
Elabela: Negative Regulation of Ferroptosis in Trophoblasts via the Ferritinophagy Pathway Implicated in the Pathogenesis of Preeclampsia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Sample Collection
2.2. Animal
2.3. Cell Culture
2.4. Transfection
2.5. Immunohistochemistry
2.6. Perls’ Prussian Blue Staining
2.7. H&E Staining
2.8. Assay for GSH, MDA and Iron
2.9. Elabela Elisa
2.10. RNA Extraction and RT-qPCR
2.11. Cell Counting Kit-8 Assay
2.12. Determination of Reactive Oxygen Species
2.13. Assessment of Lipid Peroxidation
2.14. Cellular Labile Iron Detection
2.15. Cellular Lysosomes Detection
2.16. Transmission Electron Microscopy
2.17. Autophagy Flux Analysis
2.18. Western Blotting
2.19. Matrigel Invasion Assay
2.20. Wound Healing Assay
2.21. DNA Synthesis Assay
2.22. mRNA Sequencing
2.23. Statistical Analyses
3. Results
3.1. More Severe Ferroptosis Presented in the Placentas of PE
3.2. Low Levels of Elabela in the Placenta and Circulation of the Pregnancy with Preeclampsia
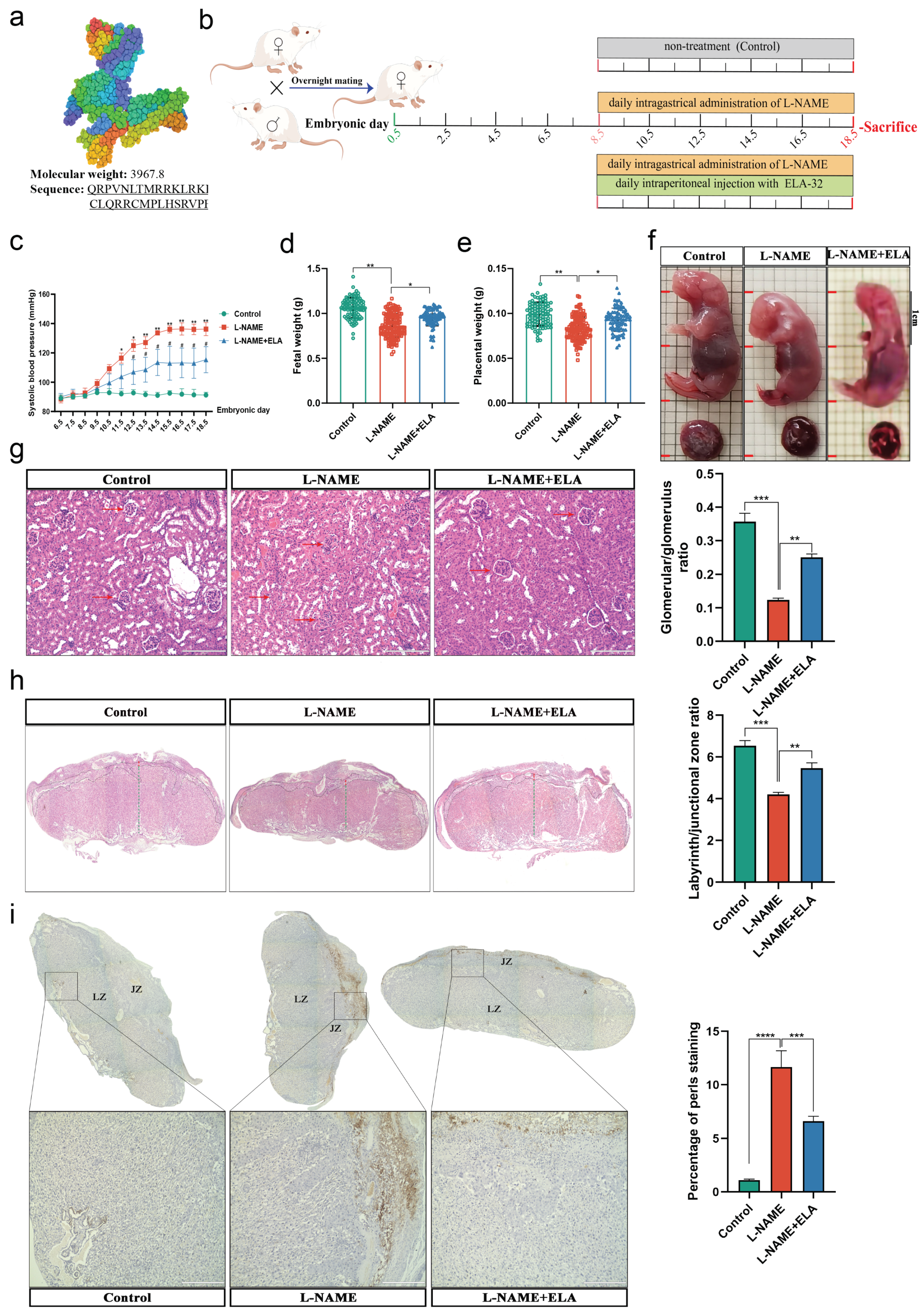
3.3. The PE-like and Ferroptosis Phenotypes in Mice Were Relieved by Elabela Administration
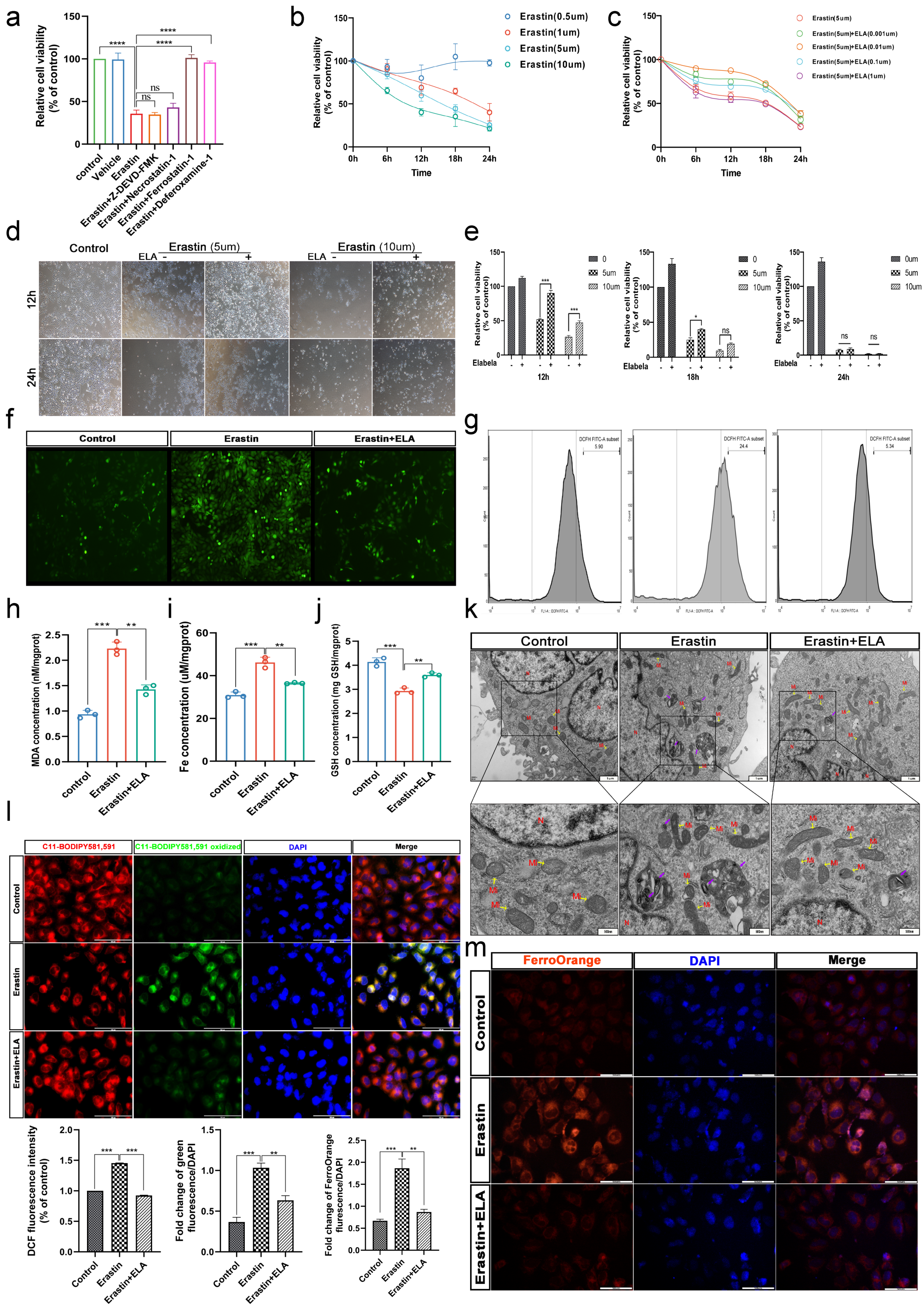
3.4. Erastin and RSL3 Induce HTR-8/SVneo Death in a Time-Dose-Dependent Manner
3.5. Elabela Can Rescue Ferroptosis Induced by Erastin
3.6. Elabela Could Reduce Erastin-Exacerbated Oxidative Stress and Lipid Peroxidation, and Rescue Cell Dysfunction
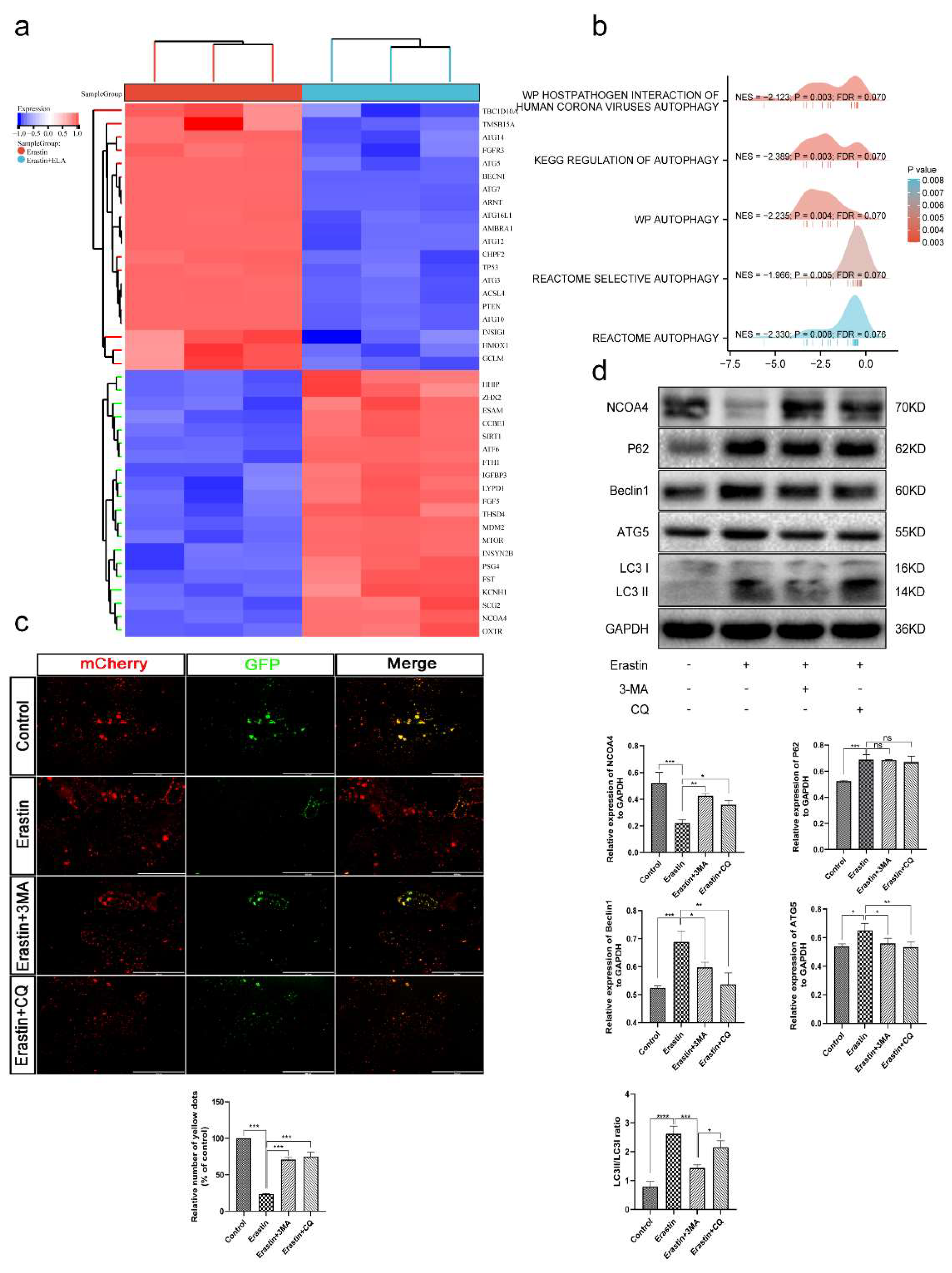
3.7. Ferroptosis Is an Autophagy-Dependent form of Cell Death
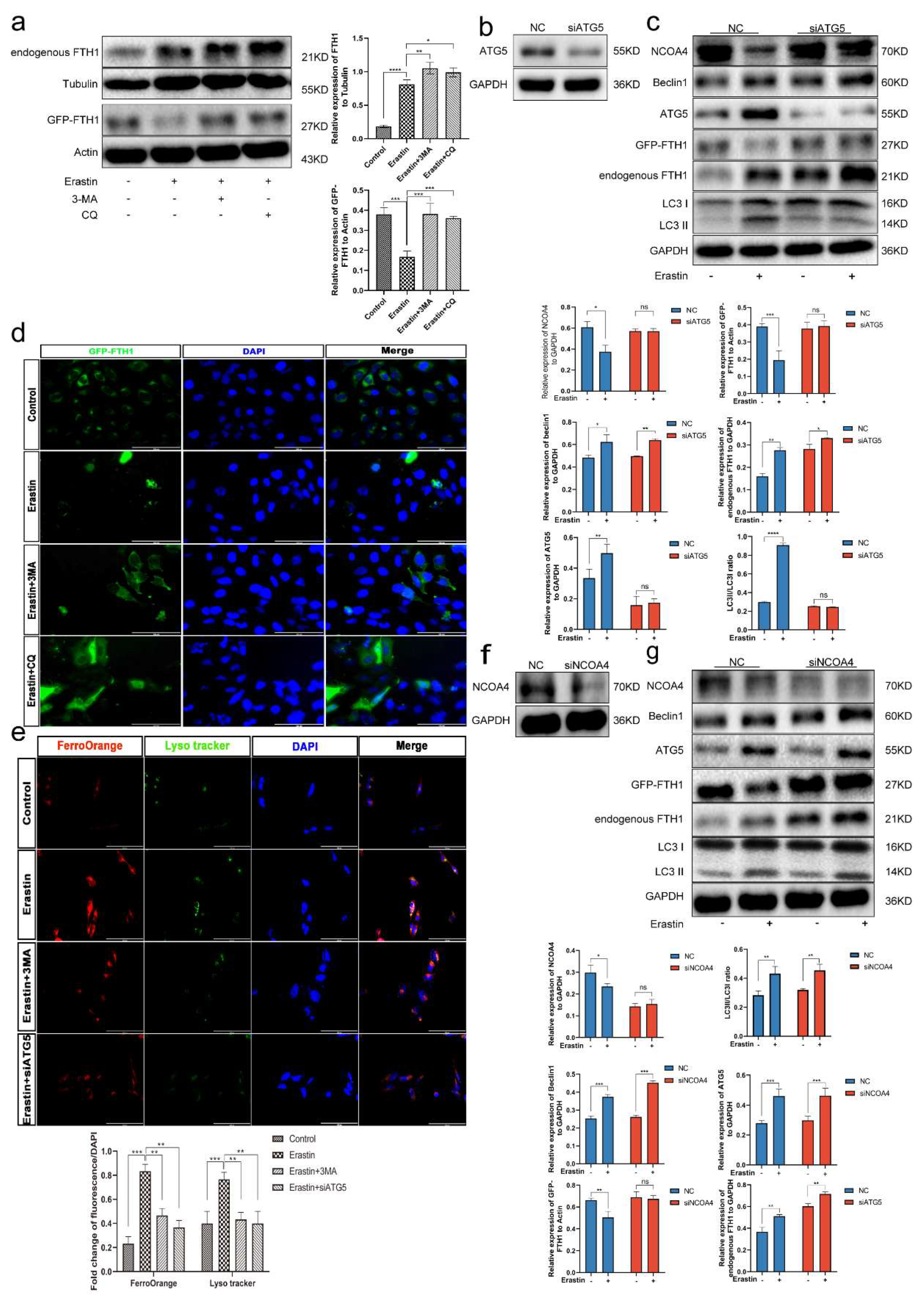
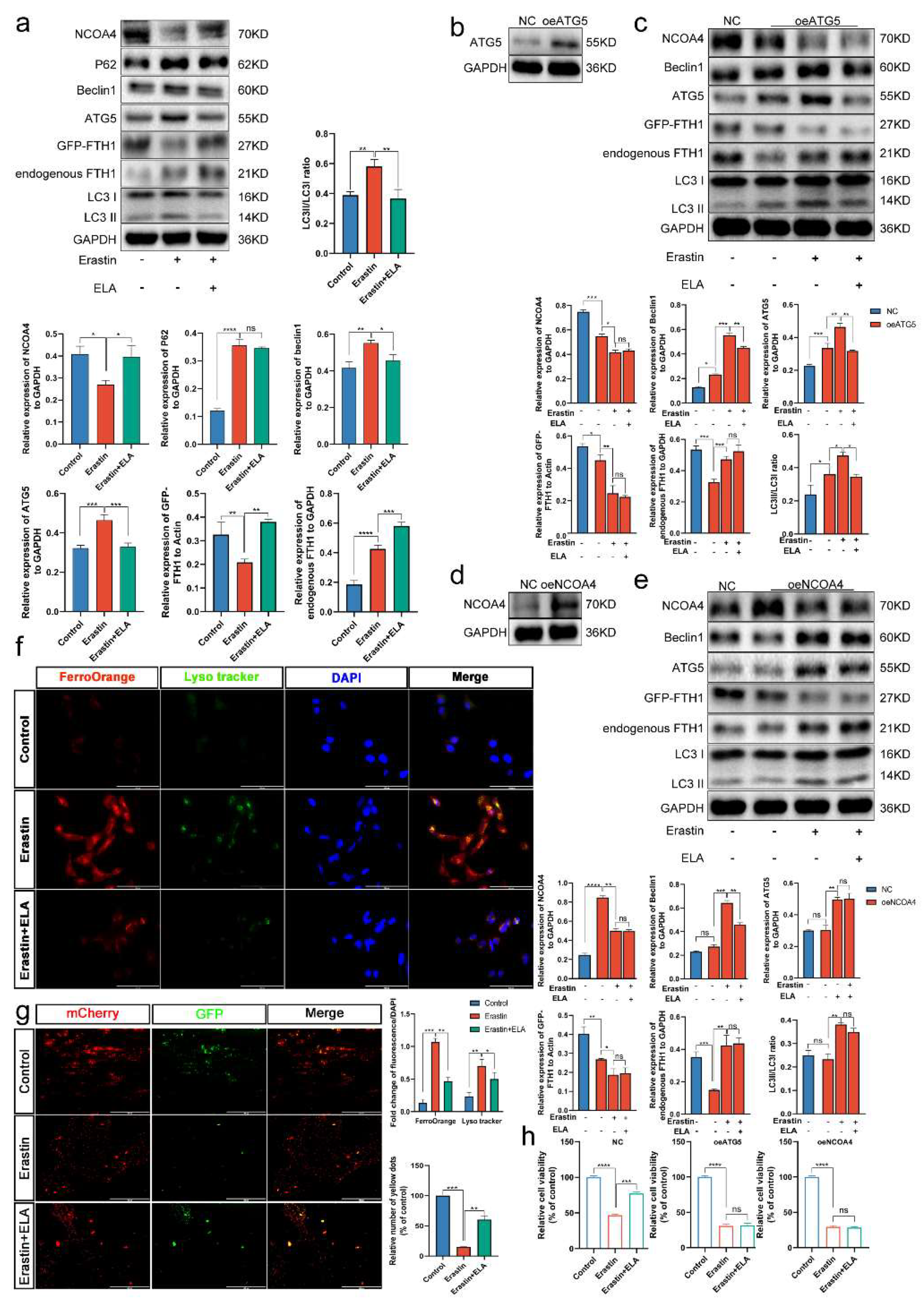
3.8. Ferritinophagy Is Involved in Ferroptosis in Trophoblasts
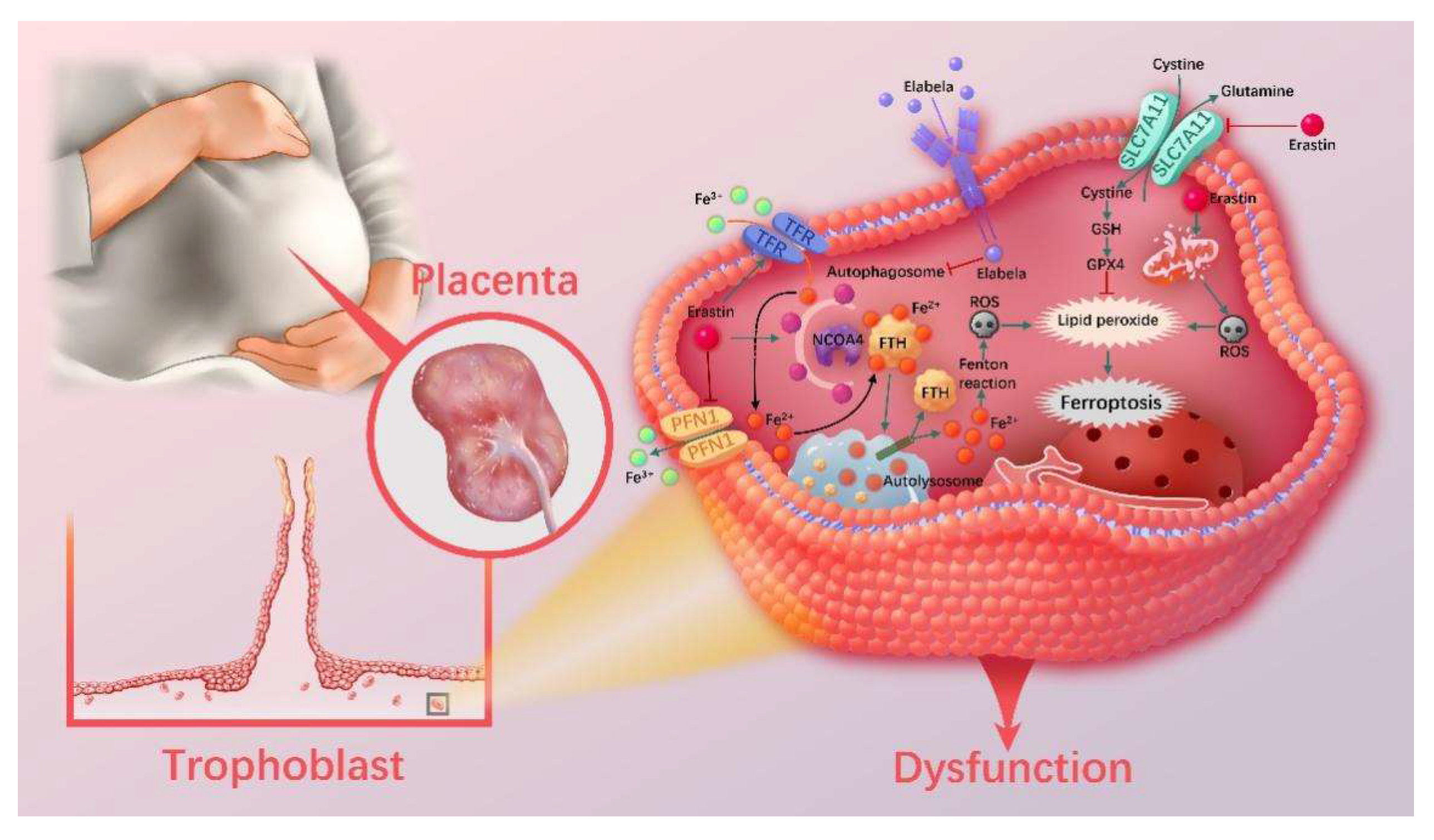
3.9. The Ferritinophagy Pathway Plays a Role in the Regulation of Ferroptosis by Elabela
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviation
| PE | Preeclampsia |
| ELA | Elabela |
| FGR | Fetal growth restriction |
| AngII | Angiotensin II |
| APJ | Angiotensin II type 1 receptor-related protein |
| PI3K | Phosphatidylinositol 3-kinase |
| AKT | Protein kinase B |
| ACOG | American College of Obstetricians and Gynecologists |
| LIP | Labile iron pool |
| GSH | Glutathione |
| MDA | Malondialdehyde |
| IHC | Immunohistochemistry |
| 4-HNE | 4-Hydroxynonenal |
| ELISA | Enzyme-linked immunosorbent assay |
| RT-qPCR | Real-time quantitative polymerase chain reaction |
| CCK8 | Cell counting kit-8 |
| ROS | Reactive oxygen species |
| WB | Western blotting |
| CTB | Cytotrophoblast |
| STB | Syncytiotrophoblast |
| iEVT | Interstitial extravillous trophoblast |
| 3-MA | 3-Methyladenine |
| CQ | Chloroquine |
| EdU | 5-Ethynyl-2’-deoxyuridine |
| MOI | Multiplicity of infection |
| Ad | Adenovirus |
| OE | Overexpression |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GSEA | Gene Set Enrichment Analysis |
References
- FIGO Working Group on Good Clinical Practice in Maternal—Fetal Medicine; Di Renzo, G.C.; Fonseca, E.; Gratacos, E.; Hassan, S.; Kurtser, M.; Malone, F.; Nambiar, S.; Nicolaides, K.; Sierra, N.; et al. Good clinical practice advice: First trimester screening and prevention of pre-eclampsia in singleton pregnancy. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 2019, 144, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Umesawa, M.; Kobashi, G. Epidemiology of hypertensive disorders in pregnancy: Prevalence, risk factors, predictors and prognosis. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2017, 40, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet 2020, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, E.; Rahbar Saadat, Y.; Hosseiniyan Khatibi, S.M.; Nariman-Saleh-Fam, Z.; Bastami, M.; Zununi Vahed, F.; Ardalan, M.; Zununi Vahed, S. Pre-Eclampsia: Microbiota possibly playing a role. Pharmacol. Res. 2020, 155, 104692. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Chattipakorn, N. Cardiac ferroptosis: New jigsaw in SCD puzzles. Blood 2022, 139, 811–812. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Xu, Y.; Li, K.; Zhou, L.; Qiao, H.; Xu, Q.; Zhao, J. The Role of Ferroptosis in Blood-Brain Barrier Injury. Cell. Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Martín-Saiz, L.; Guerrero-Mauvecin, J.; Martín-Sanchez, D.; Fresnedo, O.; Gómez, M.J.; Carrasco, S.; Cannata-Ortiz, P.; Ortiz, A.; Fernandez, J.A.; Sanz, A.B. Ferrostatin-1 modulates dysregulated kidney lipids in acute kidney injury. J. Pathol. 2022, 257, 285–299. [Google Scholar] [CrossRef]
- Xu, W.; Deng, H.; Hu, S.; Zhang, Y.; Zheng, L.; Liu, M.; Chen, Y.; Wei, J.; Yang, H.; Lv, X. Role of Ferroptosis in Lung Diseases. J. Inflamm. Res. 2021, 14, 2079–2090. [Google Scholar] [CrossRef]
- Zhang, H.; He, Y.; Wang, J.X.; Chen, M.H.; Xu, J.J.; Jiang, M.H.; Feng, Y.L.; Gu, Y.F. miR-30-5p-mediated ferroptosis of trophoblasts is implicated in the pathogenesis of preeclampsia. Redox. Biol. 2020, 29, 101402. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Wang, Q.; Ding, B.; Gong, Y.; Wu, Y.; Sun, J.; Wang, X.; Liu, L.; Zhang, F.; Du, D.; et al. Expression profiles and functions of ferroptosis-related genes in the placental tissue samples of early- and late-onset preeclampsia patients. BMC Pregnancy Childbirth 2022, 22, 87. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Zhou, X. Activation of Nrf2 signaling protects hypoxia-induced HTR-8/SVneo cells against ferroptosis. J. Obstet. Gynaecol. Res. 2021, 47, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson, L.; Masoumi, Z.; Hansson, L.R.; Hansson, S.R. The roles of free iron, heme, haemoglobin, and the scavenger proteins haemopexin and alpha-1-microglobulin in preeclampsia and fetal growth restriction. J. Intern. Med. 2021, 290, 952–968. [Google Scholar] [CrossRef]
- Ng, S.W.; Norwitz, S.G.; Norwitz, E.R. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3283. [Google Scholar] [CrossRef] [Green Version]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; St Croix, C.M.; Wallace, C.T.; Parry, S.; et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef]
- Beharier, O.; Kajiwara, K.; Sadovsky, Y. Ferroptosis, trophoblast lipotoxic damage, and adverse pregnancy outcome. Placenta 2021, 108, 32–38. [Google Scholar] [CrossRef]
- Chng, S.C.; Ho, L.; Tian, J.; Reversade, B. ELABELA: A hormone essential for heart development signals via the apelin receptor. Dev. Cell 2013, 27, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, D.; Wang, M.; Wang, Q.; Kouznetsova, J.; Yang, R.; Qian, K.; Wu, W.; Shuldiner, A.; Sztalryd, C.; et al. Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci. Rep. 2015, 5, 8170. [Google Scholar] [CrossRef] [Green Version]
- Couvineau, P.; Llorens-Cortes, C.; Iturrioz, X. Elabela/Toddler and apelin bind differently to the apelin receptor. FASEB J. 2020, 34, 7989–8000. [Google Scholar] [CrossRef]
- Yi, Y.; Tsai, S.H.; Cheng, J.C.; Wang, E.Y.; Anglesio, M.S.; Cochrane, D.R.; Fuller, M.; Gibb, E.A.; Wei, W.; Huntsman, D.G.; et al. APELA promotes tumour growth and cell migration in ovarian cancer in a p53-dependent manner. Gynecol. Oncol. 2017, 147, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Perjés, Á.; Kilpiö, T.; Ulvila, J.; Magga, J.; Alakoski, T.; Szabó, Z.; Vainio, L.; Halmetoja, E.; Vuolteenaho, O.; Petäjä-Repo, U.; et al. Characterization of apela, a novel endogenous ligand of apelin receptor, in the adult heart. Basic Res. Cardiol. 2016, 111, 2. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Tan, S.Y.; Wee, S.; Wu, Y.; Tan, S.J.; Ramakrishna, N.B.; Chng, S.C.; Nama, S.; Szczerbinska, I.; Chan, Y.S.; et al. ELABELA Is an Endogenous Growth Factor that Sustains hESC Self-Renewal via the PI3K/AKT Pathway. Cell Stem Cell 2015, 17, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; van Dijk, M.; Chye, S.T.J.; Messerschmidt, D.M.; Chng, S.C.; Ong, S.; Yi, L.K.; Boussata, S.; Goh, G.H.; Afink, G.B.; et al. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science 2017, 357, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.W.; Li, X.T.; Zhang, Z.Z.; Liu, Y.; Song, J.W.; Liu, X.M.; Chen, Y.H.; Wang, N.; Guo, Y.; Liang, L.R.; et al. Elabela blunts doxorubicin-induced oxidative stress and ferroptosis in rat aortic adventitial fibroblasts by activating the KLF15/GPX4 signaling. Cell Stress Chaperones 2022. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Q.; Chen, E.; Zhao, P.; Xu, Y. Elabela promotes the retinal angiogenesis by inhibiting ferroptosis during the vaso-obliteration phase in mouse oxygen-induced retinopathy model. FASEB J. 2022, 36, e22257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- ACOG Practice Bulletin No.202: Gestational Hypertension and Preeclampsia. Obs. Gynecol. 2019, 133, 1. [CrossRef]
- Burke, S.D.; Zsengellér, Z.K.; Khankin, E.V.; Lo, A.S.; Rajakumar, A.; DuPont, J.J.; McCurley, A.; Moss, M.E.; Zhang, D.; Clark, C.D.; et al. Soluble fms-like tyrosine kinase 1 promotes angiotensin II sensitivity in preeclampsia. J. Clin. Investig. 2016, 126, 2561–2574. [Google Scholar] [CrossRef]
- Jensen, E.C. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat. Rec. 2013, 296, 378–381. [Google Scholar] [CrossRef]
- Lee, D.C.; Romero, R.; Kim, J.S.; Tarca, A.L.; Montenegro, D.; Pineles, B.L.; Kim, E.; Lee, J.; Kim, S.Y.; Draghici, S.; et al. miR-210 targets iron-sulfur cluster scaffold homologue in human trophoblast cell lines: Siderosis of interstitial trophoblasts as a novel pathology of preterm preeclampsia and small-for-gestational-age pregnancies. Am. J. Pathol. 2011, 179, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xiong, L.; Jin, H.; Yu, J.; Li, X.; Fu, H.; Wen, L.; Qi, H.; Tong, C.; Saffery, R.; et al. Advanced maternal age causes premature placental senescence and malformation via dysregulated α-Klotho expression in trophoblasts. Aging Cell 2021, 20, e13417. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hu, H.; Lin, M.; Chen, L.; Liu, M.; Li, H.; Quan, S. ELABELA alleviates syncytiotrophoblast hypoxia/reoxygenation injury and preeclampsia-like symptoms in mice by reducing apoptosis. Placenta 2021, 106, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tao, M.; Ma, X.; Hu, Y.; Huang, G.; Qiu, A.; Zhuang, S.; Liu, N. Delayed treatment with an autophagy inhibitor 3-MA alleviates the progression of hyperuricemic nephropathy. Cell Death Dis. 2020, 11, 467. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef]
- Chen, H.; Wang, L.; Wang, W.; Cheng, C.; Zhang, Y.; Zhou, Y.; Wang, C.; Miao, X.; Wang, J.; Wang, C.; et al. ELABELA and an ELABELA Fragment Protect against AKI. J. Am. Soc. Nephrol. 2017, 28, 2694–2707. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.K.; Janowiak, M.; Badger, G.J.; Bernstein, I.M. Evidence for distinct preterm and term phenotypes of preeclampsia. J. Matern. Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obs. 2010, 23, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, J.; Homma, T.; Kobayashi, S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic. Res. 2020, 54, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Y.; Qu, H.; Xu, F.; Hu, H.; Zhang, Q.; Ye, Y. Reduced ELABELA expression attenuates trophoblast invasion through the PI3K/AKT/mTOR pathway in early onset preeclampsia. Placenta 2019, 87, 38–45. [Google Scholar] [CrossRef]
- Helker, C.S.; Schuermann, A.; Pollmann, C.; Chng, S.C.; Kiefer, F.; Reversade, B.; Herzog, W. The hormonal peptide Elabela guides angioblasts to the midline during vasculogenesis. Elife 2015, 4, e06726. [Google Scholar] [CrossRef]
- Carrasco-Wong, I.; Aguilera-Olguín, M.; Escalona-Rivano, R.; Chiarello, D.I.; Barragán-Zúñiga, L.J.; Sosa-Macías, M.; Galaviz-Hernandez, C.; San Martín, S.; Gutiérrez, J. Syncytiotrophoblast stress in early onset preeclampsia: The issues perpetuating the syndrome. Placenta 2021, 113, 57–66. [Google Scholar] [CrossRef]
- McCarthy, F.P.; Kingdom, J.C.; Kenny, L.C.; Walsh, S.K. Animal models of preeclampsia; uses and limitations. Placenta 2011, 32, 413–419. [Google Scholar] [CrossRef]
- Motta, C.; Grosso, C.; Zanuzzi, C.; Molinero, D.; Picco, N.; Bellingeri, R.; Alustiza, F.; Barbeito, C.; Vivas, A.; Romanini, M.C. Effect of Sildenafil on Pre-Eclampsia-Like Mouse Model Induced By L-Name. Reprod. Domest. Anim. Zuchthyg. 2015, 50, 611–616. [Google Scholar] [CrossRef]
- Zuo, J.; Jiang, Z. Melatonin attenuates hypertension and oxidative stress in a rat model of L-NAME-induced gestational hypertension. Vasc. Med. 2020, 25, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Przybyl, L.; Haase, N.; Golic, M.; Rugor, J.; Solano, M.E.; Arck, P.C.; Gauster, M.; Huppertz, B.; Emontzpohl, C.; Stoppe, C.; et al. CD74-Downregulation of Placental Macrophage-Trophoblastic Interactions in Preeclampsia. Circ. Res. 2016, 119, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashi, Y.; Tomita, K.; Kuwahara, Y.; Roudkenar, M.H.; Roushandeh, A.M.; Igarashi, K.; Nagasawa, T.; Nishitani, Y.; Sato, T. Mitochondrial dysfunction promotes aquaporin expression that controls hydrogen peroxide permeability and ferroptosis. Free Radic. Biol. Med. 2020, 161, 60–70. [Google Scholar] [CrossRef]
- Li, J.; Lu, K.; Sun, F.; Tan, S.; Zhang, X.; Sheng, W.; Hao, W.; Liu, M.; Lv, W.; Han, W. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J. Transl. Med. 2021, 19, 96. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryzik, M.; Asperti, M.; Denardo, A.; Arosio, P.; Poli, M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim. Et Biophys. Acta. Mol. Cell Res. 2021, 1868, 118913. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [Green Version]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; François, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef]
- Lala, P.K.; Graham, C.H. Mechanisms of trophoblast invasiveness and their control: The role of proteases and protease inhibitors. Cancer Metastasis Rev. 1990, 9, 369–379. [Google Scholar] [CrossRef]
- Chen, Y.; Yi, X.; Huo, B.; He, Y.; Guo, X.; Zhang, Z.; Zhong, X.; Feng, X.; Fang, Z.M.; Zhu, X.H.; et al. BRD4770 functions as a novel ferroptosis inhibitor to protect against aortic dissection. Pharmacol. Res. 2022, 177, 106122. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, L.; Gan, B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol. Cell. Oncol. 2019, 6, 1536845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhang, X.; Ding, Y.; Xiong, H.; Xiang, S.; Wang, Y.; Li, H.; Liu, Z.; He, J.; Tao, Y.; et al. Elabela: Negative Regulation of Ferroptosis in Trophoblasts via the Ferritinophagy Pathway Implicated in the Pathogenesis of Preeclampsia. Cells 2023, 12, 99. https://doi.org/10.3390/cells12010099
Yang H, Zhang X, Ding Y, Xiong H, Xiang S, Wang Y, Li H, Liu Z, He J, Tao Y, et al. Elabela: Negative Regulation of Ferroptosis in Trophoblasts via the Ferritinophagy Pathway Implicated in the Pathogenesis of Preeclampsia. Cells. 2023; 12(1):99. https://doi.org/10.3390/cells12010099
Chicago/Turabian StyleYang, Huan, Xuemei Zhang, Yubin Ding, Hui Xiong, Shaojian Xiang, Yang Wang, Huanhuan Li, Zheng Liu, Jie He, Yuelan Tao, and et al. 2023. "Elabela: Negative Regulation of Ferroptosis in Trophoblasts via the Ferritinophagy Pathway Implicated in the Pathogenesis of Preeclampsia" Cells 12, no. 1: 99. https://doi.org/10.3390/cells12010099