Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development


Abstract
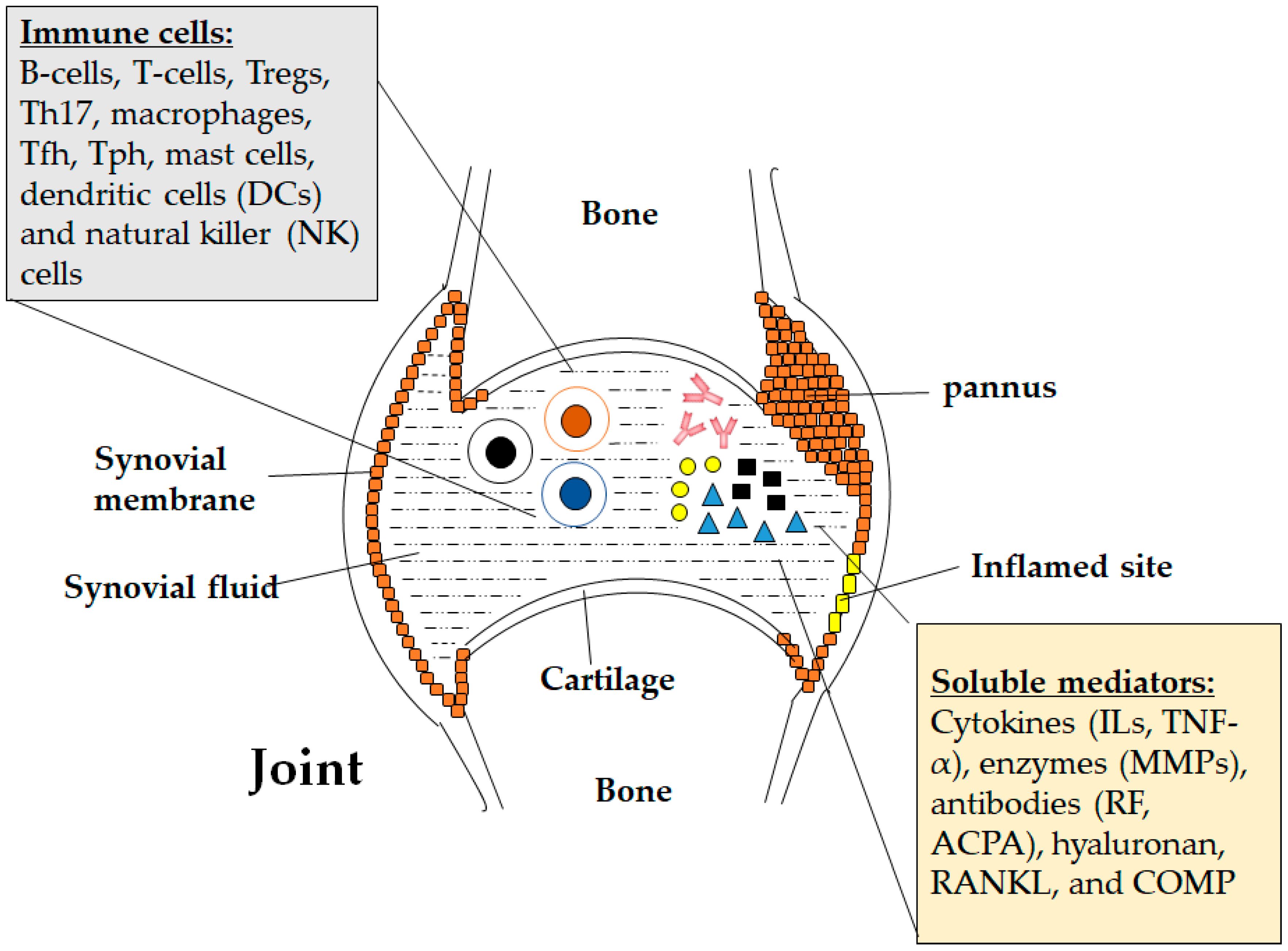
:1. Introduction
2. Pathogenic Role of Immune Cells in Rheumatoid Arthritis
2.1. B-Lymphocytes
2.2. T-Lymphocytes
2.3. Macrophages
2.4. Other Cells
3. Role of Immune-Related Secretory Molecules in Rheumatoid Arthritis
3.1. Cytokines
3.2. Antibodies
3.3. Other Rheumatoid Arthritis-Associated Soluble Mediators
4. Targeting Immunological Components for Rheumatoid Arthritis Treatment
4.1. FDA-Approved Therapies Against Rheumatoid Arthritis
4.2. Potential Rheumatoid Arthritis Therapies in Clinical Trials
5. Immune Cell Related Biomarker and Vaccine Development for Rheumatoid Arthritis
5.1. Disease Activity and Diagnostic Markers
5.2. Predictive Biomarkers for Rheumatoid Arthritis Treatment
5.3. Vaccine Development for Rheumatoid Arthritis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chronic Rheumatic Conditions. Available online: http://www.who.int/chp/topics/rheumatic/en/ (accessed on 15 February 2018).
- Garneau, E. Rheumatoid arthritis. In Ferri’s Clinical Advisor, 20th ed.; Ferri, F.F., Ed.; Elsevier: Atlanta, GA, USA, 2018; pp. 1125–1128. ISBN 978-0-323-28049-5. [Google Scholar]
- Kayanaugh, A.; Grevich, S.C. Rheumatoid arthritis. In Conn’s Current Therapy, 70th ed.; Kellerman, R.D., Bope, E.T., Eds.; Elsevier: Atlanta, GA, USA, 2018; pp. 899–903. ISBN 978-0-323-52769-9. [Google Scholar]
- Brassard, P.; Kezouh, A.; Suissa, S. Antirheumatic drugs and the risk of tuberculosis. Clin. Inf. Dis. 2006, 43, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Navarra, S.V.; Tang, B.; Lu, L.; Lin, H.Y.; Mok, C.C.; Asavatanabodee, P.; Suwannalai, P.; Hussein, H.; Rahman, M.U. Risk of tuberculosis with anti-tumor necrosis factor-α therapy: Substantially higher number of patients at risk in Asia. Int. J. Rheum. Dis. 2014, 17, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Mewar, D.; Wilson, A.G. Treatment of rheumatoid arthritis with tumour necrosis factor inhibitors. Br. J. Pharmacol. 2011, 162, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, W.G.; Hyrich, K.L.; Watson, K.D.; Lunt, M.; Galloway, J.; Ustianowski, A.; Symmons, D.P.; BSRBR Control Centre Consortium; BSR Biologics Register. Drug-specific risk of tuberculosis in patients with rheumatoid arthritis treated with anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register (BSRBR). Ann. Rheum. Dis. 2010, 69, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Rheumatoid Arthritis Treatment. Available online: http://www.hopkinsarthritis.org/arthritis-info/rheumatoid-arthritis/ra-treatment/ (accessed on 27 February 2018).
- Semerano, L.; Decker, P.; Clavel, G.; Boissier, M.C. Developments with investigational Janus kinase inhibitors for rheumatoid arthritis. Expert Opin. Investig. Drugs 2016, 25, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, S.; Montecucco, C.; Bugatti, S.; Caporali, R. Rheumatoid arthritis treatment: The earlier the better to prevent joint damage. RMD Open 2015, 1, e000057. [Google Scholar] [CrossRef] [PubMed]
- Bugatti, S.; Vitolo, B.; Caporali, R.; Montecucco, C.; Manzo, A. B cells in rheumatoid arthritis: From pathogenic players to disease biomarkers. BioMed Res. Int. 2014, 2014, 681678. [Google Scholar] [CrossRef] [PubMed]
- Giltiay, N.V.; Chappell, C.P.; Clark, E.A. B-cell selection and the development of autoantibodies. Arthritis Res. Ther. 2012, 14, S1. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P. Regulation of B-cell responses by Toll-like receptors. Immunology 2012, 136, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Samuels, J.; Ng, Y.S.; Coupillaud, C.; Paget, D.; Meffre, E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med. 2005, 201, 1659–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meffre, E. The establishment of early B cell tolerance in humans: Lessons from primary immunodeficiency diseases. Ann. N. Y. Acad. Sci. 2011, 1246, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Samuels, J.; Ng, Y.S.; Meffre, E. Inflammation-independent defective early B cell tolerance checkpoints in rheumatoid arthritis. Arthritis Rheum. 2011, 63, 1237–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Rapetti, L.; Chabele, K.M.; Evans, C.M.; Ehrenstein, M.R. B cell resistance to Fas-mediated apoptosis contributes to their ineffective control by regulatory T cells in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Aarvak, T.; Natvig, J.B. Cell-cell interactions in synovitis: Antigen presenting cells and T cell interaction in rheumatoid arthritis. Arthritis Res. 2011, 3, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, P.M.; Steiert, I.; Kötter, I.; Müller, C.A. B cell contribute to heterogeneity of IL-17 producing cells in rheumatoid arthritis and healthy controls. PLoS ONE 2013, 8, e82580. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.P.; Schulze-Koops, H.; Aringer, M. The central role of T cells in rheumatoid arthritis. Clin. Exp. Rheumatol. 2007, 25, S4–S11. [Google Scholar] [PubMed]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by memory B cells: A link between B cells and bone erosion in rheumatoid arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Molecular mechanisms of T cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol. Rev. 2009, 229, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Bevan, M.J. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007, 25, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.P. T cells in rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, S1. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Koops, H.; Kalden, J.R. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2001, 15, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Manetti, M.; Caterbi, S.; Ibba-Manneschi, L.; Bistoni, O.; Bartoloni, E.; Valentini, V.; Terenzi, R.; Gerli, R. Altered immunoregulation in rheumatoid arthritis: The role of regulatory T cells and proinflammatory Th17 cells and therapeutic implications. Mediat. Inflamm. 2015, 2015, 751793. [Google Scholar] [CrossRef] [PubMed]
- Al-Saadany, H.M.; Hussein, M.S.; Gaber, R.A.; Zaytoun, H.A. Th-17 cells and serum IL-17 in rheumatoid arthritis patients: Correlation with disease activity and severity. Egypt. Rheumatol. 2016, 38, 1–7. [Google Scholar] [CrossRef]
- Suurmond, J.; Dorjée, A.L.; Boon, M.R.; Knol, E.F.; Huizinga, T.W.; Toes, R.E.; Schuerwegh, A.J. Mast cells are the main interleukin 17-positive cells in anticitrullinated protein antibody-positive and -negative rheumatoid arthritis and osteoarthritis synovium. Arthritis Res. Ther. 2011, 13, R150. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Role of IL-17 in the pathogenesis of rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Manzo, A.; Vitolo, B.; La Manna, M.P.; Giardina, G.; Sireci, G.; Dieli, F.; Montecucco, C.M.; Alessandro, R.; Triolo, G. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2015, 54, 2264–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, K.; Kumar, U.; Das, S.; Chaudhuri, J.; Kumar, P.; Kanjilal, M.; Ghosh, P.; Sircar, G.; Basyal, R.K.; Kanga, U.; Bandyopadhaya, S.; Mitra, D.K. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooles, F.A.; Isaacs, J.D.; Anderson, A.E. Treg cells in rheumatoid arthritis: An update. Curr. Rheumatol. Rep. 2013, 15, 352. [Google Scholar] [CrossRef] [PubMed]
- Boissier, M.C.; Assier, E.; Biton, J.; Benys, A.; Falgarone, G.; Bessis, N. Regulatory T cells (Treg) in rheumatoid arthritis. Jt. Bone Spine 2009, 79, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Shima, Y.; Wing, J.B.; Sakaguchi, S.; Ogata, A.; Kumanogoh, A. The proportion of regulatory T cells in patients with rheumatoid arthritis: A meta-analysis. PLoS ONE 2016, 11, e0162306. [Google Scholar] [CrossRef] [PubMed]
- Al-Zifzaf, D.S.; El Bakry, S.A.; Mamdouh, R.; Shawarby, L.A.; Ghaffar, A.Y.A.; Amer, H.A.; Alim, A.A.; Sakr, H.M.; Rahman, R.A. FoxP3+T regulatory cells in rheumatoid arthritis and the imbalance of the Treg/TH17 cytokine axis. Egypt. Rheumatol. 2015, 37, 7–15. [Google Scholar] [CrossRef]
- Rao, D.A. T cells that help B cells in chronically inflamed tissues. Front. Immunol. 2018, 9, 1924. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017, 542, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinne, R.W.; Bräuer, R.; Stuhlmüller, B.; Palombo-Kinne, E.; Burmester, G.R. Macrophages in rheumatoid arthritis. Arthritis Res. 2000, 2, 189–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondeson, J.; Wainwright, S.D.; Lauder, S.; Amos, N.; Hughes, C.E. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res. Ther. 2006, 8, R187. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.L.; Hayder, M.; Baron, M.; Boyer, J.F.; Constantin, A.; Apparailly, F.; Poupot, R.; Cantagrel, A. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2013, 52, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Ye, C.; Kumar, P.; Chiu, I.; Subramanya, S.; Wu, H.; Shankar, P.; Manjunath, N. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol. Ther. 2010, 18, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Experimental arthritis: Anti-TNF kills the macrophage response. Nat. Rev. Rheumatol. 2018, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Nanotherapeutic approaches for the treatment of rheumatoid arthritis. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 607–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivellese, F.; Nerviani, A.; Rossi, F.W.; Marone, G.; Matucci-Cerinic, M.; de Paulis, A.; Pitzalis, C. Mast cells in rheumatoid arthritis: Friends or foes? Autoimmun. Rev. 2017, 16, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.B.; Langridge, W.H.R. The function of myeloid dendritic cells in rheumatoid arthritis. Rheumatol. Int. 2017, 37, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Greenberg, J.D.; Bhardwaj, N. Dendritic cells as targets for therapy in rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 566–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilkens, C.M.; Isaacs, J.D. Tolerogenic dendritic cell therapy for rheumatoid arthritis: Where are we now? Clin. Exp. Immunol. 2013, 172, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Shegarfi, H.; Naddafi, F.; Mirshafiey, A. Natural killer cells and their role in rheumatoid arthritis: Friend or foe? Sci. World J. 2012, 2012, 491974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996, 14, 397–440. [Google Scholar] [CrossRef] [PubMed]
- Brzustewicz, E.; Bryl, E. The role of cytokines in the pathogenesis of rheumatoid arthritis—Practical and potential application of cytokines as biomarkers and targets of personalized therapy. Cytokine 2015, 76, 527. [Google Scholar] [CrossRef] [PubMed]
- Burska, A.; Boissinot, M.; Ponchel, F. Cytokines as Biomarkers in Rheumatoid Arthritis. Med. Inflamm. 2014, 2014, 545493. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Jantan, I.; Bukhari, S.N.A. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed. Pharmacother. 2017, 92, 615–633. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Zafar, A.; Moin, S.; Khan, A.Q.; Zubair, S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 2016, 455, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Choy, E. Understanding the dynamics: Pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012, 51, v3–v11. [Google Scholar] [CrossRef] [PubMed]
- Griesmacher, A.; Peichl, P. Autoantibodies associated with rheumatic diseases. Clin. Chem. Lab. Med. 2001, 39, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.W.; Kang, E.H. Autoantibodies in rheumatoid arthritis: Rheumatoid factors and anticitrullinated protein antibodies. QJM-Int. J. Med. 2010, 103, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.Y.; Jan Wu, Y.J.; Yang, T.Y.; Chiang, N.Y.; Tsai, W.P.; Gordon, S.; Chang, G.W.; Kuo, S.F.; Lin, H.H. High levels of soluble GPR56/ADGRG1 are associated with positive rheumatoid factor and elevated tumor necrosis factor in patients with rheumatoid arthritis. J. Microbiol. Immunol. Infect. 2018, 51, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Van Boekel, M.A.; Vossenaar, E.R.; van den Hoogen, F.H.; van Venrooij, W.J. Autoantibody systems in rheumatoid arthritis: Specificity, sensitivity and diagnostic value. Arthritis Res. 2002, 4, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossenaar, E.R.; van Venrooij, W.J. Anti-CCP antibodies, a highly specific marker for (early) rheumatoid arthritis. Clin. Appl. Immunol. Rev. 2004, 4, 239–262. [Google Scholar] [CrossRef]
- Sidorov, A.; Beduleva, L.; Menshikov, I.; Terentiev, A.; Stolyarova, E.; Abisheva, N. Fc fragments of immunoglobulin G are an inductor of regulatory rheumatoid factor and a promising therapeutic agent for rheumatic diseases. Int. J. Biol. Macromol. 2017, 95, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Liao, K.; Nair, R.; Ringold, S.; Costenbader, K.H. Anti-citrullinated peptide antibody (ACPA) assays and their role in the diagnosis of rheumatoid arthritis. Arthritis Rheum. 2009, 61, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Manca, M.L.; Alunno, A.; D’Amato, C.; Bistoni, O.; Puxeddu, I.; Gerli, R.; Migliorini, P.; Pratesi, F. Anti-citrullinated peptide antibodies profiling in established rheumatoid arthritis. Jt. Bone Spine 2017, 85, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Derksen, V.F.A.M.; Huizinga, T.W.J.; van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.; Webb, T.; Seaman, A.; Infantino, M.; Meacci, F.; Manfredi, M.; Benucci, M.; Lakos, G.; Favalli, E.; Schioppo, T.; et al. Anti-CarP antibodies as promising marker to measure joint damage and disease activity in patients with rheumatoid arthritis. Immunol. Res. 2015, 61, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pangtey, G.; Gupta, R.; Rehan, H.S.; Gupta, L.K. Assessment of anti-CarP antibodies, disease activity and quality of life in rheumatoid arthritis patients on conventional and biological disease-modifying antirheumatic drugs. Reumatologia 2017, 55, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; van Steenbergen, H.W.; van Nies, J.A.B.; Levarht, E.W.N.; Huizinga, T.W.J.; van der Helm-van, A.H.M.; Toes, R.E.M.; Trouw, L.A. The specificity of anti-carbamylated protein antibodies for rheumatoid arthritis in a setting of early arthritis. Arthritis Res. Ther. 2015, 17, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, P.; Cui, J.; Yang, C.; Du, H. Keratin 8 is a novel autoantigen of rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2015, 465, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Després, N.; Lapointe, E.; van der Heijden, A.; Lora, M.; Senshu, T.; van Venrooij, W.J.; Ménard, H.A. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 2004, 6, R142–R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadda, S.; Abolkheir, E.; Afifi, R.; Gamal, M. Serum matrix metalloproteinase-3 in rheumatoid arthritis patients: Correlation with disease activity and joint destruction. Egypt. Rheumatol. 2016, 38, 153–159. [Google Scholar] [CrossRef]
- Niki, Y.; Takeuchi, T.; Nakayama, M.; Nagasawa, H.; Kurasawa, T.; Yamada, H.; Toyama, Y.; Miyamoto, T. Clinical significance of cartilage biomarkers for monitoring structural joint damage in rheumatoid arthritis patients treated with anti-TNF therapy. PLoS ONE 2012, 7, e37447. [Google Scholar] [CrossRef] [PubMed]
- Paleolog, E.M. The vasculature in rheumatoid arthritis: Cause or consequence? Int. J. Exp. Pathol. 2009, 90, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Fardellone, P.; Séjourné, A.; Paccou, J.; Goëb, V. Bone remodelling markers in rheumatoid arthritis. Mediat. Inflamm. 2014, 2014, 484280. [Google Scholar] [CrossRef] [PubMed]
- Heo, R.; Park, J.S.; Jang, H.J.; Kim, S.H.; Shin, J.M.; Suh, Y.D.; Jeong, J.H.; Jo, D.G.; Park, J.H. Hyaluronan nanoparticles bearing γ-secretase inhibitor: In vivo therapeutic effects on rheumatoid arthritis. J. Control. Release 2014, 192, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Paiva, K.B.S.; Granjeiro, J.M. Matrix metalloproteinases in bone resorption, remodelling, and repair. Prog. Mol. Biol. Transl. Sci. 2017, 148, 203–303. [Google Scholar] [PubMed]
- Darweesh, H.; Abbass, D.; Kadah, R.; Rashad, A.; Basel, M.E.; Nasr, A.S. Serum and synovial cartilage oligomeric matrix protein (COMP) in patients with rheumatoid arthritis and osteoarthritis. Ind. J. Rheumatol. 2010, 5, 112–117. [Google Scholar] [CrossRef]
- Lorenzo, P.; Aspberg, A.; Saxne, T.; Önnerfjord, P. Quantification of cartilage oligomeric matrix protein (COMP) and a COMP neoepitope in synovial fluid of patients with different joint disorders by novel automated assays. Osteoarthr. Cartil. 2017, 25, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Gorman, C.; Leandro, M.; Isenberg, D. B cell depletion in autoimmune disease. Arthritis Res. Ther. 2003, 5, S17–S21. [Google Scholar] [CrossRef] [PubMed]
- Choy, E. Interleukin 6 receptor as a target for the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, ii68–ii69. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.; Quan, J.; Totoritis, M. B cell therapy for rheumatoid arthritis: The rituximab (anti-CD20) experience. Ann. Rheum. Dis. 2003, 62, ii55–ii59. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C. Anti-cytokines and cytokines in the treatment of rheumatoid arthritis. Curr. Pharm. Des. 2003, 9, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix metalloproteinases: Role in arthritis. Front Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, K.; Müller-Ladner, U. Side effects and management of side effects of methotrexate in rheumatoid arthritis. Clin. Exp. Rheumatol. 2010, 28, S95–S101. [Google Scholar] [PubMed]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed. Rep. 2013, 1, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Perdriger, A. Infliximab in the treatment of rheumatoid arthritis. Biologics 2009, 3, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Keystone, E.C.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Teoh, L.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Haraoui, B.; Bykerk, V. Etanercept in the treatment of rheumatoid arthritis. Ther. Clin. Risk Manag. 2007, 3, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mease, P.J. Certolizumab pegol in the treatment of rheumatoid arthritis: A comprehensive review of its clinical efficacy and safety. Rheumatology (Oxford) 2011, 50, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Noorbaloochi, S.; Singh, G. Golimumab for rheumatoid arthritis: A systematic review. J. Rheumatol. 2010, 37, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Dev. Ther. 2014, 8, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, A. Tocilizumab in rheumatoid arthritis: Efficacy, safety and its place in therapy. Ther. Adv. Chronic Dis. 2013, 4, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Boyce, E.G.; Rogan, E.L.; Vyas, D.; Prasad, N.; Mai, Y. Sarilumab: Review of a second IL-6 receptor antagonist indicated for the treatment of rheumatoid arthritis. Ann. Pharmacother. 2018, 52, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Moe, S.; Myint, K.T.; Htet, A. Oral janus kinase inhibitor for the treatment of rheumatoid arthritis: Tofacitinib. ISRN Rheumatol. 2013, 2013, 357904. [Google Scholar] [CrossRef] [PubMed]
- Gras, J. Baricitinib: JAK inhibition for rheumatoid arthritis. Drugs Today 2016, 52, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Langdon, K.; Haleagrahara, N. Regulatory T-cell dynamics with abatacept treatment in rheumatoid arthritis. Int. Rev. Immunol. 2018, 37, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Australian New Zealand Clinical Trials Registry Trial Review. Available online: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=373425 (accessed on 1 August 2018).
- NIH US National Library of Medicine Clinical Trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03410056 (accessed on 3 August 2018).
- Crotti, C.; Raimondo, M.G.; Becciolini, A.; Biggioggero, M.; Favalli, E.G. Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: Evidence to date. Drug Des. Dev. Ther. 2017, 11, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Combe, B.; Dörner, T.; Kremer, J.M.; Huizinga, T.W.; Veenhuizen, M.; Gill, A.; Komocsar, W.; Berclaz, P.Y.; Ortmann, R.; Lee, C. Efficacy and safety of tabalumab, an anti-BAFF monoclonal antibody, in patients with moderate-to-severe rheumatoid arthritis and inadequate response to TNF inhibitors: Results of a randomised, double-blind, placebo-controlled, phase 3 study. RMD Open 2015, 1, e000037. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, T.W.; Batalov, A.; Stoilov, R.; Lloyd, E.; Wagner, T.; Saurigny, D.; Souberbielle, B.; Esfandiari, E. Phase 1b randomized, double-blind study of namilumab, an anti-granulocyte macrophage colony-stimulating factor monoclonal antibody, in mild-to-moderate rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.D.; Hamilton, J.A. Investigational therapies targeting the granulocyte macrophage colony-stimulating factor receptor-α in rheumatoid arthritis: Focus on mavrilimumab. Ther. Adv. Musculoskelet. Dis. 2018, 10, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kivitz, A.; Hazan, L.; Hoffman, K.; Wallin, B.A. FRI0209 MORAb-022, an anti-granulocyte macrophage-colony stimulating factor (GM-CSF) monoclonal antibody (MAB): Results of the first study in patients with mild-to-moderate rheumatoid arthritis (RA). Ann. Rheum. Dis. 2016, 75, 507. [Google Scholar] [CrossRef]
- Molfino, N.A.; Kuna, P.; Leff, J.A.; Oh, C.K.; Singh, D.; Chernow, M.; Sutton, B.; Yarranton, G. Phase 2, randomised placebo-controlled trial to evaluate the efficacy and safety of an anti-GM-CSF antibody (KB003) in patients with inadequately controlled asthma. BMJ Open 2016, 6, e007709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Weinblatt, M.E.; van der Heijde, D.; Rigby, W.F.; van Vollenhoven, R.; Bingham, C.O.; Veenhuizen, M.; Gill, A.; Zhao, F.; Komocsar, W.J.; et al. Efficacy and safety of tabalumab, an anti-B-cell-activating factor monoclonal antibody, in patients with rheumatoid arthritis who had an inadequate response to methotrexate therapy: Results from a phase III multicentre, randomised, double-blind study. Ann. Rheum. Dis. 2015, 74, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Mahtani, K.R.; Miller, A.; Rivero-Arias, O.; Heneghan, C.; Price, C.P.; Thompson, M.; Plüddemann, A.; Luqmani, R. Autoimmune markers for the diagnosis of rheumatoid arthritis in primary care: Primary care diagnostic technology update. Br. J. Gen. Pract. 2013, 63, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Lilly Press Release Archives. Available online: http://lilly.mediaroom.com/index.php?s=9042&item=136985 (accessed on 19 September 2018).
- Luime, J.J.; Colin, E.M.; Hazes, J.M.; Lubberts, E. Does anti-mutated citrullinated vimentin have additional value as a serological marker in the diagnostic and prognostic investigation of patients with rheumatoid arthritis? A systematic review. Ann. Rheum. Dis. 2010, 69, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Maksymowych, W.P.; Naides, S.J.; Bykerk, V.; Siminovitch, K.A.; van Schaardenburg, D.; Boers, M.; Landewé, R.; van der Heijde, D.; Tak, P.P.; Genovese, M.C.; Weinblatt, M.E.; et al. Serum 14-3-3η is a novel marker that complements current serological measurements to enhance detection of patients with rheumatoid arthritis. J. Rheumatol. 2014, 41, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Caplan, L.; Yazdany, J.; Robbins, M.L.; Neogi, T.; Michaud, K.; Saag, K.G.; O’Dell, J.R.; Kazi, S. Rheumatoid arthritis disease activity measures: American college of rheumatology recommendations for use in clinical practice. Arthritis Care Res. (Hoboken) 2012, 64, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Study of KB003 in Biologics-Inadequate Rheumatoid Arthritis. Available online: https://clinicaltrials.gov/ct2/results?term=NCT00995449 (accessed on 19 September 2018).
- Centola, M.; Cavet, G.; Shen, Y.; Ramanujan, S.; Knowlton, N.; Swan, K.A.; Turner, M.; Sutton, C.; Smith, D.R.; Haney, D.J.; et al. Development of a multi-biomarker disease activity test for rheumatoid arthritis. PLoS ONE 2013, 8, e60635. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, K.; Saevarsdottir, S.; Forslind, K.; Petersson, I.F.; Wallman, J.K.; Ernestam, S.; Bolce, R.J.; van Vollenhoven, R.F. A multi-biomarker disease activity score and the choice of second-line therapy in early rheumatoid arthritis after methotrexate failure. Arthritis Rheumatol. 2017, 69, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Wright, G.C.; Strand, V.; Davis, C.S.; Hitraya, E.; Sasso, E.H. Reanalysis of the multi-biomarker disease activity score for assessing disease activity in the AMPLEstudy: Comment on the article by Fleischmann et al. Arthritis Rheumatol. 2017, 69, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Chaves Chaparro, L.M.; Salvatierra Ossorio, J.; Raya Álvarez, E. Predictors of response to biologic therapies in rheumatoid arthritis. Rheumatol. Clin. 2011, 7, 141–144. [Google Scholar] [CrossRef]
- Lv, Q.; Yin, Y.; Li, X.; Shan, G.; Wu, X.; Liang, D.; Li, Y.; Zhang, X. The status of rheumatoid factor and anti-cyclic citrullinated peptide antibody are not associated with the effect of anti-TNFα agent treatment in patients with rheumatoid arthritis: A meta-analysis. PLoS ONE 2014, 9, e89442. [Google Scholar] [CrossRef] [PubMed]
- Fabris, M.; De Vita, S.; Blasone, N.; Visentini, D.; Pezzarini, E.; Pontarini, E.; Fabro, C.; Quartuccio, L.; Mazzolini, S.; Curcio, F.; et al. Serum levels of anti-CCP antibodies, anti-MCV antibodies and RF IgA in the follow-up of patients with rheumatoid arthritis treated with rituximab. Autoimmun. Highlights 2010, 1, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, A.; Maksymowych, W. SAT0070 levels of 14-3-3Eta predict good EULAR response to anti-TNF treatment in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 615–616. [Google Scholar] [CrossRef]
- Morozzi, G.; Fabbroni, M.; Bellisai, F.; Cucini, S.; Simpatico, A.; Galeazzi, M. Low serum level of COMP, a cartilage turnover marker, predicts rapid and high ACR70 response to adalimumab therapy in rheumatoid arthritis. Clin. Rheumatol. 2007, 26, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Gerlag, D.M.; Herenius, M.J.; Thurlings, R.M.; Wijbrandts, C.A.; Foell, D.; Vogl, T.; Roth, J.; Tak, P.P.; Holzinger, D. MRP8/14 serum levels as a strong predictor of response to biological treatments in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Isgren, A.; Forslind, K.; Erlandsson, M.; Axelsson, C.; Andersson, S.; Lund, A.; Bokarewa, M. High survivin levels predict poor clinical response to infliximab treatment in patients with rheumatoid arthritis. Semin. Arthritis Rheum. 2012, 41, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Nel, H.J.; Law, S.C.; Mehdi, A.M.; Street, S.; Ramnoruth, N.; Pahau, H.; Lee, B.T.; Ng, J.; Brunck, M.E.; et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci. Transl. Med. 2015, 7, 290ra87. [Google Scholar] [CrossRef] [PubMed]
- Shabahang, S.; Li, A.F.; Escher, A. Recent patents on immunoregulatory DNA vaccines for autoimmune diseases and allograft rejection. Recent Pat. DNA Gene Seq. 2010, 4, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ratsimandresy, R.A.; Duvallet, E.; Assier, E.; Semerano, L.; Delavallée, L.; Bessis, N.; Zagury, J.F.; Boissier, M.C. Active immunization against IL-23p19 improves experimental arthritis. Vaccine 2011, 29, 9329–9336. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Dore, R.K.; Lane, N.E.; Ory, P.A.; Peterfy, C.G.; Sharp, J.T.; van der Heijde, D.; Zhou, L.; Tsuji, W.; Newmark, R.; Denosumab Rheumatoid Arthritis Study Group. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: A twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008, 58, 1299–1309. [Google Scholar] [PubMed] [Green Version]
- Takeuchi, T.; Tanaka, Y.; Ishiguro, N.; Yamanaka, H.; Yoneda, T.; Ohira, T.; Okubo, N.; Genant, H.K.; van der Heijde, D. Effect of denosumab on Japanese patients with rheumatoid arthritis: A dose-response study of AMG 162 (Denosumab) in patients with rheumatoid arthritis on methotrexate to validate inhibitory effect on bone Erosion (DRIVE)-a 12-month, multicentre, randomised, double-blind, placebo-controlled, phase II clinical trial. Ann. Rheum. Dis. 2016, 75, 983–990. [Google Scholar] [PubMed]
- Feng, G.D.; Xue, X.C.; Gao, M.L.; Wang, X.F.; Shu, Z.; Mu, N.; Gao, Y.; Wang, Z.L.; Hao, Q.; Li, W.N.; et al. Therapeutic effects of PADRE-BAFF autovaccine on rat adjuvant arthritis. BioMed Res. Int. 2014, 2014, 854954. [Google Scholar] [CrossRef] [PubMed]
- Mould, A.W.; Scotney, P.; Greco, S.A.; Hayward, N.K.; Nash, A.; Kay, G.F. Prophylactic but not therapeutic activity of a monoclonal antibody that neutralizes the binding of VEGF-B to VEGFR-1 in a murine collagen-induced arthritis model. Rheumatology (Oxford) 2008, 47, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, K.S.; Mikecz, K.; Steiner, H.L.; Glant, T.T.; Finnegan, A.; Carambula, R.E.; Zimmerman, D.H. Rheumatoid arthritis vaccine therapies: Perspectives and lessons from therapeutic ligand epitope antigen presentation system vaccines for models of rheumatoid arthritis. Expert Rev. Vaccines 2015, 14, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.G.; Ritchlin, C.T. Denosumab: Targeting the RANKL pathway to treat rheumatoid arthritis. Expert Opin Biol. Ther. 2017, 17, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Vaccine development for rheumatoid arthritis. Glob. Vaccines Immunol. 2015, 1, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, K.S.; Holmdahl, R. Efficient promotion of collagen antibody induced arthritis (CAIA) using four monoclonal antibodies specific for the major epitopes recognized in both collagen induced arthritis and rheumatoid arthritis. J. Immunol. Methods 2005, 304, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Hasselberg, A.; Schön, K.; Tarkowski, A.; Lycke, N. Role of CTA1R7K-COL-DD as a novel therapeutic mucosal tolerance-inducing vector for treatment of collagen-induced arthritis. Arthritis Rheum. 2009, 60, 1672–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, D.H.; Taylor, P.; Bendele, A.; Carambula, R.; Duzant, Y.; Lowe, V.; O’Neill, S.P.; Talor, E.; Rosenthal, K.S. CEL-2000: A therapeutic vaccine for rheumatoid arthritis arrests disease development and alters serum cytokine/chemokine patterns in the bovine collagen type II induced arthritis in the DBA mouse model. Int. Immunopharmacol. 2010, 10, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Dzhambazov, B.; Nandakumar, K.S.; Kihlberg, J.; Fugger, L.; Holmdahl, R.; Vestberg, M. Therapeutic vaccination of active arthritis with a glycosylated collagen type II peptide in complex with MHC class II molecules. J. Immunol. 2006, 176, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Kochetkova, I.; Trunkle, T.; Callis, G.; Pascual, D.W. Vaccination without autoantigen protects against collagen II-induced arthritis via immune deviation and regulatory T cells. J. Immunol. 2008, 181, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Luross, J.A.; Heaton, T.; Hirst, T.R.; Day, M.J.; Williams, N.A. Escherichia coli heat-labile enterotoxin B subunit prevents autoimmune arthritis through induction of regulatory CD4+ T cells. Arthritis Rheum. 2002, 46, 1671–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Cell Type | Subtype | Pathogenic Roles | References |
|---|---|---|---|
| B-cells | - | Antibody producer, APC, T-cell activation and cytokine-producer such as IL-4 and IL-10 | [11,12,13] |
| T-cells | Th-1 | Cytokine producer, macrophage activation | [28] |
| Th-2 | Cytokine producer, B-cell activation, promote Ig class switching to IgE | [28] | |
| Th-17 | Cytokine producer, MMP stimulation, promote pannus growth, neoangiogenesis and osteoclastogenesis | [29,30,31,32] | |
| Treg | Suppress autoreactive lymphocytes | [35,36,37] | |
| Macrophage | - | APC, T-cell activation, cytokine producer, promote angiogenesis and fibroblast proliferation | [18,41,42] |
| Mast cells | - | Pro-inflammatory cytokines producer | [47] |
| Dendritic cells | - | APC and T-cell activation | [48] |
| Natural killer (NK) cells | - | Pro-inflammatory cytokines producer | [51] |
| Drug Name | Target and Action | FDA Approval Year | Reference |
|---|---|---|---|
| Etanercept | Dimeric human TNF receptor targeting TNF | 1998 | [90] |
| Infliximab | Monoclonal antibody targeting human TNFα | 2002 | [88] |
| Adalimumab | Recombinant monoclonal antibody targeting TNF | 2002 | [89] |
| Abatacept | Recombinant fusion protein targeting T-lymphocytes activation | 2005 | [98] |
| Rituximab | Chimeric monoclonal antibody that targets CD20 molecules of B-cells | 2006 | [93] |
| Certolizumab | Humanized and pegylated anti-TNFα inhibitor | 2009 | [91] |
| Golimumab | Humanized monoclonal antibody targeting TNFα | 2009 | [92] |
| Tocilizumab | Humanized monoclonal antibody that targets IL-6 receptors and blocks signaling | 2010 | [94] |
| Sarilumab | IL-6 receptor antagonist | 2017 | [95] |
| Name | Target and Mechanism | Stage | Trial Period | Reference |
|---|---|---|---|---|
| AMG 592 | Improve Treg selectivity | Phase 2 | 2018–2020 | [100] |
| DEN-181 1 | Regulate T-lymphocytes | Phase 1 | 2018 | [99] |
| Mavrilimumab | BAFF | Phase 2 | 2013–2015 | [101] |
| Namilumab (MT203) | GM-CSF ligand | Phase 2 | 2015–2016 | [103] |
| Lenzilumab/KB003 | GM-CSF | Phase 2 (terminated) | 2010–2012 | [106] |
| Tabalumab (LY2127399) | BAFF | Phase 3 (terminated) | 2011–2014 | [102,107] |
| GSK3196165 (MOR103) | GM-CSF | Phase 2 | 2015–2017 | [104] |
| MORAb-022 | GM-CSF | Phase 1 | 2013–2014 | [105] |
| AutoDECRA | Inhibit inflammation | Phase 1 | 2012–2013 | [50] |
| Biomarkers | Presence/Absence | Medication/Drug | References |
|---|---|---|---|
| Anti-CCP | Present | Rituximab | [118,119] |
| Anti-MCV | Present | Rituximab | [119] |
| 14-3-3 eta | Absent or low level | Tocilizumab, Anti-TNF drugs | [120] |
| Cartilage oligometic matrix protein (COMP) | Absent or low level | Adalimumab | [121] |
| Calprotectin | Present | Adalimumab, Infliximab, Rituximab | [122] |
| Survivin | Absent or low level | Infliximab | [123] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yap, H.-Y.; Tee, S.Z.-Y.; Wong, M.M.-T.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. https://doi.org/10.3390/cells7100161
Yap H-Y, Tee SZ-Y, Wong MM-T, Chow S-K, Peh S-C, Teow S-Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells. 2018; 7(10):161. https://doi.org/10.3390/cells7100161
Chicago/Turabian StyleYap, Hooi-Yeen, Sabrina Zi-Yi Tee, Magdelyn Mei-Theng Wong, Sook-Khuan Chow, Suat-Cheng Peh, and Sin-Yeang Teow. 2018. "Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development" Cells 7, no. 10: 161. https://doi.org/10.3390/cells7100161