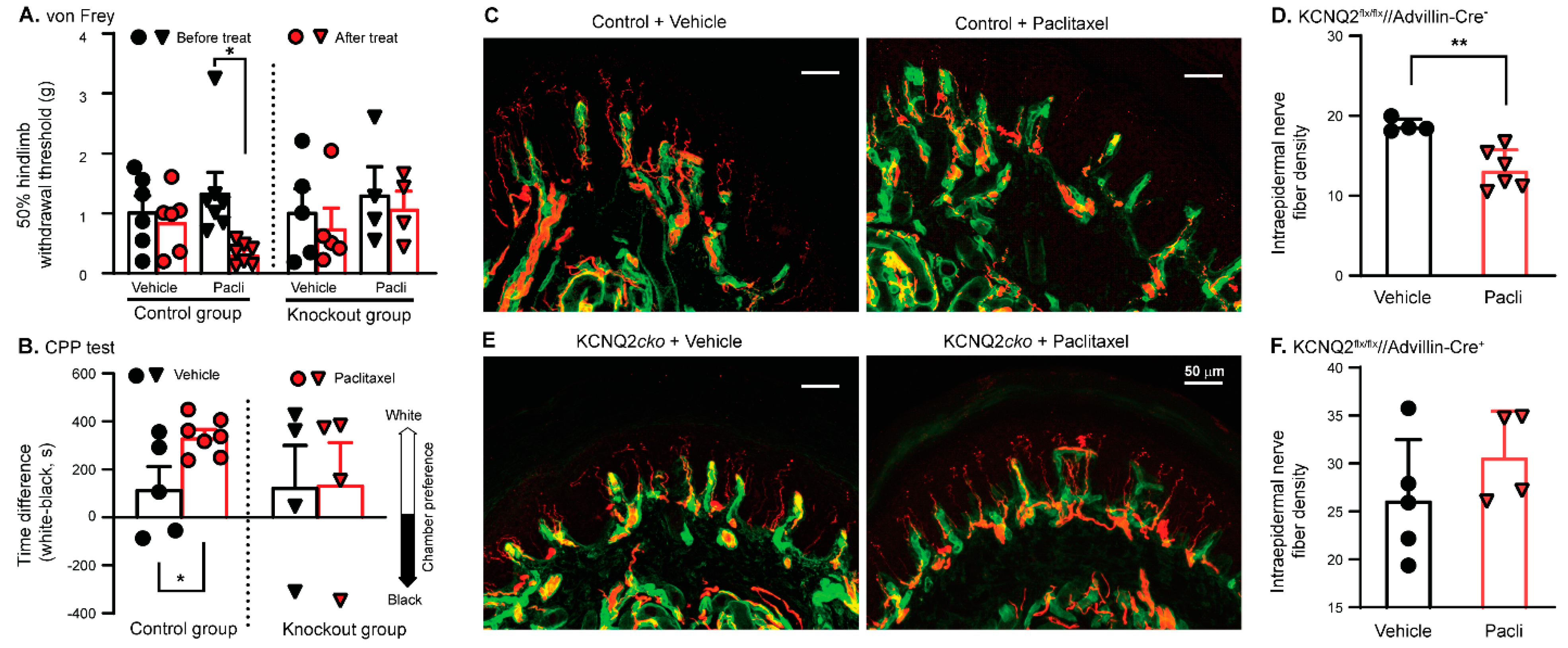
3.5. Paclitaxel Fails to Induce PIPN in KCNQ2 Conditional Knockout Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crown, J.; O’Leary, M.; Ooi, W.S. Docetaxel and paclitaxel in the treatment of breast cancer: A review of clinical experience. Oncologist 2004, 9 (Suppl. S2), 24–32. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Koltzenburg, M.; Polychronopoulos, P.; Papapetropoulos, S.; Kalofonos, H.P. Peripheral nerve damage associated with administration of taxanes in patients with cancer. Crit. Rev. Oncol. Hematol. 2008, 66, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y. Changes of fast axonal transport by taxol injected subepineurally into the rat sciatic nerve. Neurosci. Res. 1992, 14, 159–165. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, N.E.; Morfini, G.; Brady, S.T.; Feinstein, S.C.; Wilson, L.; Jordan, M.A. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 2013, 37, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Nennesmo, I.; Reinholt, F.P. Effects of intraneural injection of taxol on retrograde axonal transport and morphology of corresponding nerve cell bodies. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1988, 55, 241–246. [Google Scholar] [CrossRef]
- Theiss, C.; Meller, K. Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res. 2000, 299, 213–224. [Google Scholar] [CrossRef]
- Bobylev, I.; Joshi, A.R.; Barham, M.; Ritter, C.; Neiss, W.F.; Hoke, A.; Lehmann, H.C. Paclitaxel inhibits mRNA transport in axons. Neurobiol. Dis. 2015, 82, 321–331. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Flatters, S.J.; Bennett, G.J. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: Evidence for mitochondrial dysfunction. Pain 2006, 122, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.H.; Zheng, H.; Zheng, F.Y.; Nuydens, R.; Meert, T.F.; Bennett, G.J. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience 2011, 199, 461–469. [Google Scholar] [CrossRef]
- Zheng, H.; Xiao, W.H.; Bennett, G.J. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp. Neurol. 2011, 232, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, K.; Doyle, T.; Bryant, L.; Esposito, E.; Cuzzocrea, S.; Ryerse, J.; Bennett, G.J.; Salvemini, D. Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain 2013, 154, 2432–2440. [Google Scholar] [CrossRef] [Green Version]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Pavliv, O.; Melnyk, S.; James, S.J. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS ONE 2014, 9, e85436. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Golovoy, D.; Vincent, A.M.; Mahendru, P.; Olzmann, J.A.; Mentzer, A.; Feldman, E.L. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002, 16, 1738–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.M.; Brownlee, M.; Russell, J.W. Oxidative stress and programmed cell death in diabetic neuropathy. Ann. N. Y. Acad. Sci. 2002, 959, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Natsume, T.; Tamaoki, S.; Watanabe, J.; Asano, H.; Mikami, T.; Miyasaka, K.; Miyazaki, K.; Gondo, M.; Sakakibara, K.; et al. Antitumor activity of TZT-1027, a novel dolastatin 10 derivative. Jpn. J. Cancer Res. 1997, 88, 316–327. [Google Scholar] [CrossRef]
- Ogawa, T.; Mimura, Y.; Isowa, K.; Kato, H.; Mitsuishi, M.; Toyoshi, T.; Kuwayama, N.; Morimoto, H.; Murakoshi, M.; Nakayama, T. An antimicrotubule agent, TZT-1027, does not induce neuropathologic alterations which are detected after administration of vincristine or paclitaxel in animal models. Toxicol. Lett. 2001, 121, 97–106. [Google Scholar] [CrossRef]
- Hershman, D.L.; Unger, J.M.; Crew, K.D.; Minasian, L.M.; Awad, D.; Moinpour, C.M.; Hansen, L.; Lew, D.L.; Greenlee, H.; Fehrenbacher, L.; et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J. Clin. Oncol. 2013, 31, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Leal, A.D.; Qin, R.; Atherton, P.J.; Haluska, P.; Behrens, R.J.; Tiber, C.H.; Watanaboonyakhet, P.; Weiss, M.; Adams, P.T.; Dockter, T.J.; et al. North Central Cancer Treatment Group/Alliance trial N08CA-the use of glutathione for prevention of paclitaxel/carboplatin-induced peripheral neuropathy: A phase 3 randomized, double-blind, placebo-controlled study. Cancer 2014, 120, 1890–1897. [Google Scholar] [CrossRef] [Green Version]
- Loprinzi, C.L.; Reeves, B.N.; Dakhil, S.R.; Sloan, J.A.; Wolf, S.L.; Burger, K.N.; Kamal, A.; Le-Lindqwister, N.A.; Soori, G.S.; Jaslowski, A.J.; et al. Natural history of paclitaxel-associated acute pain syndrome: Prospective cohort study NCCTG N08C1. J. Clin. Oncol. 2011, 29, 1472–1478. [Google Scholar] [CrossRef]
- Reeves, B.N.; Dakhil, S.R.; Sloan, J.A.; Wolf, S.L.; Burger, K.N.; Kamal, A.; Le-Lindqwister, N.A.; Soori, G.S.; Jaslowski, A.J.; Kelaghan, J.; et al. Further data supporting that paclitaxel-associated acute pain syndrome is associated with development of peripheral neuropathy: North Central Cancer Treatment Group trial N08C1. Cancer 2012, 118, 5171–5178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017, 158, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossato, M.F.; Rigo, F.K.; Oliveira, S.M.; Guerra, G.P.; Silva, C.R.; Cunha, T.M.; Gomez, M.V.; Ferreira, J.; Trevisan, G. Participation of Transient Receptor Potential Vanilloid 1 in Paclitaxel-Induced Acute Visceral and Peripheral Nociception in Rodents. Eur. J. Pharmacol. 2018, 828, 4251. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Zuo, Y.; Dang, D.; Frost, J.A.; Yang, Q. Activation of KCNQ Channels Prevents Paclitaxel-Induced Peripheral Neuropathy and Associated Neuropathic Pain. J. Pain 2018, 20, 528–539. [Google Scholar] [CrossRef]
- da Silva, S.; Hasegawa, H.; Scott, A.; Zhou, X.; Wagner, A.K.; Han, B.X.; Wang, F. Proper formation of whisker barrelettes requires periphery-derived Smad4-dependent TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 3395–3400. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yoon, S.Y.; Dougherty, P.M. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. J. Pain 2012, 13, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, B.L.; Yang, Q.; Zhou, X.; Tang, S.J. Microglial ablation does not affect opioid-induced hyperalgesia in rodents. Pain 2022, 163, 508–517. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Wu, Z.; Li, L.; Xie, F.; Du, J.; Zuo, Y.; Frost, J.A.; Carlton, S.M.; Walters, E.T.; Yang, Q. Activation of KCNQ Channels Suppresses Spontaneous Activity in Dorsal Root Ganglion Neurons and Reduces Chronic Pain after Spinal Cord Injury. J. Neurotrauma 2017, 34, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Wu, Z.; Hadden, J.K.; Odem, M.A.; Zuo, Y.; Crook, R.J.; Frost, J.A.; Walters, E.T. Persistent pain after spinal cord injury is maintained by primary afferent activity. J. Neurosci. 2014, 34, 10765–10769. [Google Scholar] [CrossRef]
- Guan, D.; Lee, J.C.; Tkatch, T.; Surmeier, D.J.; Armstrong, W.E.; Foehring, R.C. Expression and biophysical properties of Kv1 channels in supragranular neocortical pyramidal neurones. J. Physiol. 2006, 571, 371–389. [Google Scholar] [CrossRef]
- Diochot, S.; Drici, M.D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diochot, S.; Schweitz, H.; Beress, L.; Lazdunski, M. Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J. Biol. Chem. 1998, 273, 6744–6749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, M.S.; Roche, J.P.; Kaftan, E.J.; Cruzblanca, H.; Mackie, K.; Hille, B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K(+) channels that underlie the neuronal M current. J. Neurosci. 2000, 20, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadley, J.K.; Passmore, G.M.; Tatulian, L.; Al-Qatari, M.; Ye, F.; Wickenden, A.D.; Brown, D.A. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J. Neurosci. 2003, 23, 5012–5019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinel, N.; Lauritzen, I.; Chouabe, C.; Lazdunski, M.; Borsotto, M. The KCNQ2 potassium channel: Splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 1998, 438, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Zaczek, R.; Chorvat, R.J.; Saye, J.A.; Pierdomenico, M.E.; Maciag, C.M.; Logue, A.R.; Fisher, B.N.; Rominger, D.H.; Earl, R.A. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: Comparison to linopirdine. J. Pharmacol. Exp. Ther. 1998, 285, 724–730. [Google Scholar] [PubMed]
- Jentsch, T.J. Neuronal KCNQ potassium channels: Physiology and role in disease. Nat. Rev. Neurosci. 2000, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Passmore, G.M.; Selyanko, A.A.; Mistry, M.; Al-Qatari, M.; Marsh, S.J.; Matthews, E.A.; Dickenson, A.H.; Brown, T.A.; Burbidge, S.A.; Main, M.; et al. KCNQ/M currents in sensory neurons: Significance for pain therapy. J. Neurosci. 2003, 23, 7227–7236. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.; Ooi, L.; Dalle, C.; Robertson, B.; Wood, I.C.; Gamper, N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 2011, 152, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.C.; Hechenberger, M.; Weinreich, F.; Kubisch, C.; Jentsch, T.J. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000, 275, 24089–24095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.P.; Kerchner, G.A. Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J. Neurosci. Res. 1998, 52, 612–617. [Google Scholar] [CrossRef]
- King, C.H.; Lancaster, E.; Salomon, D.; Peles, E.; Scherer, S.S. Kv7.2 regulates the function of peripheral sensory neurons. J. Comp. Neurol. 2014, 522, 3262–3280. [Google Scholar] [CrossRef] [Green Version]
- Suh, B.C.; Inoue, T.; Meyer, T.; Hille, B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 2006, 314, 1454–1457. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Cavanaugh, E.J.; Simkin, D. Inhibition of transient receptor potential A1 channel by phosphatidylinositol-4,5-bisphosphate. Am. J. Physiol. Cell Physiol. 2008, 295, C92–C99. [Google Scholar] [CrossRef]
- Ling, X.; Bernacki, R.J.; Brattain, M.G.; Li, F. Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J. Biol. Chem. 2004, 279, 15196–15203. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.C.; Ehrlich, B.E. Functional Interaction between Transient Receptor Potential V4 Channel and Neuronal Calcium Sensor 1 and the Effects of Paclitaxel. Mol. Pharmacol. 2021, 100, 258–270. [Google Scholar] [CrossRef]
- Taverna, E.; Francolini, M.; Jeromin, A.; Hilfiker, S.; Roder, J.; Rosa, P. Neuronal calcium sensor 1 and phosphatidylinositol 4-OH kinase beta interact in neuronal cells and are translocated to membranes during nucleotide-evoked exocytosis. J. Cell Sci. 2002, 115, 3909–3922. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Varnai, P.; Tuymetova, G.; Balla, A.; Toth, Z.E.; Oker-Blom, C.; Roder, J.; Jeromin, A.; Balla, T. Interaction of neuronal calcium sensor-1 (NCS-1) with phosphatidylinositol 4-kinase beta stimulates lipid kinase activity and affects membrane trafficking in COS-7 cells. J. Biol. Chem. 2001, 276, 40183–40189. [Google Scholar] [CrossRef]
- Stein, A.T.; Ufret-Vincenty, C.A.; Hua, L.; Santana, L.F.; Gordon, S.E. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J. Gen. Physiol. 2006, 128, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ui, M.; Okada, T.; Hazeki, K.; Hazeki, O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci. 1995, 20, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.C.; Falkenburger, B.; Shapiro, M.S. Affinity for phosphatidylinositol 4,5-bisphosphate determines muscarinic agonist sensitivity of Kv7 K+ channels. J. Gen. Physiol. 2009, 134, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shapiro, M.S. Activity-dependent transcriptional regulation of M-Type (Kv7) K(+) channels by AKAP79/150-mediated NFAT actions. Neuron 2012, 76, 1133–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.H.; Zheng, H.; Bennett, G.J. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 2012, 203, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Adamek, P.; Zhang, H.; Tatsui, C.E.; Rhines, L.D.; Mrozkova, P.; Li, Q.; Kosturakis, A.K.; Cassidy, R.M.; Harrison, D.S.; et al. The Cancer Chemotherapeutic Paclitaxel Increases Human and Rodent Sensory Neuron Responses to TRPV1 by Activation of TLR4. J. Neurosci. 2015, 35, 13487–13500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dina, O.A.; Chen, X.; Reichling, D.; Levine, J.D. Role of protein kinase Cepsilon and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience 2001, 108, 507–515. [Google Scholar] [CrossRef]
- Zhong, X.Z.; Harhun, M.I.; Olesen, S.P.; Ohya, S.; Moffatt, J.D.; Cole, W.C.; Greenwood, I.A. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J. Physiol. 2010, 588, 3277–3293. [Google Scholar] [CrossRef]
- Ishikawa, K.; Tanaka, M.; Black, J.A.; Waxman, S.G. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve 1999, 22, 502–507. [Google Scholar] [CrossRef]
- Murthy, S.E.; Loud, M.C.; Daou, I.; Marshall, K.L.; Schwaller, F.; Kuhnemund, J.; Francisco, A.G.; Keenan, W.T.; Dubin, A.E.; Lewin, G.R.; et al. The mechanosensitive ion channel Piezo2 mediates sensitivity to mechanical pain in mice. Sci. Transl. Med. 2018, 10, eaat9897. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Geng, J.; Zhou, S.; Xiao, B. Mechanically Activated Piezo Channels Mediate Touch and Suppress Acute Mechanical Pain Response in Mice. Cell Rep. 2019, 26, 1419–1431.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappia, K.J.; O’Hara, C.L.; Moehring, F.; Kwan, K.Y.; Stucky, C.L. Sensory Neuron-Specific Deletion of TRPA1 Results in Mechanical Cutaneous Sensory Deficits. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Sakamoto, K.; Ono, T.; Kimura, J. The inhibitory effect of paclitaxel on (kv2.1) k+ current in h9c2 cells. Fukushima J. Med. Sci. 2015, 61, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Tsantoulas, C.; Zhu, L.; Yip, P.; Grist, J.; Michael, G.J.; McMahon, S.B. Kv2 dysfunction after peripheral axotomy enhances sensory neuron responsiveness to sustained input. Exp. Neurol. 2014, 251, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocksteins, E.; Raes, A.L.; Van de Vijver, G.; Bruyns, T.; Van Bogaert, P.P.; Snyders, D.J. Kv2.1 and silent Kv subunits underlie the delayed rectifier K+ current in cultured small mouse DRG neurons. Am. J. Physiol. Cell Physiol. 2009, 296, C1271–C1278. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Boillat, A.; Huang, D.; Liang, C.; Peers, C.; Gamper, N. Intracellular zinc activates KCNQ channels by reducing their dependence on phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2017, 114, E6410–E6419. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Abbott, S.; Han, B.X.; Qi, Y.; Wang, F. Analyzing somatosensory axon projections with the sensory neuron-specific Advillin gene. J. Neurosci. 2007, 27, 14404–14414. [Google Scholar] [CrossRef]
- Cai, J.; Fang, D.; Liu, X.D.; Li, S.; Ren, J.; Xing, G.G. Suppression of KCNQ/M (Kv7) potassium channels in the spinal cord contributes to the sensitization of dorsal horn WDR neurons and pain hypersensitivity in a rat model of bone cancer pain. Oncol. Rep. 2015, 33, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Main, M.J.; Cryan, J.E.; Dupere, J.R.; Cox, B.; Clare, J.J.; Burbidge, S.A. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 2000, 58, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wu, Y.; Bi, Y.; Tan, L.; Gan, Y.; Wang, K. Activation of voltage-gated KCNQ/Kv7 channels by anticonvulsant retigabine attenuates mechanical allodynia of inflammatory temporomandibular joint in rats. Mol. Pain 2010, 6, 49. [Google Scholar] [CrossRef]
- Devor, M. Unexplained peculiarities of the dorsal root ganglion. Pain 1999, 82 (Suppl. S6), S27–S35. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Nelson, D.; Yudkoff, M.; Silver, I.A. Energetics of the nerve terminal in relation to central nervous system function. Biochem. Soc. Trans. 1994, 22, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erecinska, M.; Silver, I.A. Ions and energy in mammalian brain. Prog. Neurobiol. 1994, 43, 37–71. [Google Scholar] [CrossRef] [PubMed]
- Duggett, N.A.; Griffiths, L.A.; Flatters, S.J.L. Paclitaxel-induced painful neuropathy is associated with changes in mitochondrial bioenergetics, glycolysis, and an energy deficit in dorsal root ganglia neurons. Pain 2017, 158, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Trevisiol, A.; Saab, A.S.; Winkler, U.; Marx, G.; Imamura, H.; Mobius, W.; Kusch, K.; Nave, K.A.; Hirrlinger, J. Monitoring ATP dynamics in electrically active white matter tracts. Elife 2017, 6, e24241. [Google Scholar] [CrossRef]
- Neishabouri, A.M.; Faisal, A. The metabolic efficiency of myelinated vs unmyelinated axons. BMC Neurosci. 2011, 12, P100. [Google Scholar] [CrossRef] [Green Version]
- Nolano, M.; Simone, D.A.; Wendelschafer-Crabb, G.; Johnson, T.; Hazen, E.; Kennedy, W.R. Topical capsaicin in humans: Parallel loss of epidermal nerve fibers and pain sensation. Pain 1999, 81, 135–145. [Google Scholar] [CrossRef]
- Cavaletti, G.; Cavalletti, E.; Oggioni, N.; Sottani, C.; Minoia, C.; D’Incalci, M.; Zucchetti, M.; Marmiroli, P.; Tredici, G. Distribution of paclitaxel within the nervous system of the rat after repeated intravenous administration. Neurotoxicology 2000, 21, 389–393. [Google Scholar] [CrossRef]
- Carozzi, V.A.; Canta, A.; Chiorazzi, A. Chemotherapy-induced peripheral neuropathy: What do we know about mechanisms? Neurosci. Lett. 2015, 596, 90–107. [Google Scholar] [CrossRef]
- Stephens, K.E.; Zhou, W.; Ji, Z.; Chen, Z.; He, S.; Ji, H.; Guan, Y.; Taverna, S.D. Sex differences in gene regulation in the dorsal root ganglion after nerve injury. BMC Genom. 2019, 20, 147. [Google Scholar] [CrossRef]
- Hwang, B.Y.; Kim, E.S.; Kim, C.H.; Kwon, J.Y.; Kim, H.K. Gender differences in paclitaxel-induced neuropathic pain behavior and analgesic response in rats. Korean J. Anesthesiol. 2012, 62, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathias, S.V.; Abou-Khalil, B.W. Ezogabine skin discoloration is reversible after discontinuation. Epilepsy Behav. Case Rep. 2017, 7, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Reed, N.; Liu, R.; Aizenman, E.; Wipf, P.; Tzounopoulos, T. Synthesis and Evaluation of Potent KCNQ2/3-Specific Channel Activators. Mol. Pharmacol. 2016, 89, 667–677. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Toro, G.; Xu, G.; Dang, D.; Prater, C.; Yang, Q. Paclitaxel Inhibits KCNQ Channels in Primary Sensory Neurons to Initiate the Development of Painful Peripheral Neuropathy. Cells 2022, 11, 4067. https://doi.org/10.3390/cells11244067
Wu Z, Toro G, Xu G, Dang D, Prater C, Yang Q. Paclitaxel Inhibits KCNQ Channels in Primary Sensory Neurons to Initiate the Development of Painful Peripheral Neuropathy. Cells. 2022; 11(24):4067. https://doi.org/10.3390/cells11244067
Chicago/Turabian StyleWu, Zizhen, Gabor Toro, Guoying Xu, Danny Dang, Charmaine Prater, and Qing Yang. 2022. "Paclitaxel Inhibits KCNQ Channels in Primary Sensory Neurons to Initiate the Development of Painful Peripheral Neuropathy" Cells 11, no. 24: 4067. https://doi.org/10.3390/cells11244067