
Ribosomal Dysfunction Is a Common Pathomechanism in Different Forms of Trichothiodystrophy
 , ,
, , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Lentivirus Generation, Lentiviral Transfection
2.3. CRISPR CAS9 Knockout/Knockin Kit
2.4. FACs Single Cell Sorting
2.5. RNA Isolation and cDNA Synthesis
2.6. qPCR Assay Validation
2.7. Real-Time qPCR Standard Curve Analysis
2.8. Western Blot Analysis
2.9. Immunofluorescence Staining
2.10. Transfection and Luciferase Assay (Plasmids)
2.11. Luciferase Assay with mRNA Transfection
2.12. Proliferation Analysis
2.13. Protein Synthesis Assay (OPP Labeling)
2.14. BisANS Assay
2.15. Northern Blot Analysis
2.16. Ratio Analysis of Multiple Precursors (RAMP)
2.17. Polysomal Profiling
2.18. Carbonylation Assay
2.19. Statistical Analysis
3. Results
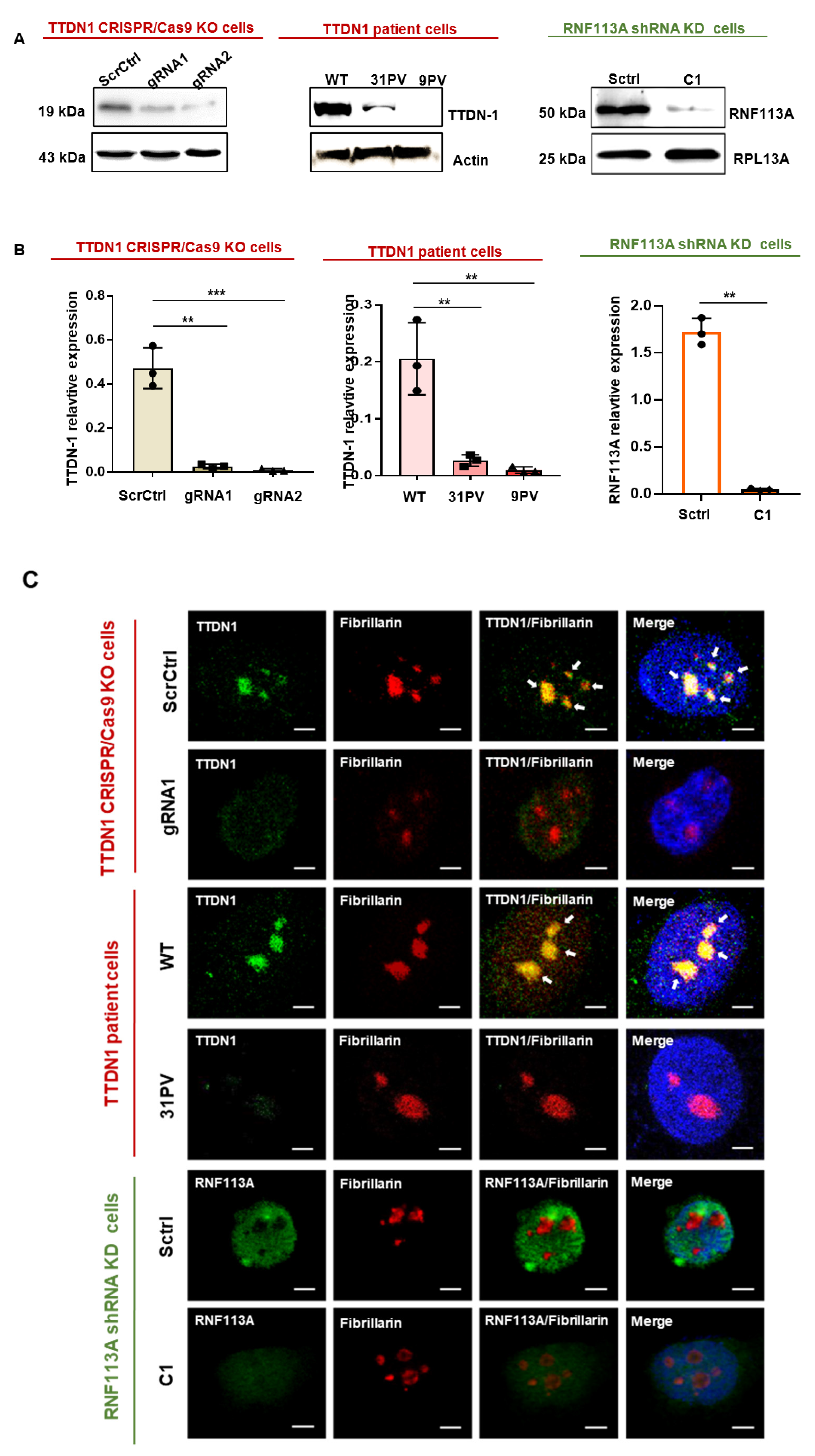
3.1. Generation of Knockout and Knockdown Cells
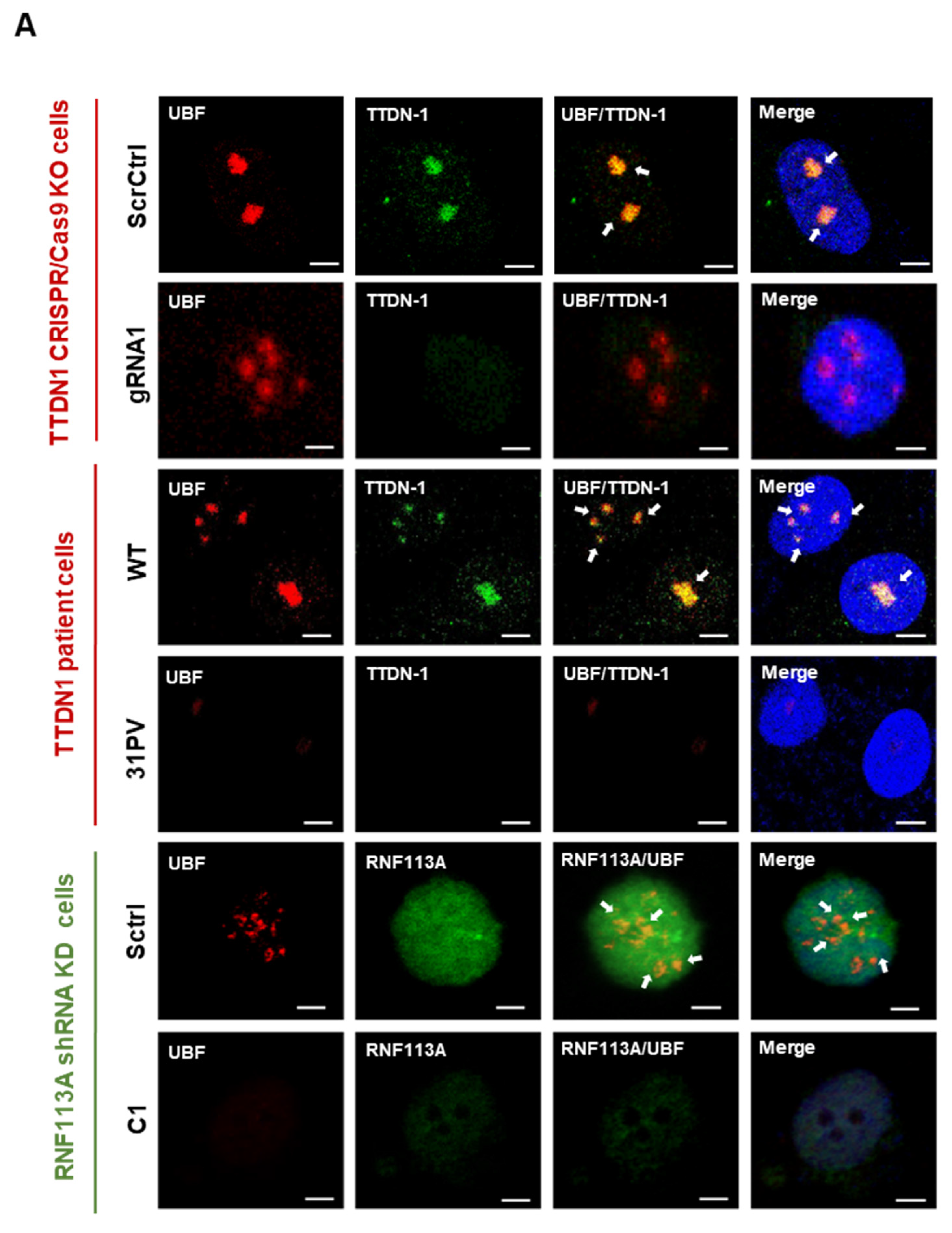
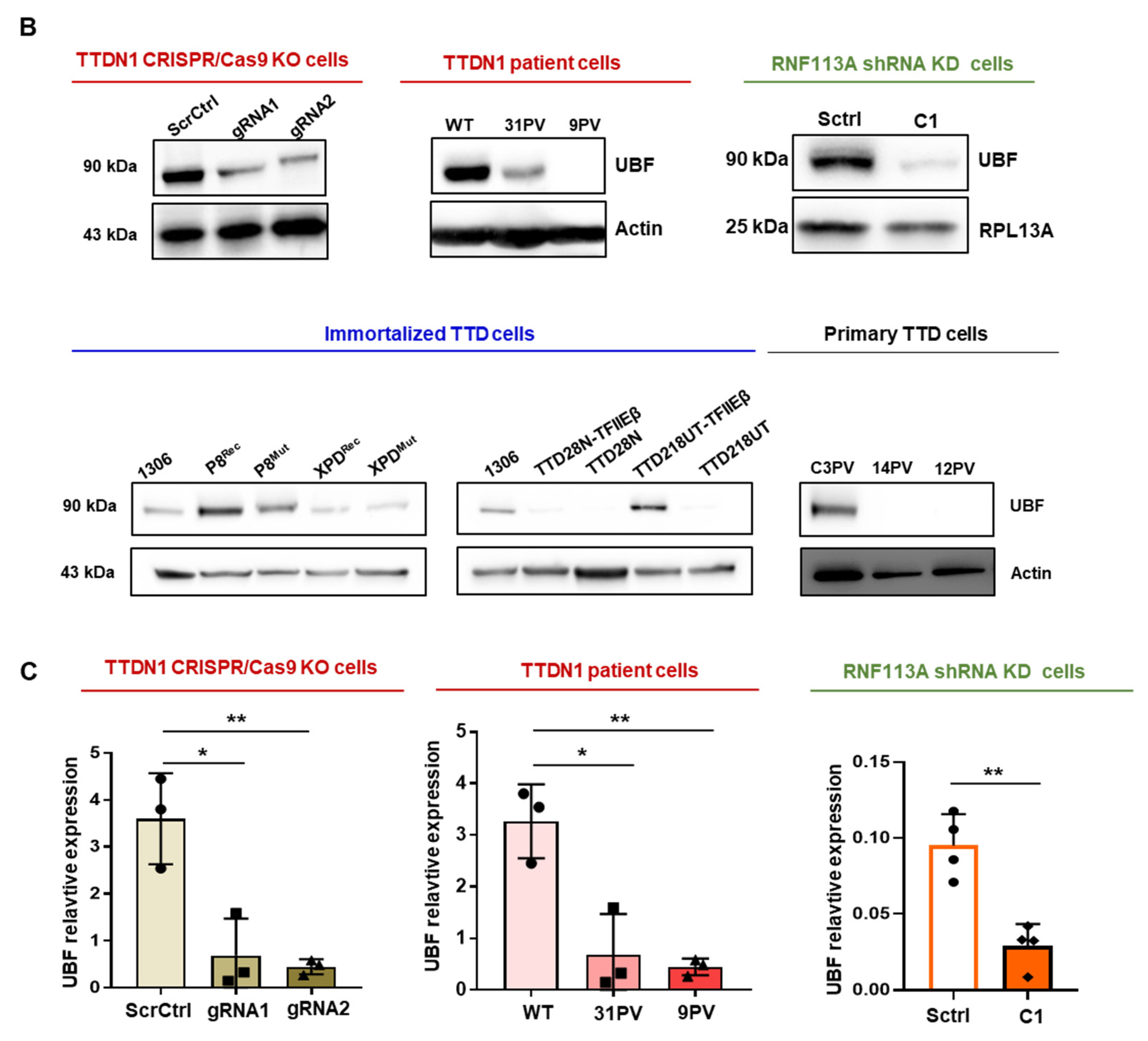
3.2. Reduced TTDN1 and RNF113A Impact on the RNA Polymerase I Transcription Factor UBF
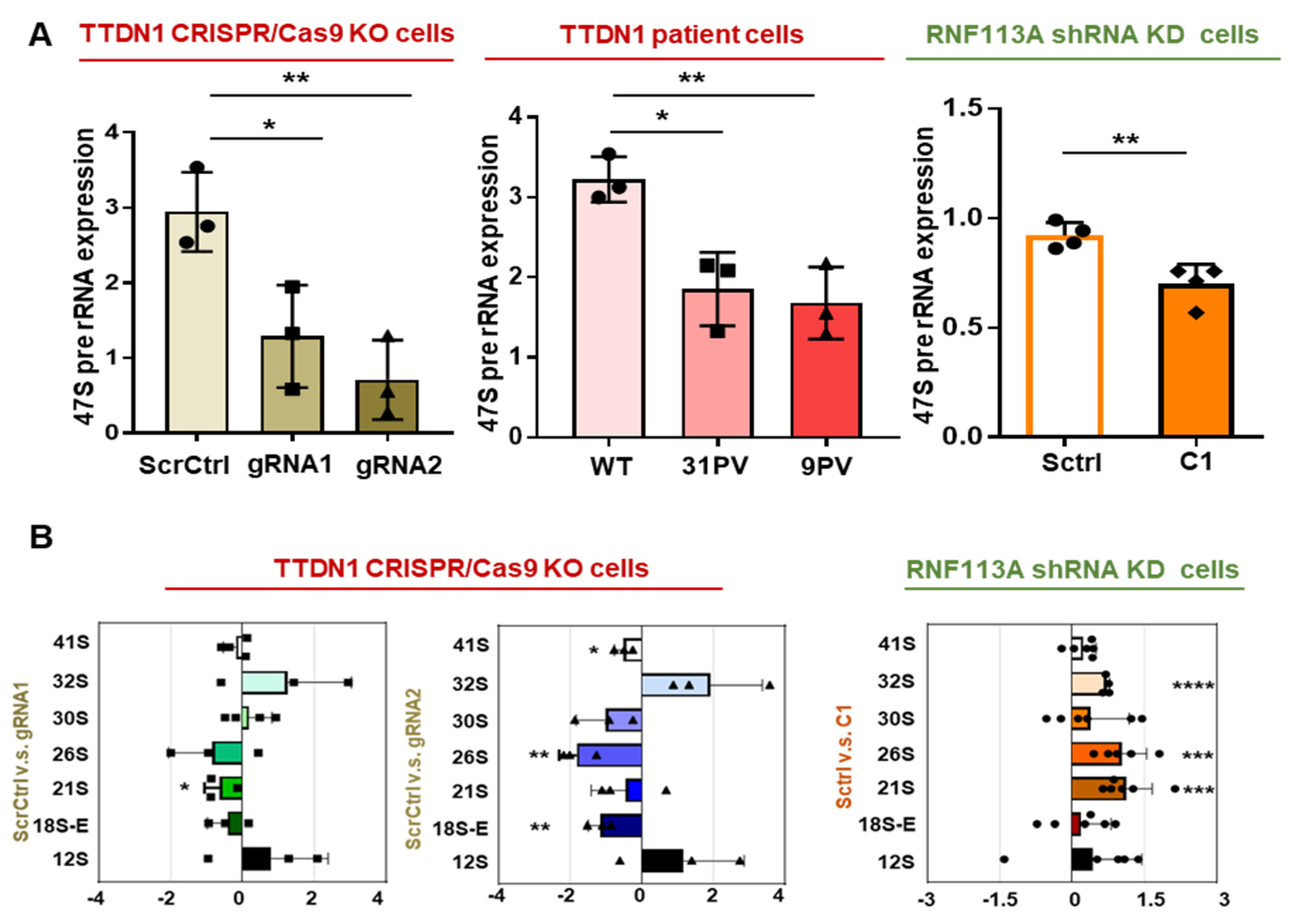
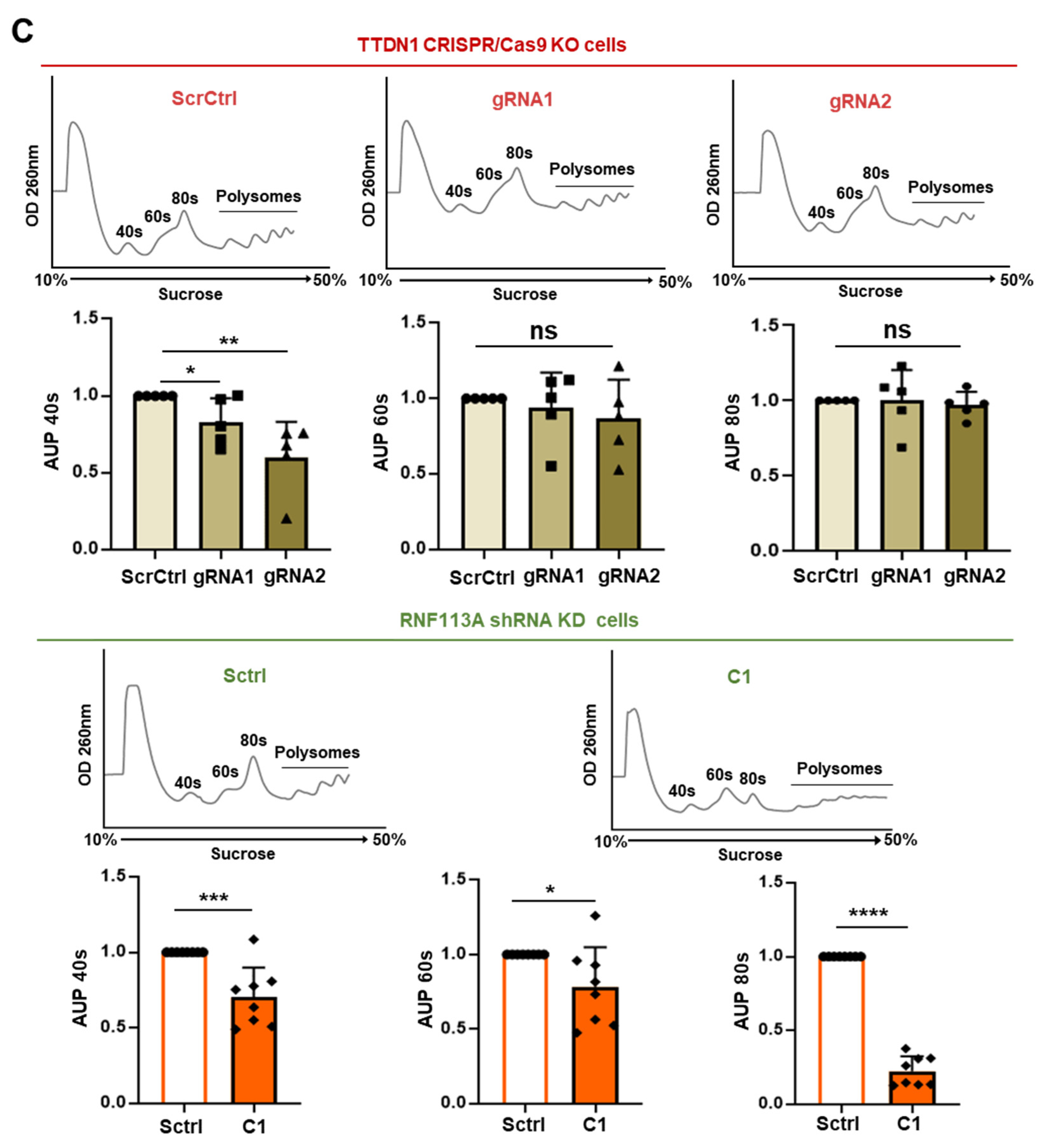
3.3. rRNA Transcription and Ribosomal Subunits Are Differentially Affected in the TTDN1 Knockout and RNF113A Knockdown Cells
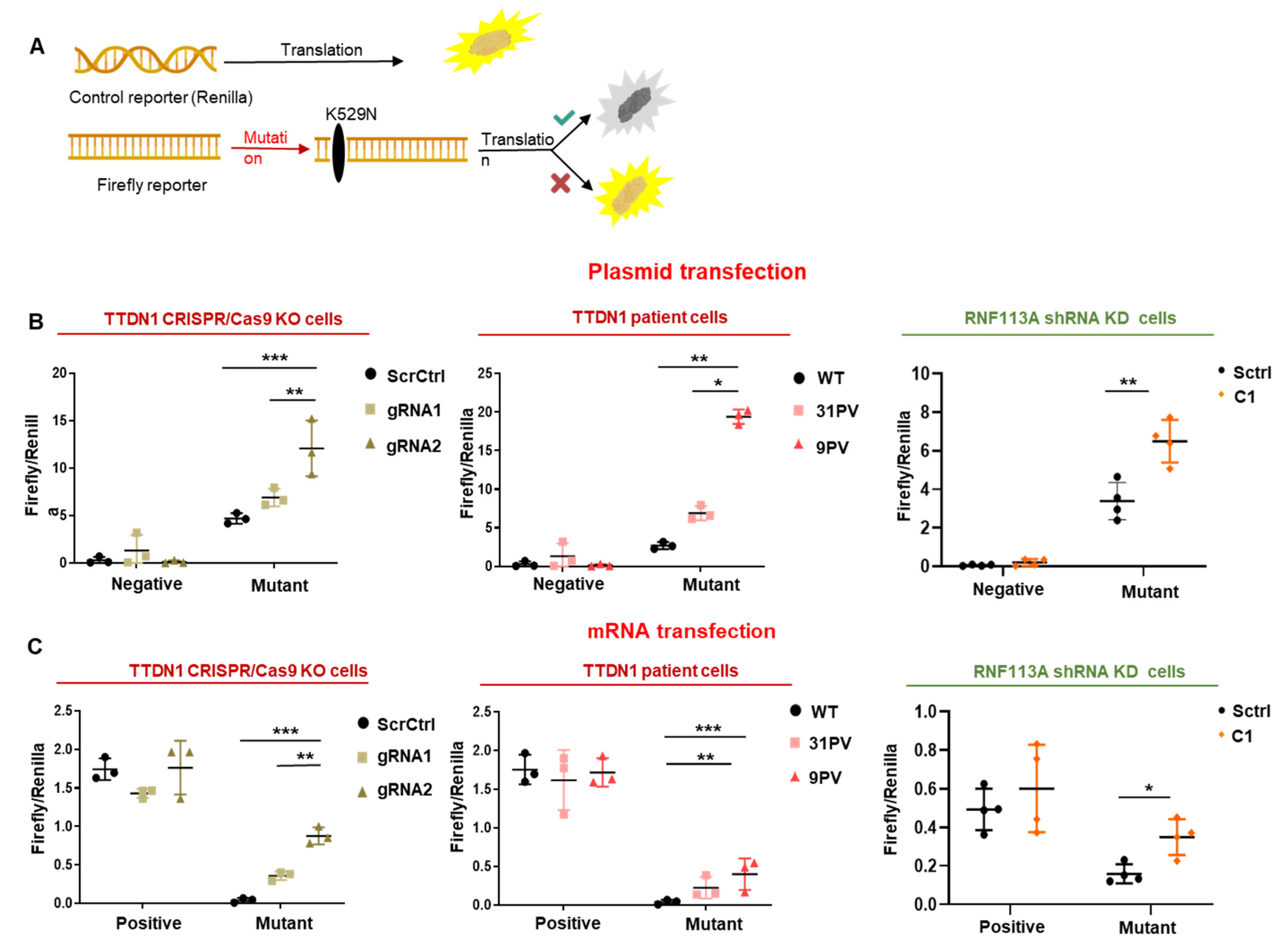
3.4. The Accuracy of the Translation Process at the Ribosome Is Decreased in TTDN1 and RNF113A Cells
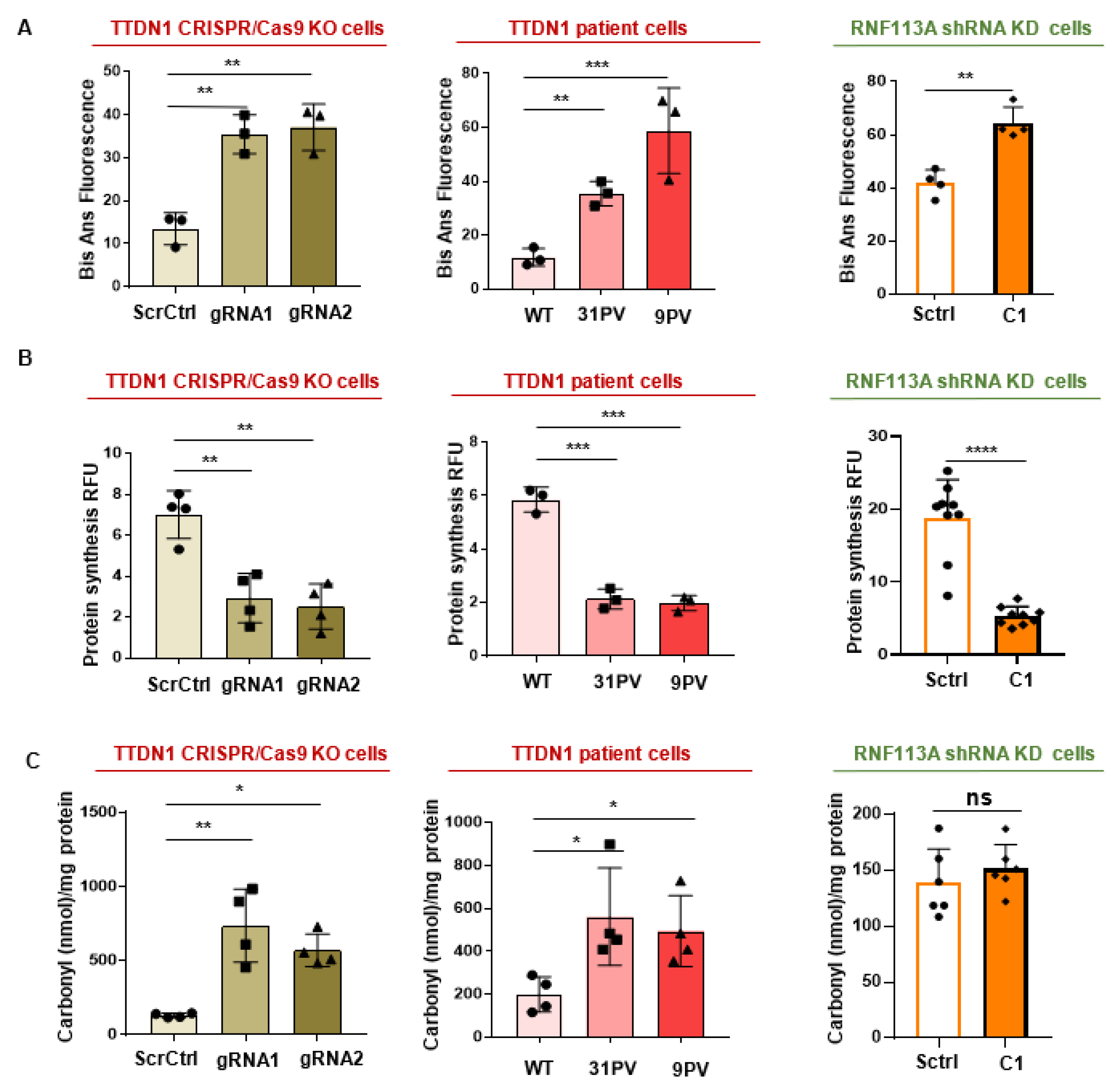
3.5. Elevated Misfolded Proteins and a Reduced Protein Synthesis in the Affected Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stefanini, M.; Botta, E.; Lanzafame, M.; Orioli, D. Trichothiodystrophy: From basic mechanisms to clinical implications. DNA Repair 2010, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Faghri, S.; Tamura, D.; Kraemer, K.H.; Digiovanna, J.J. Trichothiodystrophy: A systematic review of 112 published cases char-acterises a wide spectrum of clinical manifestations. J. Med. Genet. 2008, 45, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Ferri, D.; Orioli, D.; Botta, E. Heterogeneity and overlaps in nucleotide excision repair disorders. Clin. Genet. 2020, 97, 12–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, E.; Egly, J.M. Trichothiodystrophy, a transcription syndrome. Trends Genet. 2001, 17, 279–286. [Google Scholar] [CrossRef]
- Kuschal, C.; Botta, E.; Orioli, D.; Digiovanna, J.J.; Seneca, S.; Keymolen, K.; Tamura, D.; Heller, E.; Khan, S.G.; Caligiuri, G.; et al. GTF2E2 Mutations Destabilize the General Transcription Factor Complex TFIIE in Individuals with DNA Repair-Proficient Trichothiodystrophy. Am. J. Hum. Genet. 2016, 98, 627–642. [Google Scholar] [CrossRef] [Green Version]
- Theil, A.F.; Mandemaker, I.K.; van den Akker, E.; Swagemakers, S.M.A.; Raams, A.; Wust, T.; Marteijn, J.A.; Giltay, J.C.; Co-lombijn, R.M.; Moog, U.; et al. Trichothiodystrophy causative TFIIEbeta mutation affects transcription in highly differentiated tissue. Hum. Mol. Genet. 2017, 26, 4689–4698. [Google Scholar] [CrossRef] [Green Version]
- Theil, A.F.; Botta, E.; Raams, A.; Smith, D.E.C.; Mendes, M.I.; Caligiuri, G.; Giachetti, S.; Bione, S.; Carriero, R.; Liberi, G.; et al. Bi-Allelic TARS Mutations Are Associated with Brittle Hair Phenotype. Am. J. Hum. Genet. 2019, 105, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Botta, E.; Theil, A.F.; Raams, A.; Caligiuri, G.; Giachetti, S.; Bione, S.; Accadia, M.; Lombardi, A.; Smith, D.E.C.; Mendes, M.I.; et al. Protein instability associated with AARS1 and MARS1 mutations causes trichothiodystrophy. Hum. Mol. Genet. 2021, 30, 1711–1720. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Amann, D.; Ren, Y.; Saarialho-Kere, U.; Avidan, N.; Gentles, S.; MacDonald, J.R.; Puffenberger, E.G.; Chris-tiano, A.M.; Martinez-Mir, A.; et al. Identification of C7orf11 (TTDN1) gene mutations and genetic heterogeneity in nonpho-tosensitive trichothiodystrophy. Am. J. Hum. Genet. 2005, 76, 510–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, M.A.; Dudding-Byth, T.; Crock, P.A.; Botta, E.; Christie, L.M.; Nardo, T.; Caligiuri, G.; Hobson, L.; Boyle, J.; Mansour, A.; et al. A novel X-linked trichothiodystrophy associated with a nonsense mutation in RNF113A. J. Med. Genet. 2015, 52, 269–274. [Google Scholar] [CrossRef]
- Khalid, F.; Phan, T.; Qiang, M.; Maity, P.; Lasser, T.; Wiese, S.; Penzo, M.; Alupei, M.; Orioli, D.; Scharffetter-Kochanek, K.; et al. TFIIH mutations can impact on translational fidelity of the ribosome. Hum. Mol. Genet. 2022, 32, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Maity, P.; Ludwig, C.; Streit, L.; Michaelis, J.; Tsesmelis, M.; Scharffetter-Kochanek, K.; Iben, S. Nucleolar TFIIE plays a role in ribosomal biogenesis and performance. Nucleic Acids Res. 2021, 49, 11197–11210. [Google Scholar] [CrossRef] [PubMed]
- Assfalg, R.; Lebedev, A.; Gonzalez, O.G.; Schelling, A.; Koch, S.; Iben, S. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012, 40, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Nonnekens, J.; Perez-Fernandez, J.; Theil, A.F.; Gadal, O.; Bonnart, C.; Giglia-Mari, G. Mutations in TFIIH causing trichothi-odystrophy are responsible for defects in ribosomal RNA production and processing. Hum. Mol. Genet. 2013, 22, 2881–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, E.R.; Khan, S.G.; Kuschal, C.; Tamura, D.; DiGiovanna, J.J.; Kraemer, K.H. Mutations in the TTDN1 gene are associated with a distinct trichothiodystrophy phenotype. J. Investig. Dermatol. 2015, 135, 734–741. [Google Scholar] [CrossRef] [Green Version]
- Townley, A.B.; Luke, B.; Albino, B.; Timur, R.; Nicolas, S.; Sridhar, N.S.; Nathanial, E.C.; Fadhel, M.; Reilly, A.S.; Joshua, R.; et al. A condensate forming tether for lariat debranching enzyme is defective in non-photosensitive trichothiodystrophy. bioRxiv 2022. [Google Scholar]
- Zhang, Y.; Tian, Y.; Chen, Q.; Chen, D.; Zhai, Z.; Shu, H.B. TTDN1 is a Plk1-interacting protein involved in maintenance of cell cycle integrity. Cell. Mol. Life Sci. 2007, 64, 632–640. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef]
- Brickner, J.R.; Soll, J.M.; Lombardi, P.M.; Vagbo, C.B.; Mudge, M.C.; Oyeniran, C.; Rabe, R.; Jackson, J.; Sullender, M.E.; Blazosky, E.; et al. A ubiquitin-dependent signalling axis specific for ALKBH-mediated DNA dealkylation repair. Nature 2017, 551, 389–393. [Google Scholar] [CrossRef]
- Tsampoula, M.; Tarampoulous, I.; Manolakou, T.; Ninou, E.; Politis, P.K. The Neurodevelopmental Disorders Associated Gene Rnf113a Regulates Survival and Differentiation Properties of Neural Stem Cells. Stem Cells 2022, 40, 678–690. [Google Scholar] [CrossRef]
- Russell, J.; Zomerdijk, J.C. The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 2006, 73, 203–216. [Google Scholar] [CrossRef]
- Potapova, T.A.; Gerton, J.L. Ribosomal DNA and the nucleolus in the context of genome organization. Chromosome Res. 2019, 27, 109–127. [Google Scholar] [CrossRef]
- Bassler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu. Rev. Biochem. 2019, 88, 281–306. [Google Scholar] [CrossRef]
- Alupei, M.C.; Maity, P.; Esser, P.R.; Krikki, I.; Tuorto, F.; Parlato, R.; Penzo, M.; Schelling, A.; Laugel, V.; Montanaro, L.; et al. Loss of Proteostasis Is a Pathomechanism in Cockayne Syndrome. Cell Rep. 2018, 23, 1612–1619. [Google Scholar] [CrossRef]
- Qiang, M.; Khalid, F.; Phan, T.; Ludwig, C.; Scharffetter-Kochanek, K.; Iben, S. Cockayne Syndrome-Associated CSA and CSB Mutations Impair Ribosome Biogenesis, Ribosomal Protein Stability, and Global Protein Folding. Cells 2021, 10, 1616. [Google Scholar] [CrossRef]
- Salas-Marco, J.; Bedwell, D.M. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 2005, 348, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Treaster, S.B.; Ridgway, I.D.; Richardson, C.A.; Gaspar, M.B.; Chaudhuri, A.R.; Austad, S.N. Superior proteome stability in the longest lived animal. Age 2014, 36, 9597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukan, S.; Farewell, A.; Ballesteros, M.; Taddei, F.; Radman, M.; Nystrom, T. Protein oxidation in response to increased tran-scriptional or translational errors. Proc. Natl. Acad. Sci. USA 2000, 97, 5746–5749. [Google Scholar] [CrossRef]
- Horibata, K.; Iwamoto, Y.; Kuraoka, I.; Jaspers, N.G.; Kurimasa, A.; Oshimura, M.; Ichihashi, M.; Tanaka, K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15410–15415. [Google Scholar] [CrossRef] [PubMed]
- Nardo, T.; Oneda, R.; Spivak, G.; Vaz, B.; Mortier, L.; Thomas, P.; Orioli, D.; Laugel, V.; Stary, A.; Hanawalt, P.C.; et al. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 6209–6214. [Google Scholar] [CrossRef]
- Spivak, G.; Hanawalt, P.C. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syn-drome but normal in UV-sensitive syndrome fibroblasts. DNA Repair 2006, 5, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Theil, A.F.; Nonnekens, J.; Steurer, B.; Mari, P.O.; de Wit, J.; Lemaitre, C.; Marteijn, J.A.; Raams, A.; Maas, A.; Vermeij, M.; et al. Disruption of TTDA results in complete nucleotide excision repair deficiency and embryonic lethality. PLoS Genet. 2013, 9, e1003431. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Compe, E.; Le May, N.; Egly, J.M. TFIIH subunit alterations causing xeroderma pigmentosum and trichothi-odystrophy specifically disturb several steps during transcription. Am. J. Hum. Genet. 2015, 96, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, K.L.; van der Woude, M.; Davo-Martinez, C.; Dekkers, D.H.W.; Sabatella, M.; Demmers, J.A.A.; Vermeulen, W.; Lans, H.C. elegans TFIIH subunit GTF-2H5/TTDA is a non-essential transcription factor indispensable for DNA repair. Commun. Biol. 2021, 4, 1336. [Google Scholar] [CrossRef] [PubMed]
- Coin, F.; Proietti De Santis, L.; Nardo, T.; Zlobinskaya, O.; Stefanini, M.; Egly, J.M. p8/TTD-A as a repair-specific TFIIH subunit. Mol. Cell 2006, 21, 215–226. [Google Scholar] [CrossRef]
- Lee, J.W.; Beebe, K.; Nangle, L.A.; Jang, J.; Longo-Guess, C.M.; Cook, S.A.; Davisson, M.T.; Sundberg, J.P.; Schimmel, P.; Ackerman, S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006, 443, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Brilkova, M.; Nigri, M.; Kumar, H.S.; Moore, J.; Mantovani, M.; Keller, C.; Grimm, A.; Eckert, A.; Shcherbakov, D.; Akber-genov, R.; et al. Error-prone protein synthesis recapitulates early symptoms of Alzheimer disease in aging mice. Cell Rep. 2022, 40, 111433. [Google Scholar] [CrossRef]
- Sanij, E.; Hannan, R.D. The role of UBF in regulating the structure and dynamics of transcriptionally active rDNA chromatin. Epigenetics 2009, 4, 374–382. [Google Scholar] [CrossRef]
- Sanij, E.; Diesch, J.; Lesmana, A.; Poortinga, G.; Hein, N.; Lidgerwood, G.; Cameron, D.P.; Ellul, J.; Goodall, G.J.; Wong, L.H.; et al. A novel role for the Pol I transcription factor UBTF in maintaining genome stability through the regulation of highly tran-scribed Pol II genes. Genome Res. 2015, 25, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Chi, C.S.; Lee, H.F.; Tsai, C.R. Clinico-Radiological Phenotype of UBTF c.628G>A Pathogenic Variant-Related Neurodegenera-tion in Childhood: A Case Report and Literature Review. Brain Sci. 2022, 12, 1262. [Google Scholar] [CrossRef]
- Tremblay, M.G.; Sibai, D.S.; Valere, M.; Mars, J.C.; Lessard, F.; Hori, R.T.; Khan, M.M.; Stefanovsky, V.Y.; LeDoux, M.S.; Moss, T. Ribosomal DNA promoter recognition is determined in vivo by cooperation between UBTF1 and SL1 and is compromised in the UBTF-E210K neuroregression syndrome. PLoS Genet. 2022, 18, e1009644. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Nicolae, C.M.; Agrawal, P.B.; Mignot, C.; Payne, K.; Prasad, A.N.; Prasad, C.; Sadler, L.; Nava, C.; Mullen, T.E.; et al. Heterozygous De Novo UBTF Gain-of-Function Variant Is Associated with Neurodegeneration in Childhood. Am. J. Hum. Genet. 2017, 101, 267–273. [Google Scholar] [CrossRef]
- Hamdane, N.; Stefanovsky, V.Y.; Tremblay, M.G.; Nemeth, A.; Paquet, E.; Lessard, F.; Sanij, E.; Hannan, R.; Moss, T. Condi-tional inactivation of Upstream Binding Factor reveals its epigenetic functions and the existence of a somatic nucleolar pre-cursor body. PLoS Genet. 2014, 10, e1004505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, R.T.; Moshahid Khan, M.; Xiao, J.; Hargrove, P.W.; Moss, T.; LeDoux, M.S. Behavioral and molecular effects of Ubtf knockout and knockdown in mice. Brain Res. 2022, 1793, 148053. [Google Scholar] [CrossRef]
- Hernandez-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M. Transcription and genome integrity. DNA Repair 2022, 118, 103373. [Google Scholar] [CrossRef]
- Assfalg, R.; Alupei, M.C.; Wagner, M.; Koch, S.; Gonzalez, O.G.; Schelling, A.; Scharffetter-Kochanek, K.; Iben, S. Cellular sensi-tivity to UV-irradiation is mediated by RNA polymerase I transcription. PLoS ONE 2017, 12, e0179843. [Google Scholar] [CrossRef] [Green Version]
- Goulielmaki, E.; Tsekrekou, M.; Batsiotos, N.; Ascensao-Ferreira, M.; Ledaki, E.; Stratigi, K.; Chatzinikolaou, G.; Topalis, P.; Kosteas, T.; Altmuller, J.; et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat. Com-mun. 2021, 12, 3153. [Google Scholar] [CrossRef]
- Donnio, L.M.; Cerutti, E.; Magnani, C.; Neuillet, D.; Mari, P.O.; Giglia-Mari, G. XAB2 dynamics during DNA dam-age-dependent transcription inhibition. Elife 2022, 11, e77094. [Google Scholar] [CrossRef]
- Sawyer, I.A.; Sturgill, D.; Dundr, M. Membraneless nuclear organelles and the search for phases within phases. Wiley Interdis-cip. Rev. RNA 2019, 10, e1514. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Arseni, L.; Lanzafame, M.; Compe, E.; Fortugno, P.; Afonso-Barroso, A.; Peverali, F.A.; Zambruno, G.; Egly, J.M.; Stefanini, M.; Orioli, D. TFIIH-dependent MMP-1 overexpression in trichothiodystrophy leads to extracellular matrix alterations in patient skin. Pnas 2015, 112, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Botta, E.; Offman, J.; Nardo, T. Mutations in the C7orf11 (TTDN1) gene in six nonphotosensitive trichothiodystrophy patients: No obvious genotype-phenotype relationships. Hum. Mutat. 2007, 28, 92–96. [Google Scholar] [CrossRef]
- Botta, E.; Nardo, T.; Broughton, B.; Marinoni, S.; Lehmann, A.R.; Stefanini, M. Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: Site of mutation correlates with repair deficiency but gene dosage appears to determine clinical severity. Am. J. Hum. Genet. 1998, 63, 1036–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botta, E.; Nardo, T.; Lehmann, A.R.; Egly, J.M.; Pedrini, A.M.; Stefanini, M. Reduced level of the repair/transcription factor TFIIH in trichothiodystrophy. Hum Mol Genet. 2002, 11, 2919–2928. [Google Scholar] [CrossRef] [Green Version]
- Broughton, R.; Billings, R.; Cartwright, R. Homicidal somnambulism: A case report. Sleep 1994, 17, 253–264. [Google Scholar] [PubMed]
- Giglia-Mari, G.; Coin, F.; Ranish, J.A. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat. Genet. 2004, 36, 714–719. [Google Scholar] [CrossRef]
- King, M.D.; Dudgeon, J.; Stephenson, J.B. Joubert’s syndrome with retinal dysplasia: Neonatal tachypnoea as the clue to a genetic brain-eye malformation. Arch. Dis. Child. 1984, 59, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, A.; Arseni, L.; Carriero, R. Reduced levels of prostaglandin I2 synthase: A distinctive feature of the cancer-free trichothiodystrophy. Proc. Natl. Acad. Sci. USA 2021, 118, e2024502118. [Google Scholar] [CrossRef]
- Rizzo, R.; Pavone, L.; Micali, G.; Calvieri, S.; Di Gregorio, L. Trichothiodystrophy: Report of a new case with severe nervous system impairment. J. Child Neurol. 1992, 7, 300–303. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalog | Source Clone ID | Sequence in 5′-3′ | |
|---|---|---|---|
| RNF113A-shRNA1 | V3SH11240-226743083 | V3SVHS00_6680731 | CAATGGCGTCTTCAATCCA |
| RNF113A-shRNA2 | V3SH11240-230736642 | V3SVHS00_10674292 | GCGAAAGAATTGATTGCTA |
| RNF113A-shRNA3 | V3SH11240-230369254 | V3SVHS00_10306903 | CCCGAGCATCTACGTGCCA |
| Scrambled control | SO-2952232G | SVC17010402 | - |
| Reagents | Well 1 | Well 2 | Well 3 |
|---|---|---|---|
| Donor | 1 µg | 1 µg | 1 µg |
| gRNA (1 µg each) | gRNA1 | gRNA2 | Scramble Control |
| Opti-MEM | 250 µL | 250 µL | 250 µL |
| TurboFectin | 6 µL | 6 µL | 6 µL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, G.; Khalid, F.; Zhang, D.; Cao, Z.; Maity, P.; Kestler, H.A.; Orioli, D.; Scharffetter-Kochanek, K.; Iben, S. Ribosomal Dysfunction Is a Common Pathomechanism in Different Forms of Trichothiodystrophy. Cells 2023, 12, 1877. https://doi.org/10.3390/cells12141877
Zhu G, Khalid F, Zhang D, Cao Z, Maity P, Kestler HA, Orioli D, Scharffetter-Kochanek K, Iben S. Ribosomal Dysfunction Is a Common Pathomechanism in Different Forms of Trichothiodystrophy. Cells. 2023; 12(14):1877. https://doi.org/10.3390/cells12141877
Chicago/Turabian StyleZhu, Gaojie, Fatima Khalid, Danhui Zhang, Zhouli Cao, Pallab Maity, Hans A. Kestler, Donata Orioli, Karin Scharffetter-Kochanek, and Sebastian Iben. 2023. "Ribosomal Dysfunction Is a Common Pathomechanism in Different Forms of Trichothiodystrophy" Cells 12, no. 14: 1877. https://doi.org/10.3390/cells12141877