Development of Cellular Models to Study Efficiency and Safety of Gene Edition by Homologous Directed Recombination Using the CRISPR/Cas9 System

 , , ,
, , ,  and
and 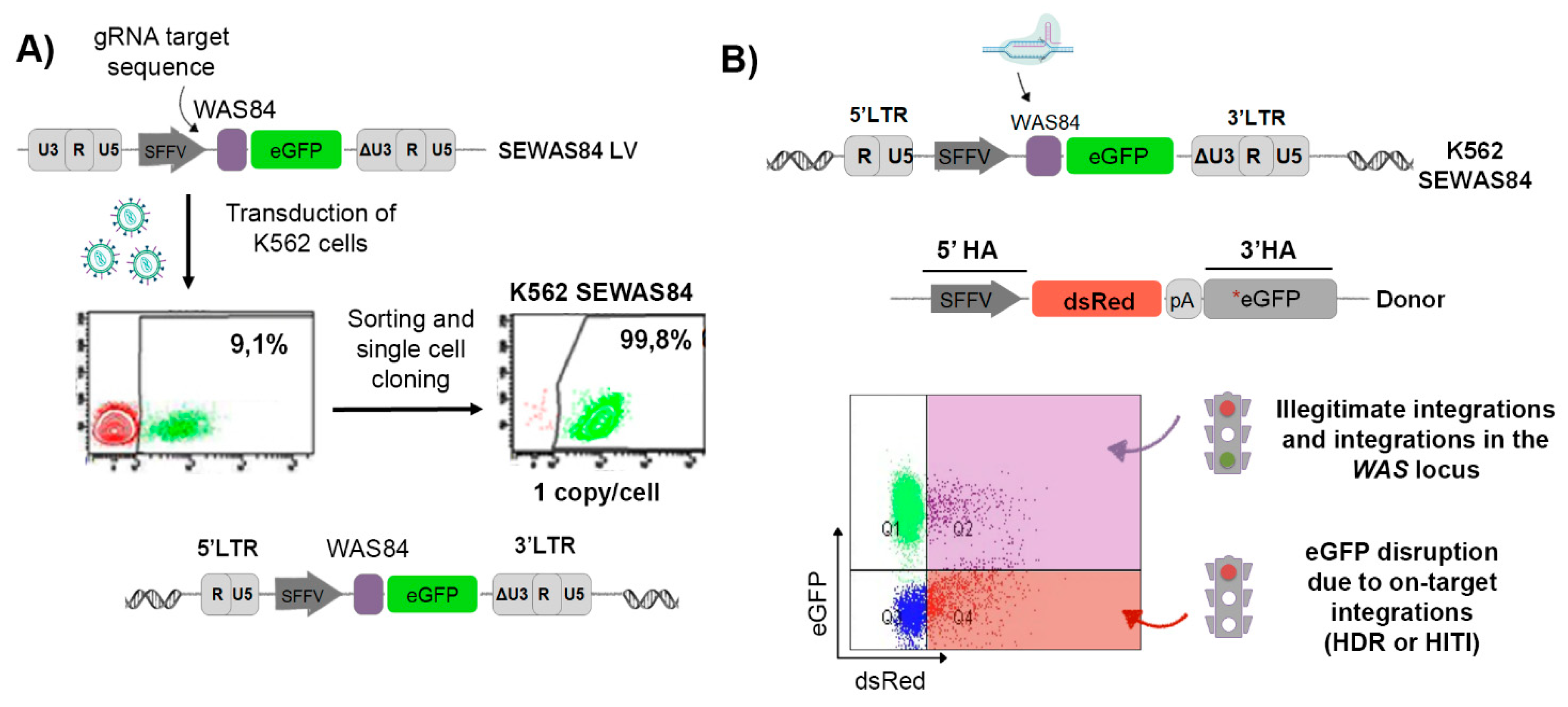{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Lines and Culture Media
2.2. Lentiviral Constructions
2.3. Donor Constructions
2.4. CRISPR/Cas9 Constructions
2.5. Vector Production and Virus Titration
2.6. Cell Transduction
2.7. Copy Number (qPCR)
2.8. Single Cell Cloning
2.9. Cas9 RNP Assembly
2.10. Nucleofection
2.11. Flow Cytometry
2.12. Statistical Analyses
3. Results
3.1. Generation of the eGFP-OFF/dsRED-ON Cellular Model
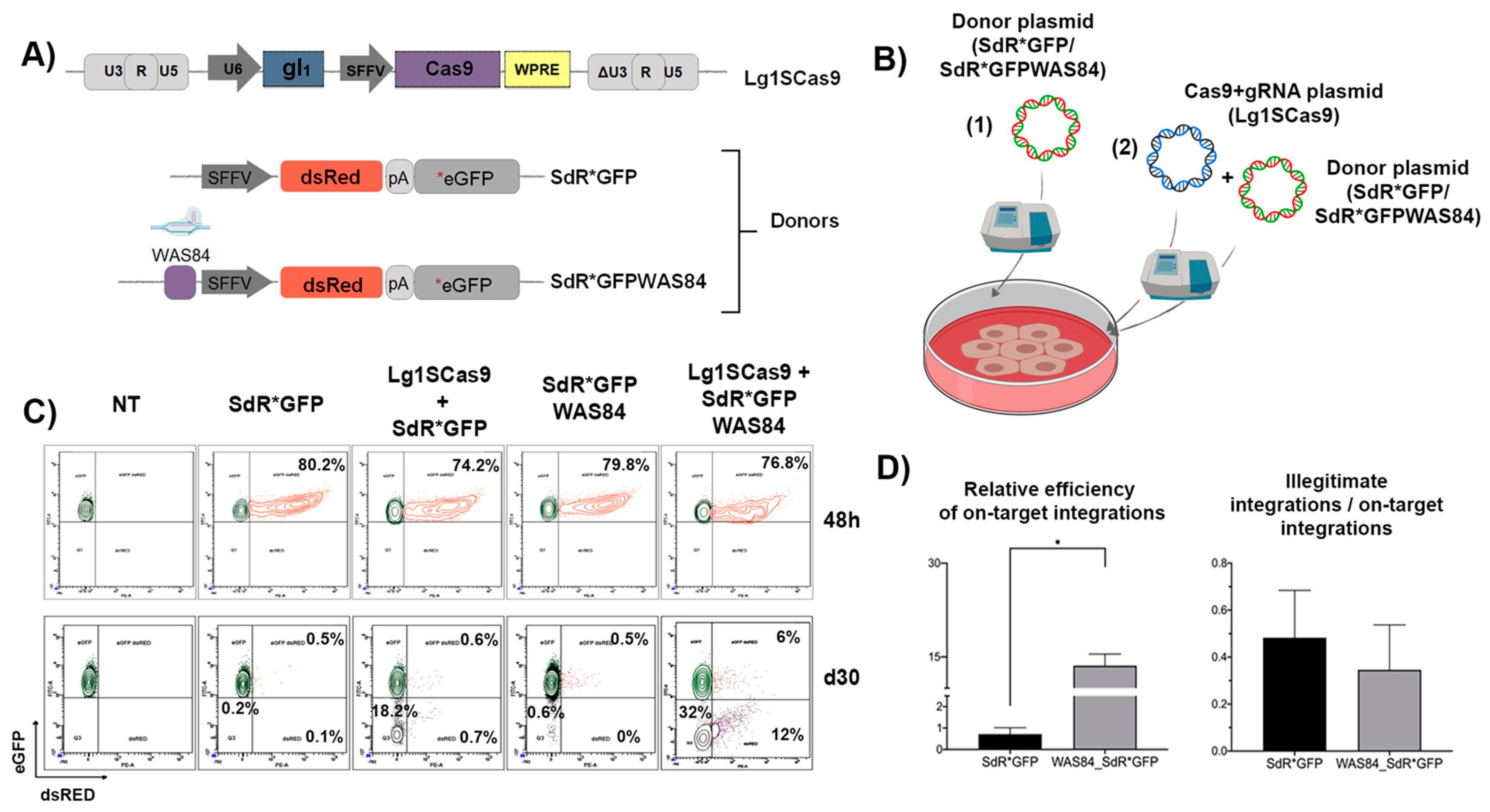
3.2. The Inclusion of the CRISPR/Cas9 Target Sequence into the Donor Template Increases the HDR Efficiency in K562 SEWAS84 Cells without Compromising Specificity
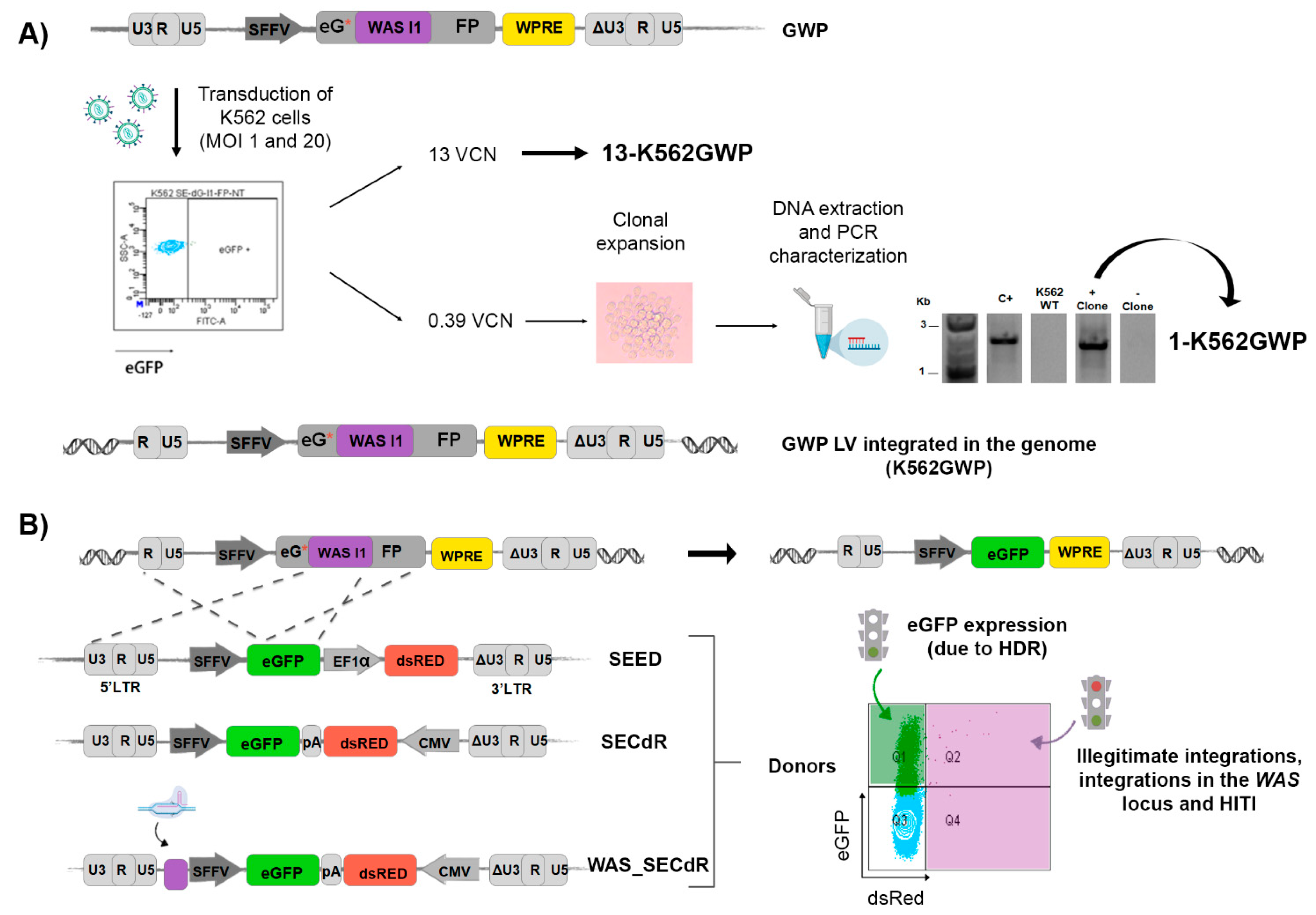
3.3. Generation of Cellular Models to Detect HDR-GE: eGFP-ON/dsRED-OFF Reporter
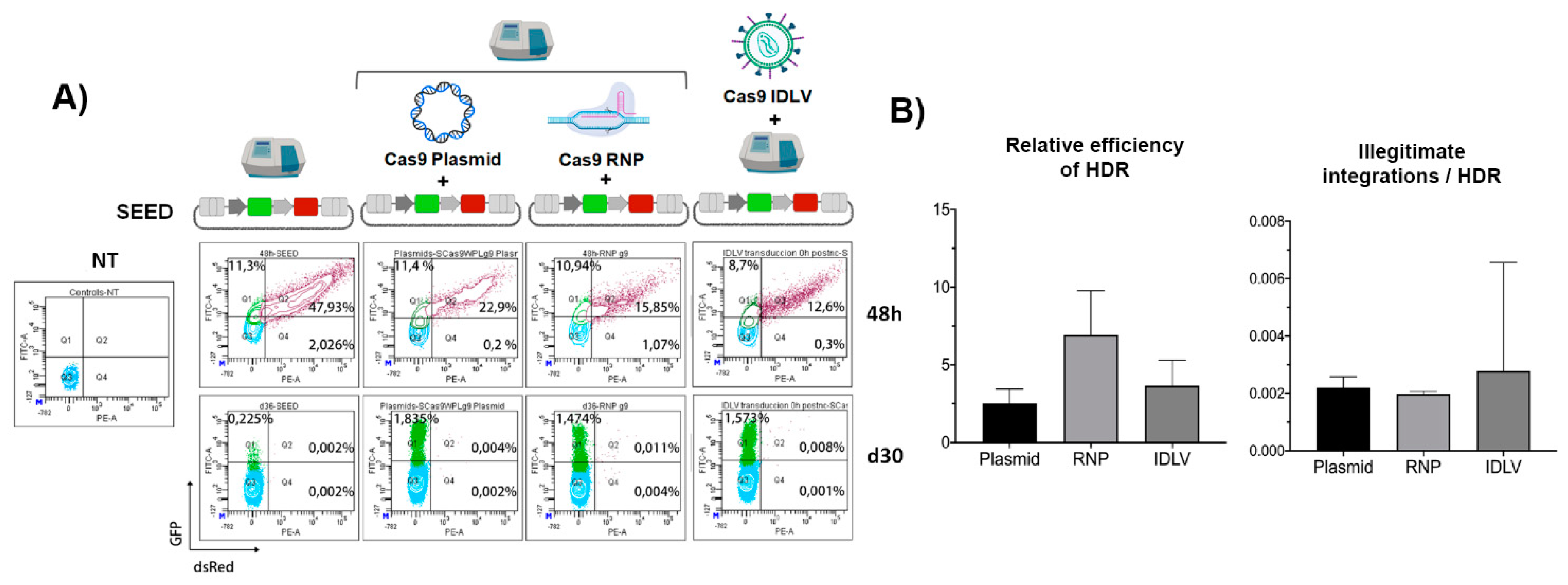
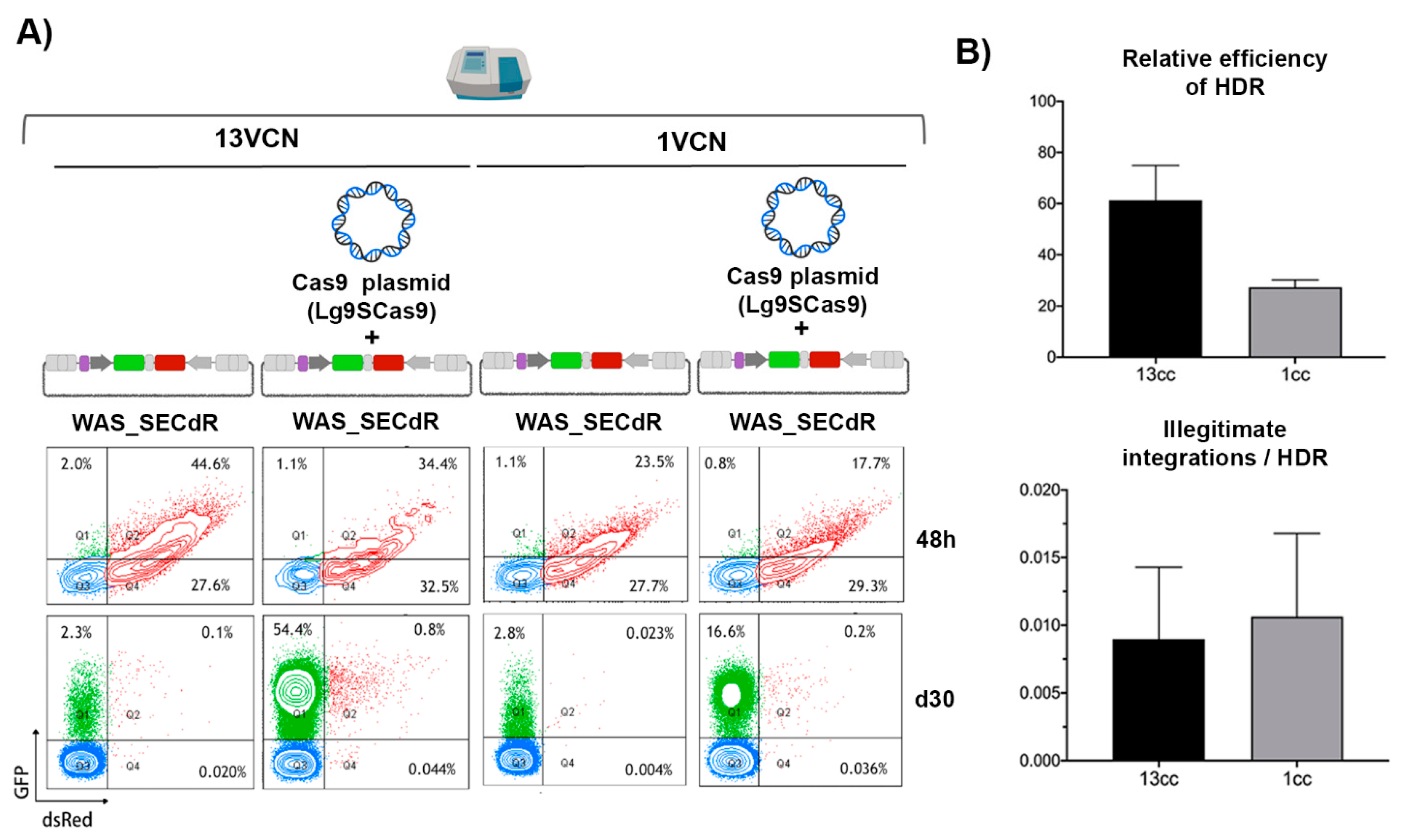
3.4. The Efficiency and Specificity of HDR-GE Is Independent on the CRISPR/Cas Delivery Method
3.5. Analysis of GE Efficacy and Specificity of Different DNA Donor Design Using 13-K562GWP Reporter Cells
3.6. The Efficacy of GE Correlates with the Frequency of Target Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broeders, M.; Herrero-Hernandez, P.; Ernst, M.P.T.; van der Ploeg, A.T.; Pijnappel, W. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. Iscience 2020, 23, 100789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benabdellah, K.; Sanchez-Hernandez, S.; Aguilar-Gonzalez, A.; Maldonado-Perez, N.; Gutierrez-Guerrero, A.; Cortijo-Gutierrez, M.; Ramos-Hernandez, I.; Tristan-Manzano, M.; Galindo-Moreno, P.; Herrera, C.; et al. Genome-edited adult stem cells: Next-generation advanced therapy medicinal products (ATMPs). Stem Cells Transl. Med. 2020, 9, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepek, M.; Brondani, V.; Buchel, J.; Serrano, L.; Segal, D.J.; Cathomen, T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007, 25, 786–793. [Google Scholar] [CrossRef]
- Paschon, D.E.; Lussier, S.; Wangzor, T.; Xia, D.F.; Li, P.W.; Hinkley, S.J.; Scarlott, N.A.; Lam, S.C.; Waite, A.J.; Truong, L.N.; et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun. 2019, 10, 1133. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Huang, S.; Jiang, W.Z.; Wright, D.; Spalding, M.H.; Weeks, D.P.; Yang, B. TAL nucleases (TALNs): Hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011, 39, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Mahfouz, M.M.; Li, L.; Shamimuzzaman, M.; Wibowo, A.; Fang, X.; Zhu, J.K. De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc. Natl. Acad. Sci. USA 2011, 108, 2623–2628. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.G. Genome editing-based HIV therapies. Trends Biotechnol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman-Rodriguez, F.J.; Ugalde, L.; Alvarez, L.; Diez, B.; Ramirez, M.J.; Risueno, C.; Corton, M.; Bogliolo, M.; Bernal, S.; March, F.; et al. NHEJ-Mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell 2019, 25, 607–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Pesch, T.; Bonati, L.; Kelton, W.; Parola, C.; Ehling, R.A.; Csepregi, L.; Kitamura, D.; Reddy, S.T. Molecular Design, Optimization, and Genomic Integration of Chimeric B Cell Receptors in Murine B Cells. Front. Immunol. 2019, 10, 2630. [Google Scholar] [CrossRef] [Green Version]
- Puthenveetil, G.; Malik, P. Gene therapy for hemoglobinopathies: Are we there yet? Curr. Hematol. Rep. 2004, 3, 298–305. [Google Scholar]
- Park, S.H.; Lee, C.M.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H.; et al. Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef]
- Pavel-Dinu, M.; Wiebking, V.; Dejene, B.T.; Srifa, W.; Mantri, S.; Nicolas, C.E.; Lee, C.; Bao, G.; Kildebeck, E.J.; Punjya, N.; et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019, 10, 1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.Y.; Thrasher, A.J.; Zhang, F. Gene therapy and genome editing for primary immunodeficiency diseases. Genes Dis. 2020, 7, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Poletti, V.; Biffi, A. Gene-Based Approaches to Inherited Neurometabolic Diseases. Hum. Gene Ther. 2019, 30, 1222–1235. [Google Scholar] [CrossRef] [PubMed]
- Poletto, E.; Baldo, G.; Gomez-Ospina, N. Genome Editing for Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Wurtele, H.; Little, K.C.; Chartrand, P. Illegitimate DNA integration in mammalian cells. Gene Ther. 2003, 10, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Guerrero, A.; Sanchez-Hernandez, S.; Galvani, G.; Pinedo-Gomez, J.; Martin-Guerra, R.; Sanchez-Gilabert, A.; Aguilar-Gonzalez, A.; Cobo, M.; Gregory, P.; Holmes, M.; et al. Comparison of Zinc Finger Nucleases Versus CRISPR-Specific Nucleases for Genome Editing of the Wiskott-Aldrich Syndrome Locus. Hum. Gene Ther. 2018, 29, 366–380. [Google Scholar] [CrossRef]
- Olsen, P.A.; Gelazauskaite, M.; Randol, M.; Krauss, S. Analysis of illegitimate genomic integration mediated by zinc-finger nucleases: Implications for specificity of targeted gene correction. BMC Mol. Biol. 2010, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.; Sanchez-Hernandez, S.; Gutierrez-Guerrero, A.; Pinedo-Gomez, J.; Benabdellah, K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int. J. Mol. Sci. 2016, 17, 1507. [Google Scholar] [CrossRef] [PubMed]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv. Sci. 2020, 7, 1902312. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Song, C.Q.; Suresh, S.; Wu, Q.; Walsh, S.; Rhym, L.H.; Mintzer, E.; Bolukbasi, M.F.; Zhu, L.J.; Kauffman, K.; et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017, 35, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.B.; Kim, D.Y.; Ko, J.H.; Kim, Y.S. Recent advances in the CRISPR genome editing tool set. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A.M.; et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat. Commun. 2020, 11, 2109. [Google Scholar] [CrossRef]
- Paulsen, B.S.; Mandal, P.K.; Frock, R.L.; Boyraz, B.; Yadav, R.; Upadhyayula, S.; Gutierrez-Martinez, P.; Ebina, W.; Fasth, A.; Kirchhausen, T.; et al. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR-Cas9 genome editing. Nat. Biomed. Eng. 2017, 1, 878–888. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019, 10, 2866. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Stieger, K. Optimizing the DNA Donor Template for Homology-Directed Repair of Double-Strand Breaks. Mol. Ther. Nucleic Acids 2017, 7, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Kalkan, B.M.; Kala, E.Y.; Yuce, M.; Karadag Alpaslan, M.; Kocabas, F. Development of gene editing strategies for human beta-globin (HBB) gene mutations. Gene 2020, 734, 144398. [Google Scholar] [CrossRef]
- Zhang, J.P.; Li, X.L.; Li, G.H.; Chen, W.; Arakaki, C.; Botimer, G.D.; Baylink, D.; Zhang, L.; Wen, W.; Fu, Y.W.; et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Nami, F.; Basiri, M.; Satarian, L.; Curtiss, C.; Baharvand, H.; Verfaillie, C. Strategies for In Vivo Genome Editing in Nondividing Cells. Trends Biotechnol. 2018, 36, 770–786. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gerbi, S.A. Making ends meet: Targeted integration of DNA fragments by genome editing. Chromosoma 2018, 127, 405–420. [Google Scholar] [CrossRef]
- Dever DP, Porteus MH: The changing landscape of gene editing in hematopoietic stem cells: A step towards Cas9 clinical translation. Curr. Opin. Hematol. 2017, 24, 481–488. [CrossRef]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Bartholomae, C.C.; Gabriel, R.; Deichmann, A.; Schmidt, M. The LAM-PCR Method to Sequence LV Integration Sites. Methods Mol. Biol. 2016, 1448, 107–120. [Google Scholar] [PubMed]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef] [PubMed]
- de Vree, P.J.; de Wit, E.; Yilmaz, M.; van de Heijning, M.; Klous, P.; Verstegen, M.J.; Wan, Y.; Teunissen, H.; Krijger, P.H.; Geeven, G.; et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat. Biotechnol. 2014, 32, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Melamed, A.; Bangham, C.R. High-Throughput Mapping and Clonal Quantification of Retroviral Integration Sites. Methods Mol. Biol. 2017, 1582, 127–141. [Google Scholar] [PubMed]
- Dawes, J.C.; Webster, P.; Iadarola, B.; Garcia-Diaz, C.; Dore, M.; Bolt, B.J.; Dewchand, H.; Dharmalingam, G.; McLatchie, A.P.; Kaczor, J.; et al. LUMI-PCR: An Illumina platform ligation-mediated PCR protocol for integration site cloning, provides molecular quantitation of integration sites. Mobile DNA 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP: Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. 2014, 15, 121–132. [CrossRef]
- Xu, W.; Fu, W.; Zhu, P.; Li, Z.; Wang, C.; Wang, C.; Zhang, Y.; Zhu, S. Comprehensive Analysis of CRISPR/Cas9-Mediated Mutagenesis in Arabidopsis thaliana by Genome-wide Sequencing. Int. J. Mol. Sci. 2019, 20, 4125. [Google Scholar] [CrossRef] [Green Version]
- Certo, M.T.; Ryu, B.Y.; Annis, J.E.; Garibov, M.; Jarjour, J.; Rawlings, D.J.; Scharenberg, A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 2011, 8, 671–676. [Google Scholar] [CrossRef]
- Kuhar, R.; Gwiazda, K.S.; Humbert, O.; Mandt, T.; Pangallo, J.; Brault, M.; Khan, I.; Maizels, N.; Rawlings, D.J.; Scharenberg, A.M.; et al. Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease-induced breaks. Nucleic Acids Res. 2014, 42, e4. [Google Scholar] [CrossRef] [Green Version]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.; Toscano, M.G.; Blundell, M.; Frecha, C.; Srivastava, G.K.; Santamaria, M.; Thrasher, A.J.; Molina, I.J. Lentiviral vectors transcriptionally targeted to hematopoietic cells by WASP gene proximal promoter sequences. Gene Ther. 2005, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Benabdellah, K.; Cobo, M.; Munoz, P.; Toscano, M.G.; Martin, F. Development of an all-in-one lentiviral vector system based on the original TetR for the easy generation of Tet-ON cell lines. PLoS ONE 2011, 6, e23734. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Suzuki, K.; Izpisua Belmonte, J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018, 63, 157–164. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Hernández, S.; Aguilar-González, A.; Guijarro-Albaladejo, B.; Maldonado-Pérez, N.; Ramos-Hernández, I.; Cortijo-Gutiérrez, M.; Sánchez Martín, R.M.; Benabdellah, K.; Martin, F. Development of Cellular Models to Study Efficiency and Safety of Gene Edition by Homologous Directed Recombination Using the CRISPR/Cas9 System. Cells 2020, 9, 1492. https://doi.org/10.3390/cells9061492
Sánchez-Hernández S, Aguilar-González A, Guijarro-Albaladejo B, Maldonado-Pérez N, Ramos-Hernández I, Cortijo-Gutiérrez M, Sánchez Martín RM, Benabdellah K, Martin F. Development of Cellular Models to Study Efficiency and Safety of Gene Edition by Homologous Directed Recombination Using the CRISPR/Cas9 System. Cells. 2020; 9(6):1492. https://doi.org/10.3390/cells9061492
Chicago/Turabian StyleSánchez-Hernández, Sabina, Araceli Aguilar-González, Beatriz Guijarro-Albaladejo, Noelia Maldonado-Pérez, Iris Ramos-Hernández, Marina Cortijo-Gutiérrez, Rosario María Sánchez Martín, Karim Benabdellah, and Francisco Martin. 2020. "Development of Cellular Models to Study Efficiency and Safety of Gene Edition by Homologous Directed Recombination Using the CRISPR/Cas9 System" Cells 9, no. 6: 1492. https://doi.org/10.3390/cells9061492