L-Selectin/CD62L Is a Key Driver of Non-Alcoholic Steatohepatitis in Mice and Men

 , , ,
, , ,  and
and
Abstract
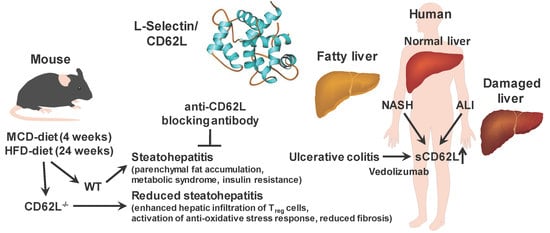
:
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Animal Studies
2.2.1. Animal Welfare
2.2.2. Housing and Generation of Mice
2.2.3. Dietary Treatments
2.2.4. Therapeutic Antibody Treatments
2.2.5. Glucose and Pyruvat Tolerance Test
2.2.6. μ Computer Tomography (μCT) Imaging
2.2.7. Blood Collection
2.3. Human CD62L Serum Elisa
2.4. AST/ALT Analysis
2.5. NAFLD Activity Score
2.6. Insulin Measurement/Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) Calculation
2.7. Hepatic Triglycerides
2.8. Hepatic Free Fatty Acids and Hepatic Cholesterol
2.9. Hydroxyproline
2.10. Histology, Sirius Red, and Oil Red O Staining
2.11. Immunofluorescence Staining
2.12. Gene Expression Analysis by Real-Time PCR
2.13. Isolation of Cells and Flow Cytometry
2.14. Isolation of Primary Liver Sinusoidal Endothelial Cells
2.15. Cell Culture Experiments with Primary Liver Sinusoidal Endothelial Cells and Hepa1-6 Cells
2.16. SDS PAGE and Western Blot
2.17. Quantification and Statistics
3. Results
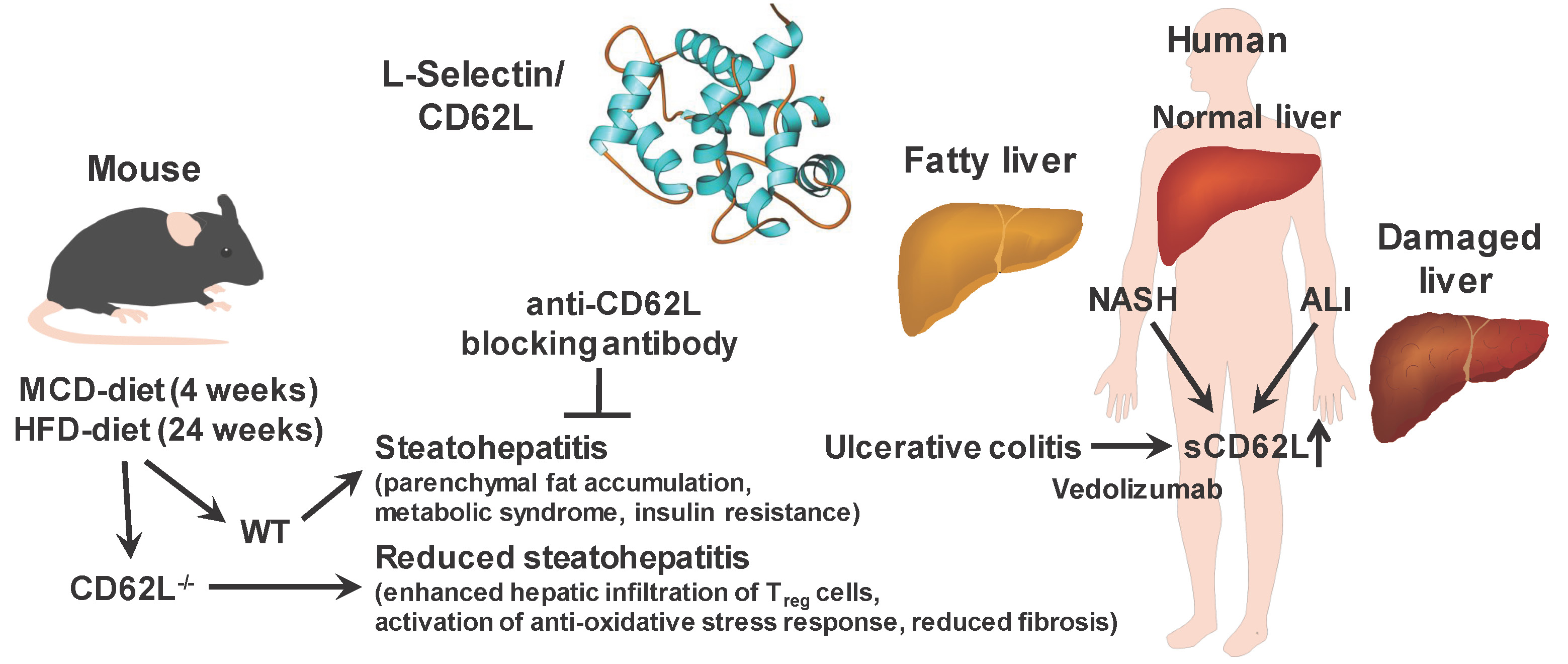
3.1. L-Selectin/CD62L is Increased in Patients With Acute and Chronic Liver Injury
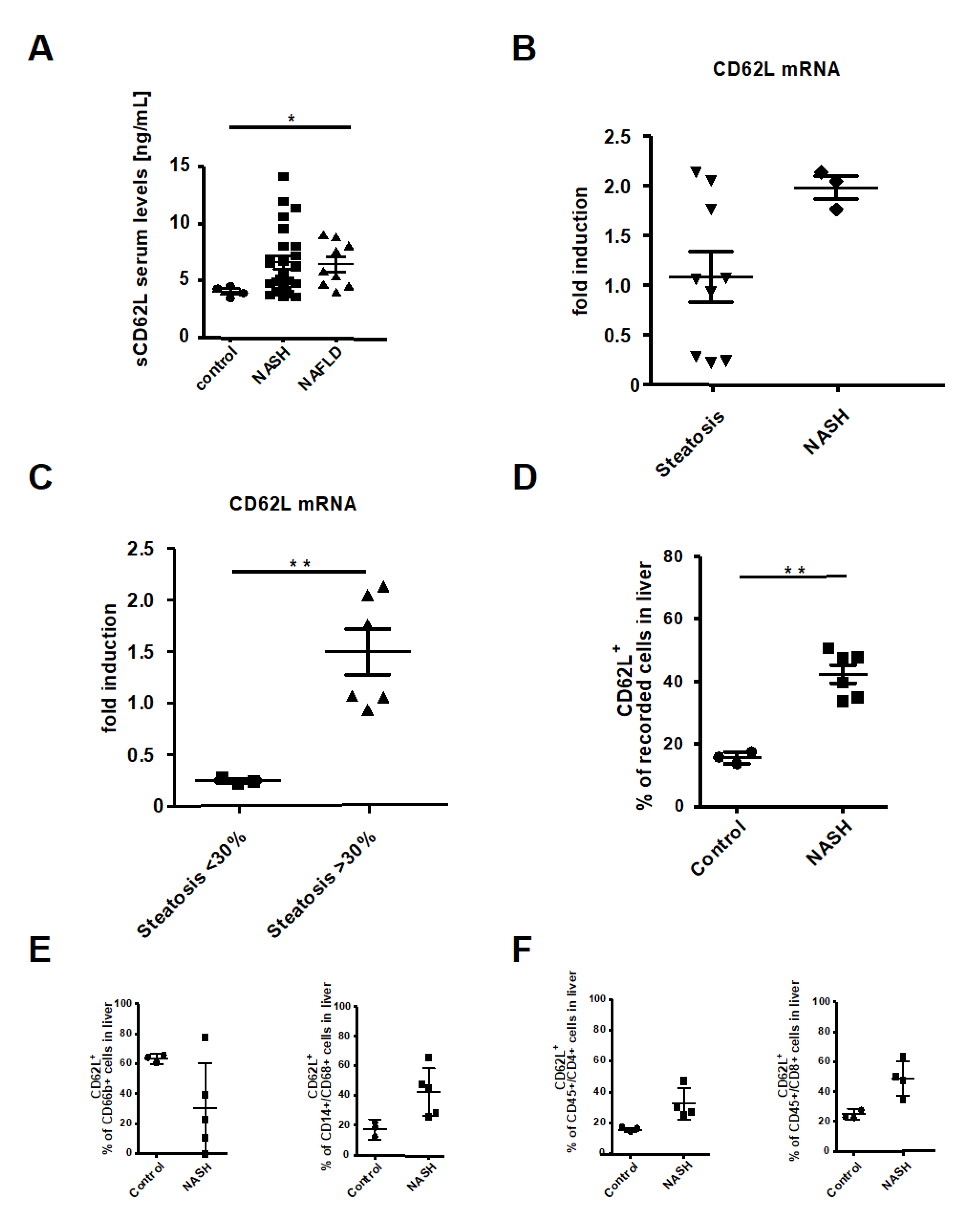
3.2. Deletion of CD62L Improves Metabolic Disorders and Steatosis in Diet-Induced Steatohepatitis
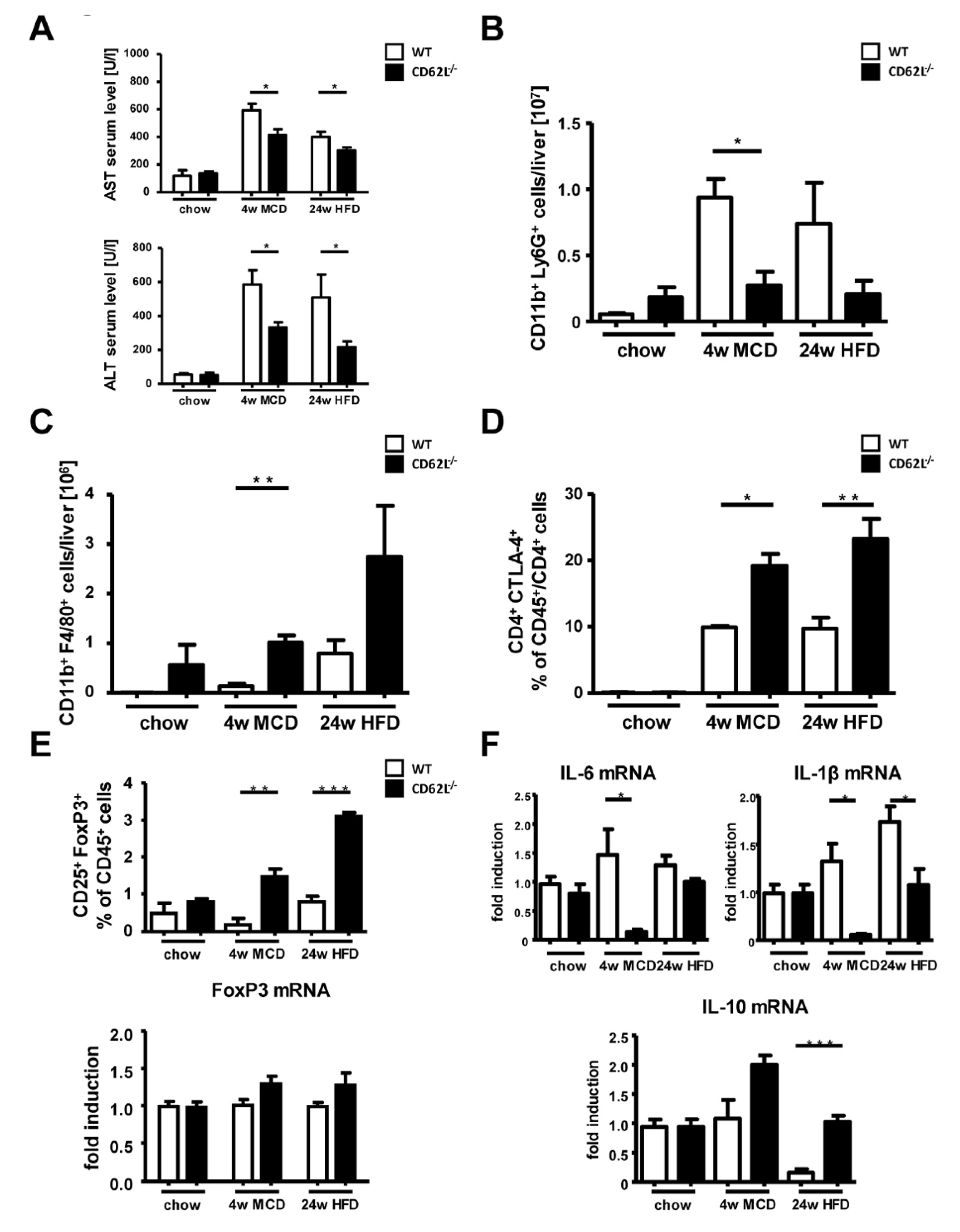
3.3. CD62L Deficiency Strongly Activates the Anti-Inflammatory Immune Response
3.4. Less Steatohepatitis Progression in CD62L−/− Mice is Associated with Reduced Oxidative Stress Response
3.5. Deletion of CD62L Does Not Lead to Compensatory Upregulation of Adhesion Molecules in the Liver
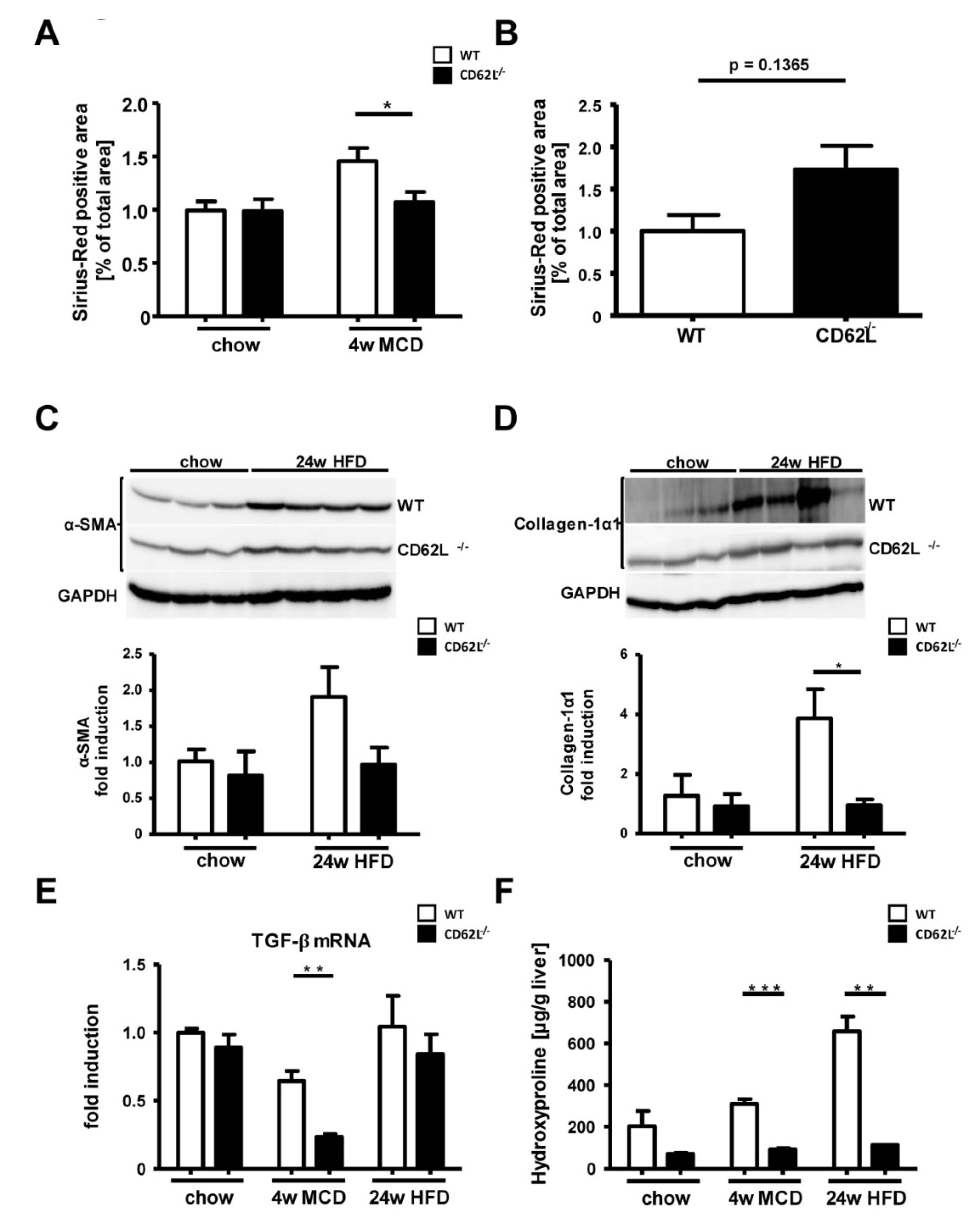
3.6. Loss of CD62L Reduces Fibrosis Progression after MCD and HFD Feeding
3.7. CD62L Deficiency Leads to Increased Activation of the Anti-Oxidative Stress Response in Hepatocytes
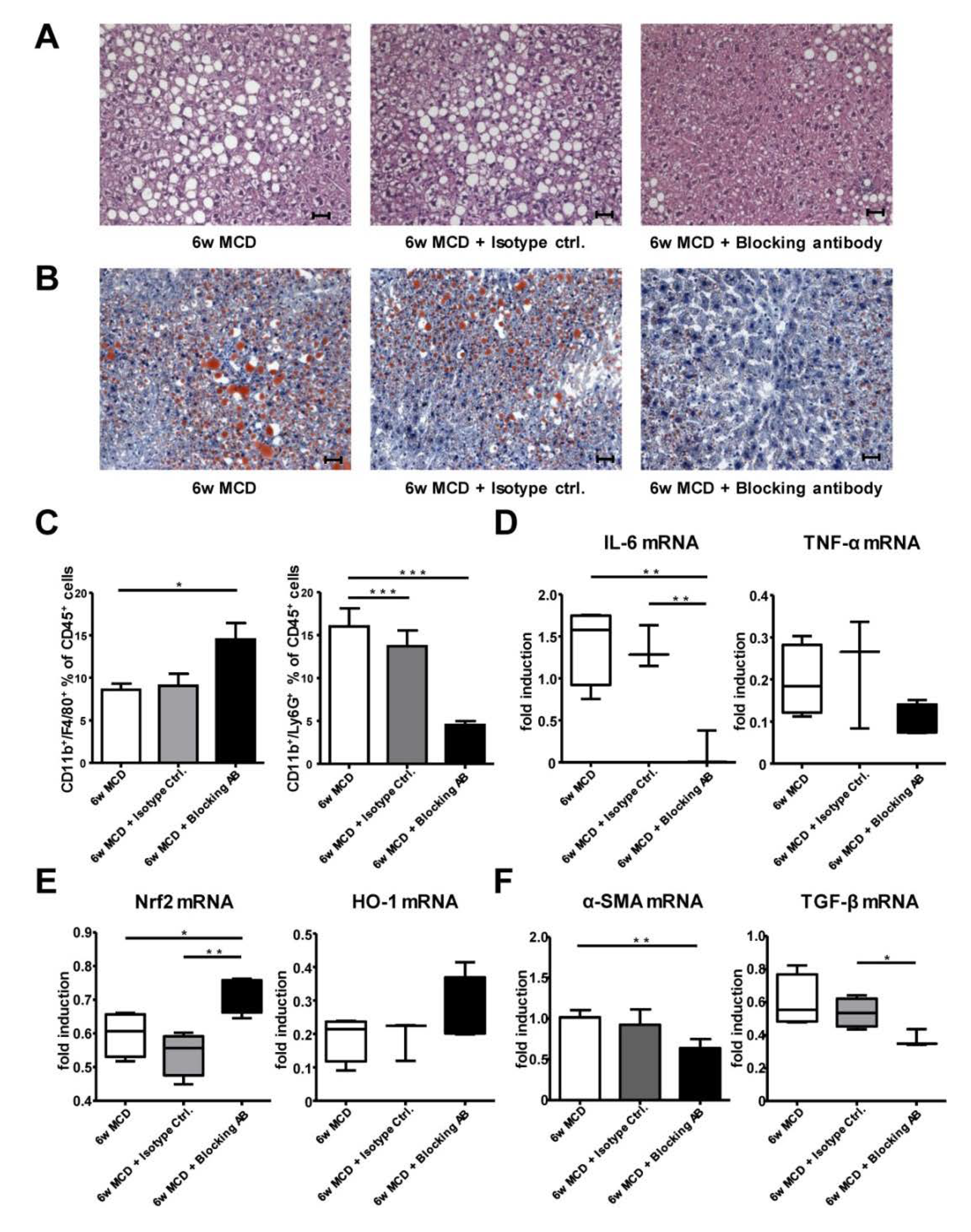
3.8. Therapeutic CD62L Intervention Leads to Improved Disease Outcome in Diet-Induced Steatohepatitis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global perspectives on non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M. The epidemiology of nonalcoholic fatty liver disease in adults. J. Clin. Gastroenterol. 2006, 40 (Suppl. 1), S5–S10. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Wong Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Agopian, V.G.; Kaldas, F.M.; Hong, J.C.; Whittaker, M.; Holt, C.; Rana, A.; Zarrinpar, A.; Petrowsky, H.; Farmer, D.; Yersiz, H.; et al. Liver transplantation for nonalcoholic steatohepatitis: The new epidemic. Ann. Surg. 2012, 256, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.N.; Han, X.; Munro, H.M.; Gross, A.L.; Shu, X.O.; Hargreaves, M.K.; Zhen, W.; Powers, A.C.; Blot, W.J. The obesity epidemic and rising diabetes incidence in a low-income racially diverse southern US cohort. PLoS ONE 2018, 13, e0190993. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Angulo, P. GI epidemiology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2008, 19, 295–300. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers. 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.A.; Newsome, P.N. Non-alcoholic fatty liver disease in 2016. Br. Med. Bull. 2016, 119, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalanasi, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A. Lifestyle modification as the primary treatment of NASH. Clin. Liver. Dis. 2009, 13, 649–665. [Google Scholar] [CrossRef]
- Soriano, A.; Salas, A.; Sans, M.; Gironella, M.; Elena, M.; Anderson, D.C.; Pique, J.M.; Panes, J. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab. Investig. 2000, 80, 1541–1551. [Google Scholar] [CrossRef]
- Wagner, N.; Lohler, J.; Tedder, T.F.; Rajewsky, K.; Müller, W.; Steeber, D.A. L-selectin and beta7 integrin synergistically mediate lymphocyte migration to mesenteric lymph nodes. Eur. J. Immunol. 1998, 28, 3832–3839. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boenge, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Fotis, L.; Giannakopoulos, D.; Stamogiannou, L.; Xatzipsalti, M. Intercellular cell adhesion molecule-1 and vascular cell adhesion molecule-1 in children. Do they play a role in the progression of atherosclerosis? Hormones (Athens) 2012, 11, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleiffenbaum, B.; Spertini, O.; Tedder, T.F. Soluble L-selectin is present in human plasma at high levels and retains functional activity. J. Cell Biol. 1992, 119, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Arbonés, M.L.; Ord, D.C.; Ley, K.; Ratech, H.; Maynard-Curry, C.; Otten, G.; Capon, D.J.; Tedder, T.F. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity 1994, 1, 247–260. [Google Scholar] [CrossRef]
- Wedepohl, S.; Beceren-Braun, F.; Riese, S.; Buscher, K.; Enders, S.; Bernhard, G.; Kilian, K.; Blanchard, V.; Dernedde, J.; Tauber, R. L-selectin—A dynamic regulator of leukocyte migration. Eur. J. Cell Biol. 2012, 91, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ivetic, A. Signals regulating L-selectin-dependent leucocyte adhesion and transmigration. Int. J. Biochem. Cell Biol. 2013, 45, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yago, T.; Zhang, N.; Panicker, S.R.; Wang, Y.; Yao, L.; Mahta-D’souza, P.; Xia, L.; Zhu, C.; McEver, R.P. L-selectin mechanochemistry restricts neutrophil priming in vivo. Nat. Commun. 2017, 8, 15196. [Google Scholar] [CrossRef] [Green Version]
- Rzeniewicz, K.; Newe, A.; Rey Gallardo, A.; Davies, J.; Holt, M.R.; Patel, A.; Charras, G.T.; Stramer, B.; Molenaar, C.; Tedder, T.F.; et al. L-selectin shedding is activated specifically within transmigrating pseudopods of monocytes to regulate cell polarity in vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E1461–E1470. [Google Scholar] [CrossRef] [Green Version]
- Allport, J.R.; Ding, H.T.; Ager, A.; Steeber, D.A.; Tedder, T.F.; Luscinskas, F.W. L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in vitro. J. Immunol. 1997, 158, 4365–4372. [Google Scholar] [PubMed]
- von Andrian, U.H.; Chambers, J.D.; Berg, E.L.; Michie, S.A.; Brown, D.A.; Karolak, D.; Ramezani, L.; Berger, E.M.; Arfons, K.E.; Butcher, E.C. L-selectin mediates neutrophil rolling in inflamed venules through sialyl LewisX-dependent and -independent recognition pathways. Blood 1993, 82, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Smalley, D.M.; Ley, K. L-selectin: Mechanisms and physiological significance of ectodomain cleavage. J. Cell Mol. Med. 2005, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Schultz, J.B.; Knauf, P.A.; King, M.R. Mechanical shedding of L-selectin from the neutrophil surface during rolling on sialyl Lewis x under flow. J. Biol. Chem. 2007, 282, 4812–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafezi-Moghadam, A.; Thomas, K.L.; Prorock, A.J.; Huo, Y.; Ley, K. L-selectin shedding regulates leukocyte recruitment. J. Exp. Med. 2001, 193, 863–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Liu, F.; Wang, Q.J.; Rosenberg, S.A.; Morgan, R.A. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS ONE 2011, 6, e22560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Font, J.; Pizcueta, P.; Ramos-Casals, M.; Cervera, R.; Garcia-Carrasco, M.; Navarro, M.; Ingelmo, M.; Engel, P. Increased serum levels of soluble L-selectin (CD62L) in patients with active systemic lupus erythematosus (SLE). Clin. Exp. Immunol. 2000, 119, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, S.; Silverman, M.; Shaw, R.J.; Klein, N. Soluble L-selectin concentration in bronchoalveolar lavage fluid obtained from infants who develop chronic lung disease of prematurity. Arch. Dis. Child Fetal. Neonatal. Ed. 1998, 78, F143–F147. [Google Scholar] [CrossRef]
- Martinez-Mier, G.; Toledo-Pereyra, L.H.; McDuffie, E.; Warner, R.L.; Ward, P.A. L-Selectin and chemokine response after liver ischemia and reperfusion. J. Surg. Res. 2000, 93, 156–162. [Google Scholar] [CrossRef]
- Faleck, D.M.; Winters, A.; Chablaney, S.; Shashi, P.; Meserve, J.; Weiss, A.; Aniwan, S.; Koliani-Pace, J.L.; Kochhar, G.; Boland, B.S.; et al. Shorter disease duration is associated with higher rates of response to Vedolizumab in patients with Crohn’s disease but not ulcerative colitis. Clin. Gastroenterol. Hepatol. 2019, 17, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Gremse, F.; Stark, M.; Ehling, J.; Menzel, J.R.; Lammers, T.; Kiessling, F. Imalytics Preclinical: Interactive analysis of biomedical volume data. Theranostics 2016, 6, 328–341. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Hubscher, S.G. Histological assessment of non-alcoholic fatty liver disease. Histopathology 2006, 49, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Qayyum, A.; Nystrom, M.; Noworolski, S.M.; Chu, P.; Mohanty, A.; Merriman, R. MRI steatosis grading: Development and initial validation of a color mapping system. AJR Am. J. Roentgenol. 2012, 198, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Herrick, A.; Jayson, M.I. Altered levels of soluble adhesion molecules in rheumatoid arthritis, vasculitis and systemic sclerosis. Br. J. Rheumatol. 1995, 34, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Machold, K.P.; Kiener, H.P.; Graninger, W.; Graninger, W.B. Soluble intercellular adhesion molecule-1 (sICAM-1) in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin. Immunol. Immunopathol. 1993, 68, 74–78. [Google Scholar] [CrossRef]
- Pino-Otin, M.R.; Vinas, O.; de la Fuente, M.A.; Juan, M.; Font, J.; Torradeflot, M.; Pallares, L.; Lozano, F.; Alberola-Ila, J.; Martorell, J. Existence of a soluble form of CD50 (intercellular adhesion molecule-3) produced upon human lymphocyte activation. Present in normal human serum and levels are increased in the serum of systemic lupus erythematosus patients. J. Immunol. 1995, 154, 3015–3024. [Google Scholar] [PubMed]
- Belmont, B.J.; Giorno, R.; Abramson, S. Up-regulation of endothelial cell adhesion molecules characterizes disease activity in systemic lupus erythematosus. The Shwartzman phenomenon revisited. Arthritis Rheum. 1994, 37, 376–383. [Google Scholar] [CrossRef]
- Lefere, S.; Van de Velde, F.; Devisscher, L.; Bekaert, M.; Raevens, S.; Verheist, X.; Van Nieuwenhove, Y.; Praet, M.; Hoorens, A.; Van Steenkiste, C.; et al. Serum vascular cell adhesion molecule-1 predicts significant liver fibrosis in non-alcoholic fatty liver disease. Int. J. Obes. (Lond.) 2017, 41, 1207–1213. [Google Scholar] [CrossRef] [Green Version]
- Patsenker, E.; Stickel, F. Role of integrins in fibrosing liver diseases. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 301, G425–G434. [Google Scholar] [CrossRef]
- Lard, L.R.; Mul, F.P.; de Haas, M.; Roos, D.; Duits, A.J. Neutrophil activation in sickle cell disease. J. Leukoc. Biol. 1999, 66, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.I.; Burns, A.R.; Taylor, A.D.; Gopalan, P.K.; Lynam, E.B.; Sklar, L.A.; Smith, C.W. L-selectin (CD62L) cross-linking signals neutrophil adhesive functions via the Mac-1 (CD11b/CD18) beta 2-integrin. J. Immunol. 1995, 155, 1502–1514. [Google Scholar] [PubMed]
- Nijhuis, J.; Rensen, S.S.; Slaats, Y.; van Dielen, F.M.; Buurman, W.A.; Greve, J.M. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity 2009, 17, 2014–2018. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 688. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; Lopez-Posadas, R.; Watson, A.; Neufert, C.; Atreya, I.; Neurath, M.F. Blockade of alphaEbeta7 integrin suppresses accumulation of CD8(+) and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyara, M.; Sakaguchi, S. Natural regulatory T cells: Mechanisms of suppression. Trends Mol. Med. 2007, 13, 108–116. [Google Scholar] [CrossRef]
- Takai, S.; Kimura, K.; Nagaki, M.; Satake, S.; Kakimi, K.; Moriwaki, H. Blockade of neutrophil elastase attenuates severe liver injury in hepatitis B transgenic mice. J. Virol. 2005, 79, 15142–15150. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Uria, G.; Day, J.N.; Nasir, A.J.; Russell, S.K.; Vilar, F.J. Reduction in neutrophil count during hepatitis C treatment: Drug toxicity or predictor of good response? Dig. Dis. Sci. 2010, 55, 2058–2062. [Google Scholar] [CrossRef]
- Rensen, S.S.; Bieghs, V.; Xanthoulea, S.; Arfianti, E.; Bakker, J.A.; Shiri-Sverdlov, R.; Hofker, M.H.; Greve, J.W.; Buurman, W.A. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PLoS ONE 2012, 7, e52411. [Google Scholar] [CrossRef] [Green Version]
- Pillay, J.; Kamp, V.M.; van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, R.; Buerger, L.; Ibrahim, S.; Melter, M.; Weiss, T.S. Augmenter of liver regeneration (ALR) exhibits a dual signaling impact on hepatic acute-phase response. Exp. Mol. Pathol. 2017, 102, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, G.; Behrend, M.; Schrader, B.; Duijvestijn, A.M.; Wonigeit, K. Expression patterns of leukocyte adhesion ligand molecules on human liver endothelia. Lack of ELAM-1 and CD62 inducibility on sinusoidal endothelia and distinct distribution of VCAM-1, ICAM-1, ICAM-2, and LFA-3. Am. J. Pathol. 1993, 142, 481–488. [Google Scholar] [PubMed]
- Apostolopoulou, M.; Gordillo, R.; Koliaki, C.; Gancheva, S.; Jelenik, T.; De Filippo, E.; Herder, C.; Markgraf, D.; Jankowiak, F.; Esposito, J.; et al. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care 2018, 41, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadori, P.; Drescher, H.; Erschfeld, S.; Schumacher, F.; Berger, C.; Fragoulis, A.; Schenkel, J.; Kensler, T.W.; Wruck, C.J.; Trautwein, C.; et al. Hepatocyte-specific Keap1 deletion reduces liver steatosis but not inflammation during non-alcoholic steatohepatitis development. Free Radic. Biol. Med. 2016, 91, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Sacerdoti, D.; Singh, S.P.; Schragenheim, J.; Bellner, L.; Vanella, L.; Raffaele, M.; Meissner, A.; Grant, I.; Favero, G.; Rezzani, R.; et al. Development of NASH in Obese Mice is Confounded by Adipose Tissue Increase in Inflammatory NOV and Oxidative Stress. Int. J. Hepatol. 2018, 2018, 3484107. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.M.; Pae, H.O.; Jeong, Y.R.; Kim, Y.M.; Chung, H.T. Critical role of heme oxygenase-1 in Foxp3-mediated immune suppression. Biochem. Biophys. Res. Commun. 2005, 327, 1066–1071. [Google Scholar] [CrossRef]
- Maretti-Mira, A.C.; Wang, X.; Wang, L.; DeLeve, L.D. Incomplete differentiation of engrafted bone marrow endothelial progenitor cells initiates hepatic fibrosis in the rat. Hepatology 2019, 69, 1259–1272. [Google Scholar] [CrossRef]
- Ibusuki, R.; Uto, H.; Arima, S.; Mawatari, S.; Setoguchi, Y.; Iwashita, Y.; Hashimoto, S.; Maeda, T.; Tanoue, S.; Kanmura, S.; et al. Transgenic expression of human neutrophil peptide-1 enhances hepatic fibrosis in mice fed a choline-deficient, L-amino acid-defined diet. Liver Int. 2013, 33, 1549–1556. [Google Scholar] [CrossRef]
- Alkhouri, N.; Morris-Stiff, G.; Campbell, C.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Zein, N.N.; Feldstein, A.E. Neutrophil to lymphocyte ratio: A new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int. 2012, 32, 297–302. [Google Scholar] [CrossRef]
- Kolachala, V.L.; Palle, S.; Shen, M.; Feng, A.; Shayakhmetov, D.; Gupta, N.A. Loss of L-selectin-guided CD8(+), but not CD4(+), cells protects against ischemia reperfusion injury in a steatotic liver. Hepatology 2017, 66, 1258–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunoreactivity | Dilution | Species | Manufacturer | Cat. No. |
|---|---|---|---|---|
| Ly6G | 1:100 | Rat | BD Pharmingen, Heidelberg, Germany | 550291 |
| F4/80 | 1:100 | Rat | Biorad, Hercules, CA USA | MCA497GA |
| DHE | 1:500 | - | Invitrogen, Carlsbad, CA, USA | D23107 |
| VCAM-1 | 1:100 | Rat | BD Pharmingen | 553329 |
| ICAM-1 | 1:100 | Rat | eBioscience, Santa Clara, CA, USA | 14-0541-81 |
| Gene | Species | Sequence |
|---|---|---|
| GAPDH | Mouse | for: 5′-TGT TGA AGT CAC AGG AGA CAA CCT-3′ rev: 5′-AAC CTG CCA AGT ATG ATG ACA TCA-3′ |
| TGF-β1 | Mouse | for: 5′-ATA CGC CTG AGT GGC TGT CT -3′ rev: 5′-TCA TGG ATG GTG CCC AGG TC-3′ |
| Nrf2 | Mouse | for: 5′-CCG AGA TAT ACG CAG GAG AGG TA-3′ rev: 5′-GCT CGA CAA TGT TCT CCA GCT T-3′ |
| HO-1 | Mouse | for: 5′-TGG TGG CCC ACG CAT ATA C-3′ rev: 5′-CAT GGC CTT CTG TGC AAT CTT-3′ |
| IL-10 | Mouse | for: 5′-GAT GCC CCA GGC AGA GAA-3′ rev: 5′-CAC CCA GGG AAT TCA AAT GC-3′ |
| FoxP3 | Mouse | for: 5′-GGC AAA TGG AGT CTG CAA GTG-3′ rev: 5′-CAG GAG ATG ATC TGC TTG GCA-3′ |
| TNF-α | Mouse | for: 5′-AGC TCG TAG CAA ACC ACC AA-3′ rev: 5′-GAG AAC CTG GGA GTA GAC AAG G-3′ |
| IL-1β | Mouse | for: 5′-GGA GAA CCA AGC AAC GAC AAA-3′ rev: 5′-GGG TGT GCC GTC TTT CAT TAC-3′ |
| ADAM-17 | Mouse | for: 5′-CAC AAA ACT TGA GAG TCG TGG T-3′ rev: 5′-GCT AGA ACC CTA GAG TCA GGC-3′ |
| IL-6 | Mouse | for: 5′-CTG CAA GAG ACT TCC ATC CAG-3′ rev: 5′-AGT GGT ATA GAC AGG TCT GTT GG-3′ |
| Plin2 | Mouse | for: 5′-GTC CAC CTG ATT GAA TTC GC-3′ rev: 5′-CGA TGT GCT CAA CAC AGT G-3′ |
| G0S2 | Mouse | for: 5′-ACT GCA CCC TAG GCC CAG-3′ rev: 5′-GTC TCA ACT AGG CCG AGC AC-3′ |
| CD36 | Mouse | for: 5′-CAA ATG CAA AGA AGG AAA GCC-3′ rev: 5′-AAT GGT CCC AGT CTC ATT TAG C-3′ |
| Fabp1 | Mouse | for: 5′-GCA GAG CCA GGA GAA CTT TG-3′ rev: 5′-GGG TCC ATA GGT GAT GGT GAG- 3′ |
| β-Actin | Human | for: 5′-TCC ATC ATG AAG TGT GAC GT-3′ reverse: TAC TCC TGC TTG CTG ATC CAC-3′ |
| RPLP0 | Human | for: 5′-ACT GTG CCA GCC CAG AAC A-3′ rev: 5′-AGC CTG GAA AAA GGA GGT CTT C-3′ |
| CD62L | Human | for: 5′-GGA CTG CGT GGA GAT CTA TAT CAA-3′ rev: 5′-TGG CAG GCG TCA TCG TT-3′ |
| β7-Integrin | Human | for: 5′-GGA CTC CAG CAA CGT GGT ACA-3′ rev: 5′-TCA CGG TGG AAG ACA GGC TAT-3′ |
| MAdCAM-1 | Human | for: 5′-TGA GTG GCC AGC CTT TCC-3′ rev: 5′-CCC TGA CCA GTT CTC AAC TTG AA-3′ |
| ICAM-1 | Human | for: 5′-TGG CCC TCC ATA GAC ATG TGT-3′ rev: 5′-TGG CAT CCG TCA GGA AGT G-3′ |
| VCAM-1 | Human | for: 5′-CAA AGG CAG AGT ACG CAA ACA C-3′ rev: 5′-GCT GAC CAA GAC GGT TGT ATC TC-3′ |
| Cross-Reactivity (Panel) | Antibody | Conjugate | Manufacturer | Cat. No. |
|---|---|---|---|---|
| Mouse (1) | CD45 | APC-Cy7 | BD Pharmingen, Heidelberg, Germany | 557659 |
| F4/80 | APC | eBioscience, Santa Clara, CA, USA | 47-4801 | |
| CD11b | PE | eBioscience | 12-0112-82 | |
| CD11c | PE-Cy7 | eBioscience | 25-0114-81 | |
| Ly6G/Ly6C | PerCP-Cy5.5 | Biolegend, San Diego, CA, USA | 127615 | |
| Ly6G | FITC | eBioscience | 17-5931-81 | |
| Hoechst 33258 | - | Sigma Aldrich, Merck, Taufkirchen, Germany | B2883 | |
| Mouse (2) | CD45 | APC-Cy7 | BD Pharmingen | 557659 |
| CD3 | PE-Cy7 | Biolegend | 100219 | |
| CD4 | FITC | eBioscience | 11-0042-82 | |
| CTLA4 | PE | eBioscience | 12-1522-81 | |
| CD8 | PerCP-Cy5.5 | BD Pharmingen | 551162 | |
| CD25 | PE | eBioscience | 12-0025-81 | |
| FoxP3 | APC | eBioscience | 17-5773-80 | |
| Hoechst 33258 | - | Sigma Aldrich | B2883 | |
| Human (1) | CD45 | BV510 | BD Pharmingen | 563204 |
| CD4 | APC-Cy7 | BD Pharmingen | 561839 | |
| CD8 | AF700 | BD Pharmingen | 557945 | |
| CD62L | PE-Cy7 | BD Pharmingen | 565535 | |
| CD66b | APC | eBioscience | 17-0666-42 | |
| CD14 | VioGreen | Milteny Biotech, Bergisch Gladbach, Germany | 130-096-875 | |
| CD68 | PE-eFluor610 | eBioscience | 61-0689-41 | |
| Hoechst 33258 | - | Sigma Aldrich | B2883 |
| Chemical | SC1 | SC2 | GBSS-A | GBSS-B |
|---|---|---|---|---|
| NaCl | 8000 mg/L | 8000 mg/L | - | 8000 mg/L |
| KCl | 400 mg/L | 400 mg/L | 370 mg/L | 370 mg/L |
| NaH2PO4 × H2O | 88.17 mg/L | 88.17 mg/L | - | - |
| Na2HPO4 | 120.45 mg/L | 120.45 mg/L | 59.6 mg/L | 59.6 mg/L |
| HEPES | 2380 mg/L | 2380 mg/L | - | - |
| NaHCO3 | 350 mg/L | 350 mg/L | 227 mg/L | 227 mg/L |
| EGTA | 190 mg/L | - | - | - |
| Glucose | 900 mg/L | - | 991 mg/L | 991 mg/L |
| CaCl2 × 2H2O | - | 560 mg/L | 225 mg/L | 225 mg/L |
| MgCl2 × 6H2O | - | - | 210 mg/L | 210 mg/L |
| MgSO4 × 7H2O | - | - | 70 mg/L | 70 mg/L |
| KH2PO4 | - | - | 30 mg/L | 30 mg/L |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drescher, H.K.; Schippers, A.; Rosenhain, S.; Gremse, F.; Bongiovanni, L.; de Bruin, A.; Eswaran, S.; Gallage, S.U.; Pfister, D.; Szydlowska, M.; et al. L-Selectin/CD62L Is a Key Driver of Non-Alcoholic Steatohepatitis in Mice and Men. Cells 2020, 9, 1106. https://doi.org/10.3390/cells9051106
Drescher HK, Schippers A, Rosenhain S, Gremse F, Bongiovanni L, de Bruin A, Eswaran S, Gallage SU, Pfister D, Szydlowska M, et al. L-Selectin/CD62L Is a Key Driver of Non-Alcoholic Steatohepatitis in Mice and Men. Cells. 2020; 9(5):1106. https://doi.org/10.3390/cells9051106
Chicago/Turabian StyleDrescher, Hannah K., Angela Schippers, Stefanie Rosenhain, Felix Gremse, Laura Bongiovanni, Alain de Bruin, Sreepradha Eswaran, Suchira U. Gallage, Dominik Pfister, Marta Szydlowska, and et al. 2020. "L-Selectin/CD62L Is a Key Driver of Non-Alcoholic Steatohepatitis in Mice and Men" Cells 9, no. 5: 1106. https://doi.org/10.3390/cells9051106