The Secretome Analysis of Activated Human Renal Fibroblasts Revealed Beneficial Effect of the Modulation of the Secreted Peptidyl-Prolyl Cis-Trans Isomerase A in Kidney Fibrosis

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell line and Culture Procedure
2.2. Cell Culture of Renal Fibroblasts on Type I Collagen Gels
2.3. FCS-Free Cell Culture and Cytokine/Hormone Treatment Experiments
2.4. Protein Extraction and Precipitation
2.5. Two-Dimensional Gel Electrophoresis (2-DE)
2.6. Image Analysis
2.7. In-Gel Digestion and Mass Spectrometry Analysis of Protein Spots
2.8. Bioinformatics
2.9. Western Blot Analysis
2.10. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Cell Viability Assay
2.11. Analysis of Apoptosis by Flow Cytometry
2.12. Indirect Immunofluorescence Staining
2.13. Immunofluorescence Staining of the Kidney Sections
2.14. siRNA Mediated Knockdown of PPIA
2.15. Protein Co-Immunoprecipitation
2.16. In-Gel Digestion, Mass Spectrometric Analysis, and Identification of the Potential Interaction Partners of PPIA
2.17. Data Analysis
3. Results
3.1. Enrichment of Secretome Proteins: Protocol Optimization
3.2. Transformation of Renal Cell Fibroblast toward Fibrosis Phenotype Results in Significant Alteration of Cell Secretome
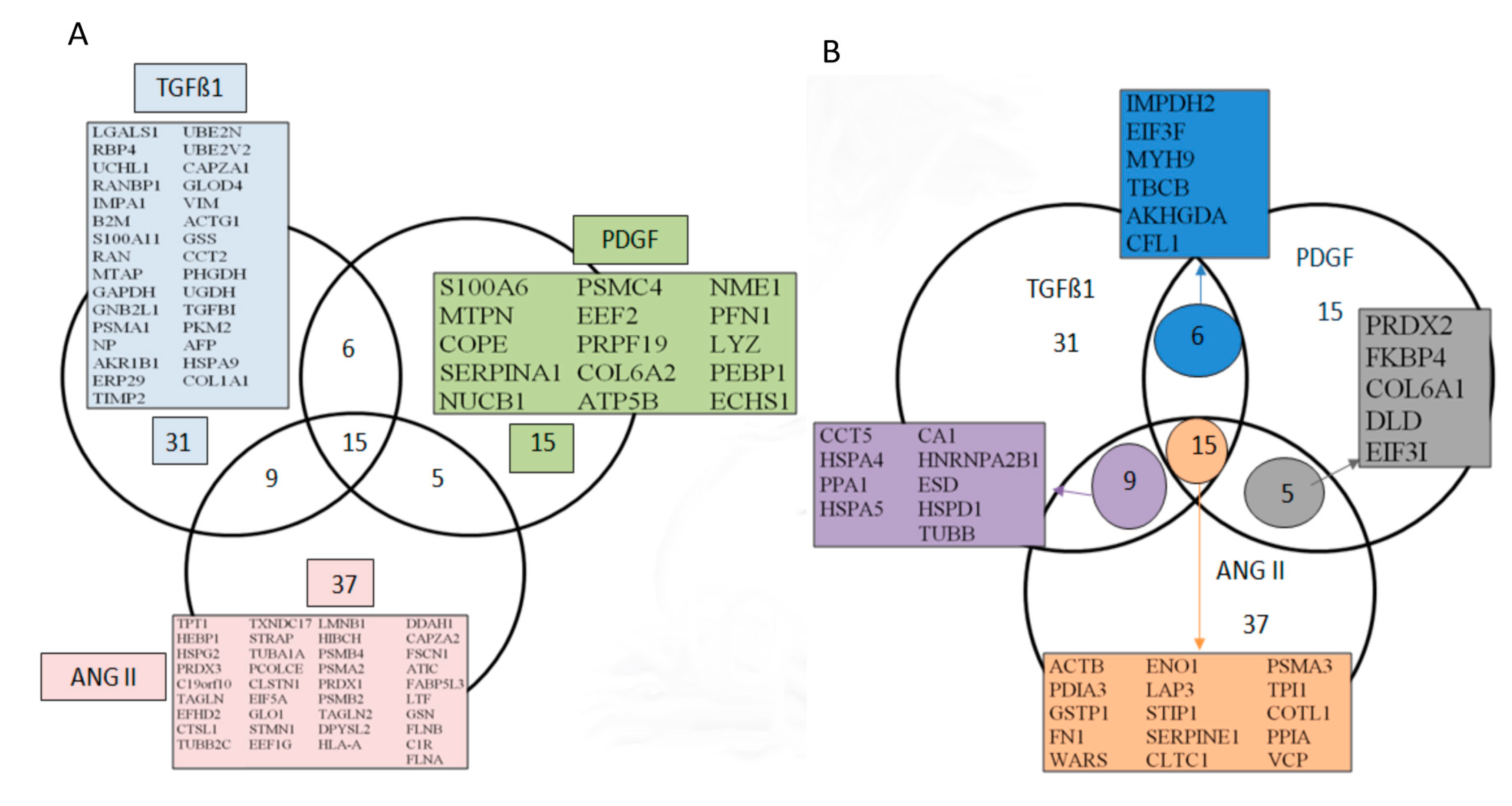
3.3. Bioinformatics Analyses of the Secreted Proteins
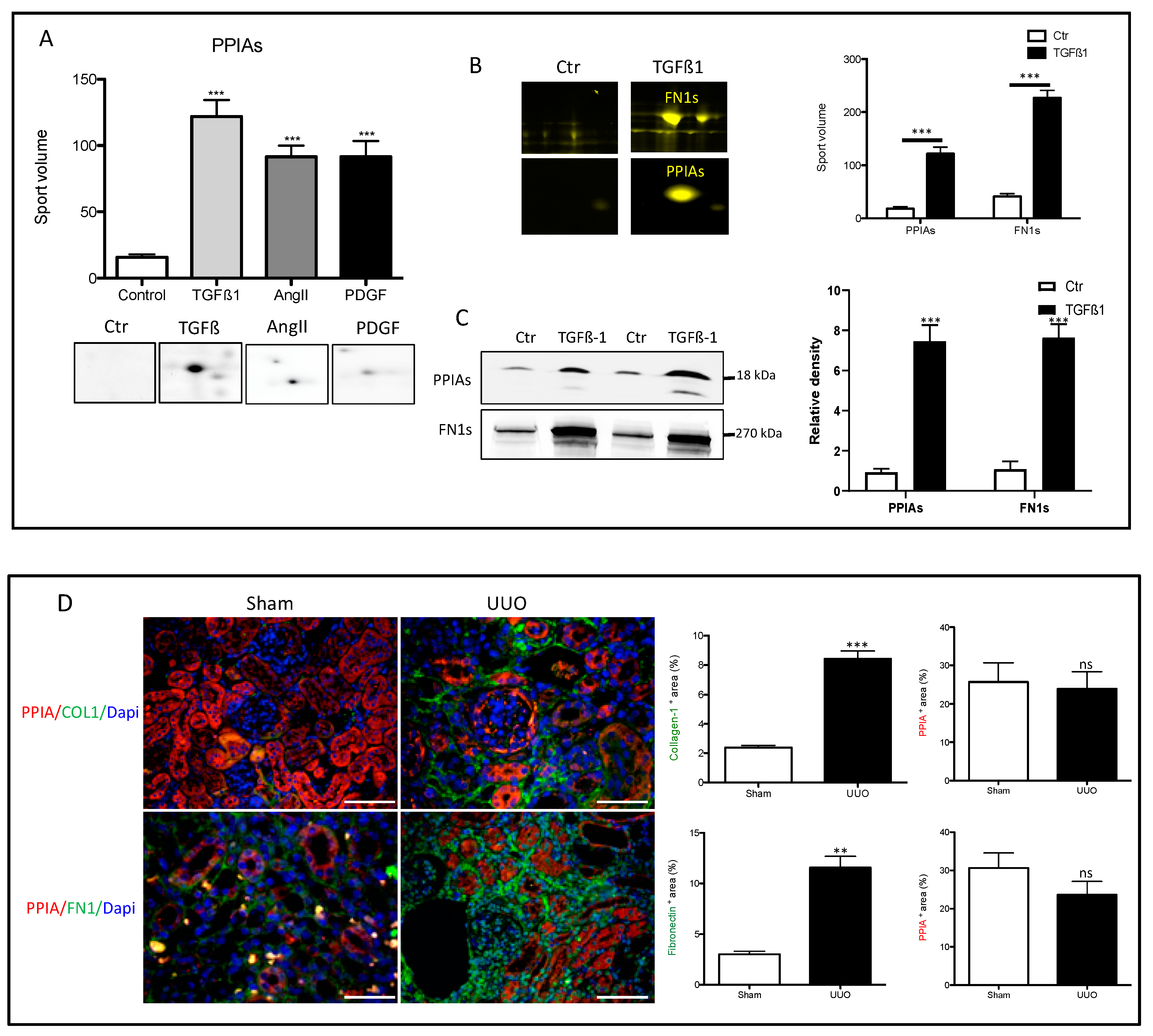
3.4. Secretion of Peptidyl-Prolyl Cis-Trans Isomerase A as a Common Reaction in Activated Renal Fibroblasts
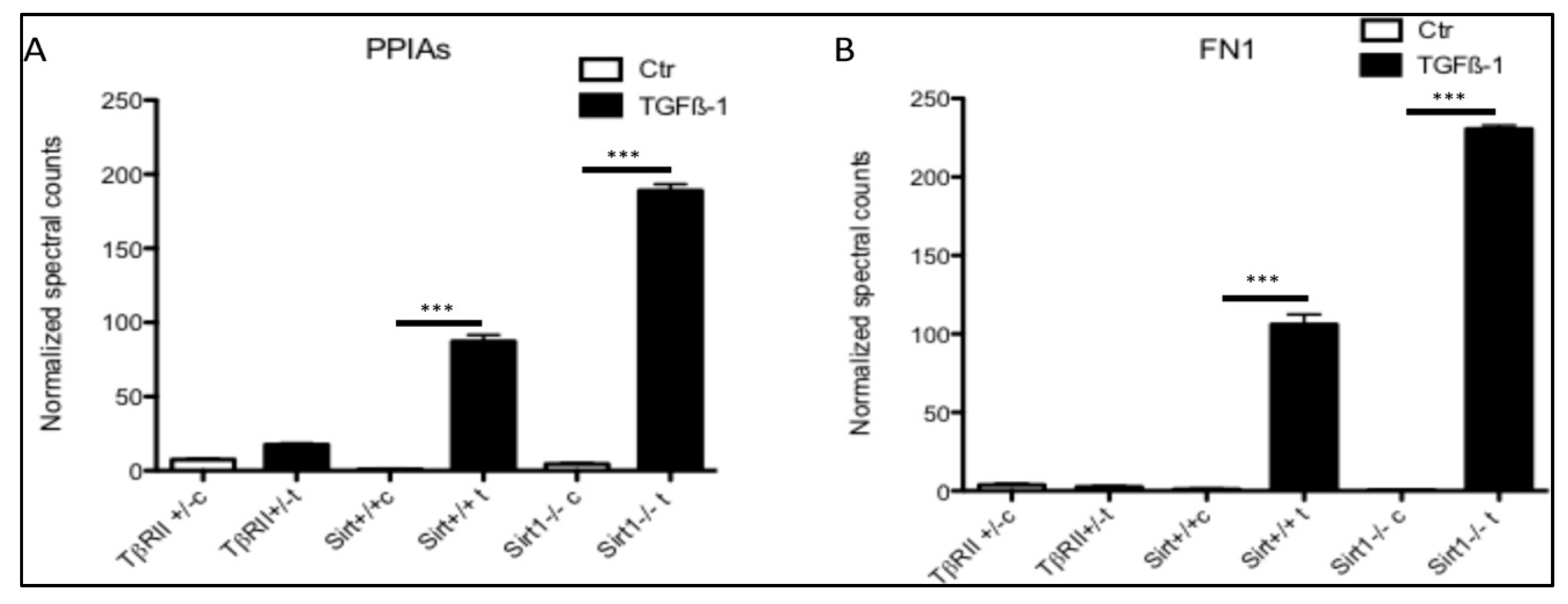
3.5. Secretion of PPIA Increased Significantly during Fibrosis Progression
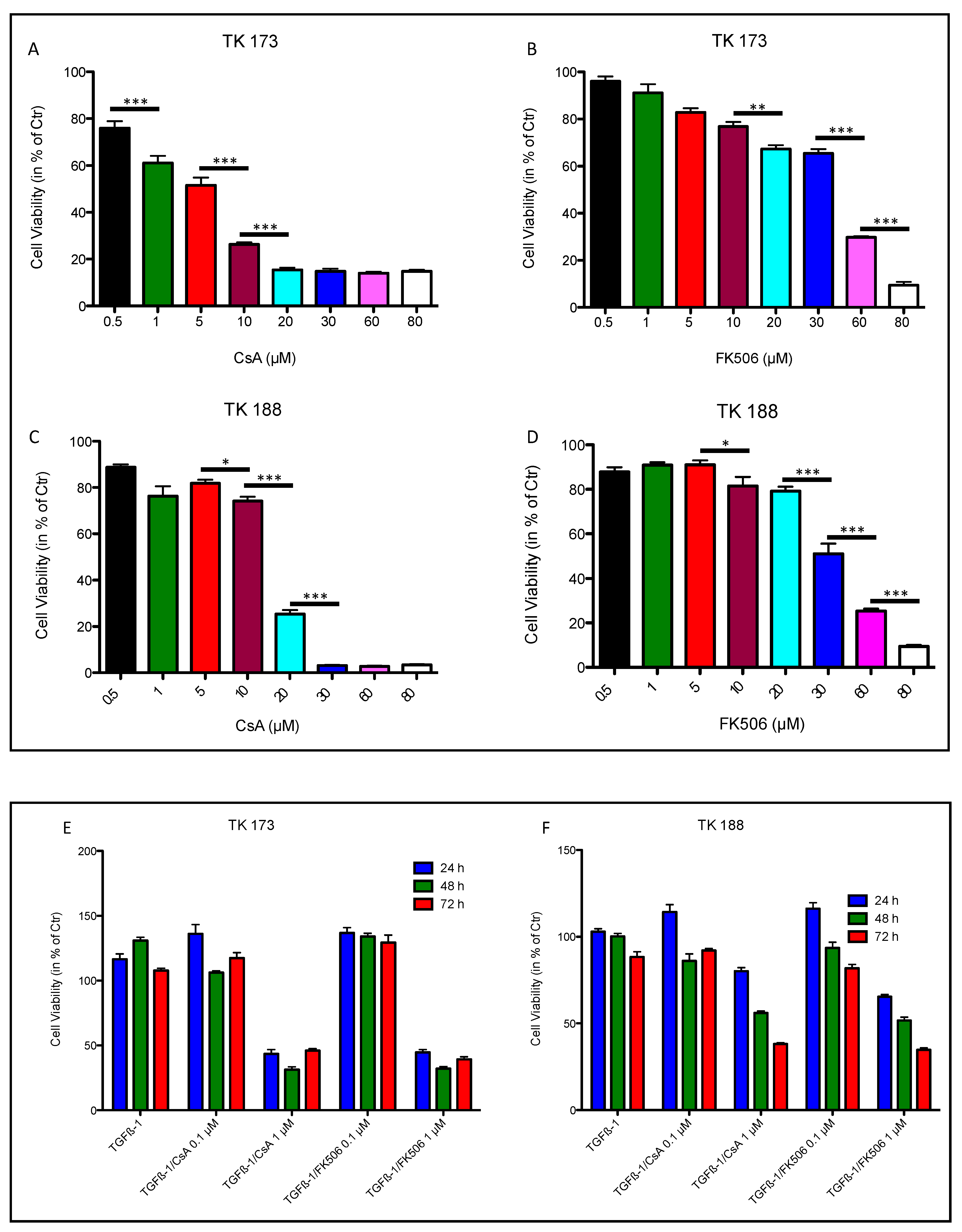
3.6. Effect of Ciclosporin A (CsA) and FK506 on Fibroblasts Cell Viability and Survival
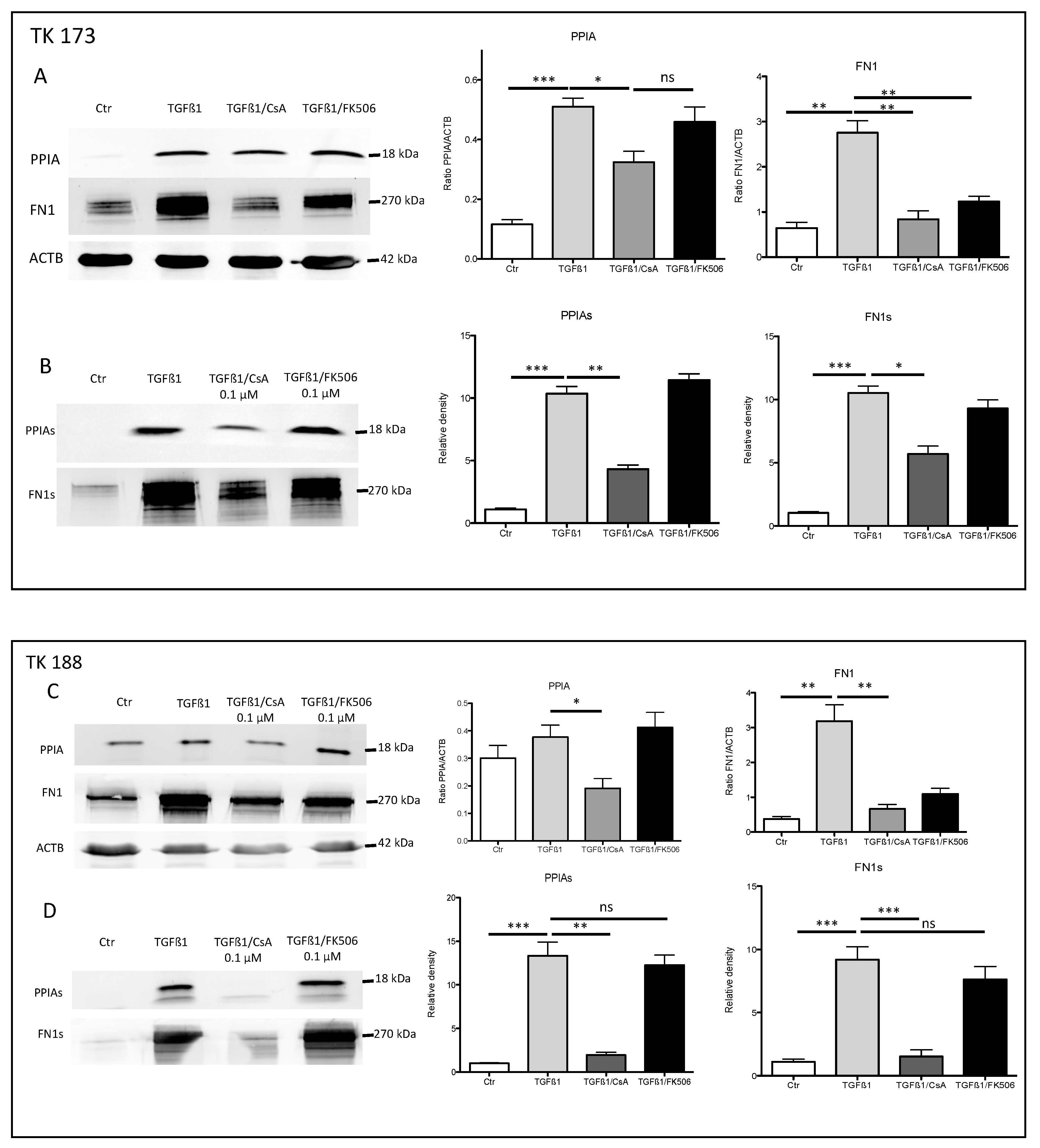
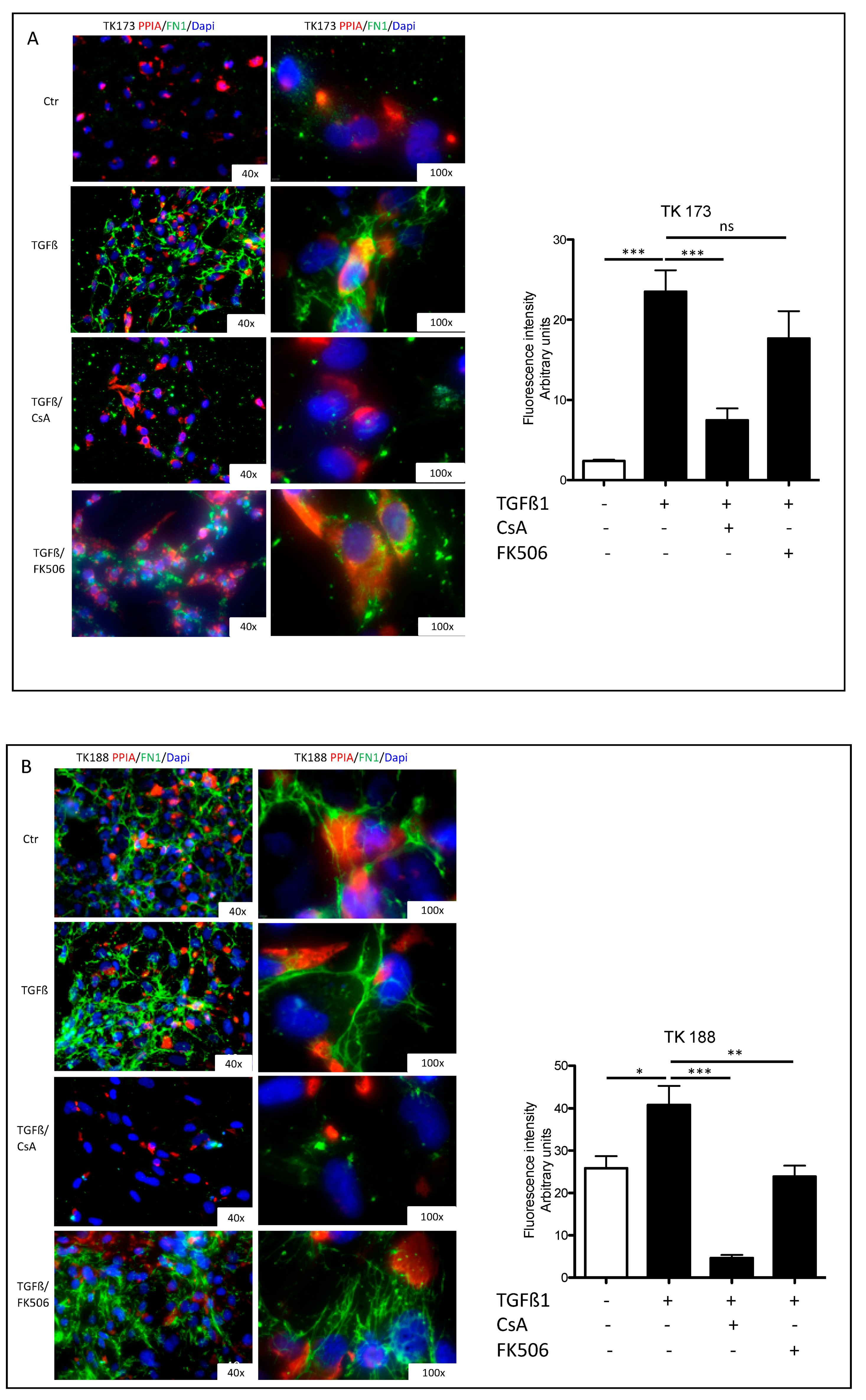
3.7. Impact of PPIA Inhibition on ECM Production and Accumulation
3.8. Co-Immunoprecipitation Revealed Interaction of PPIA with ECM and Inflammatory Proteins
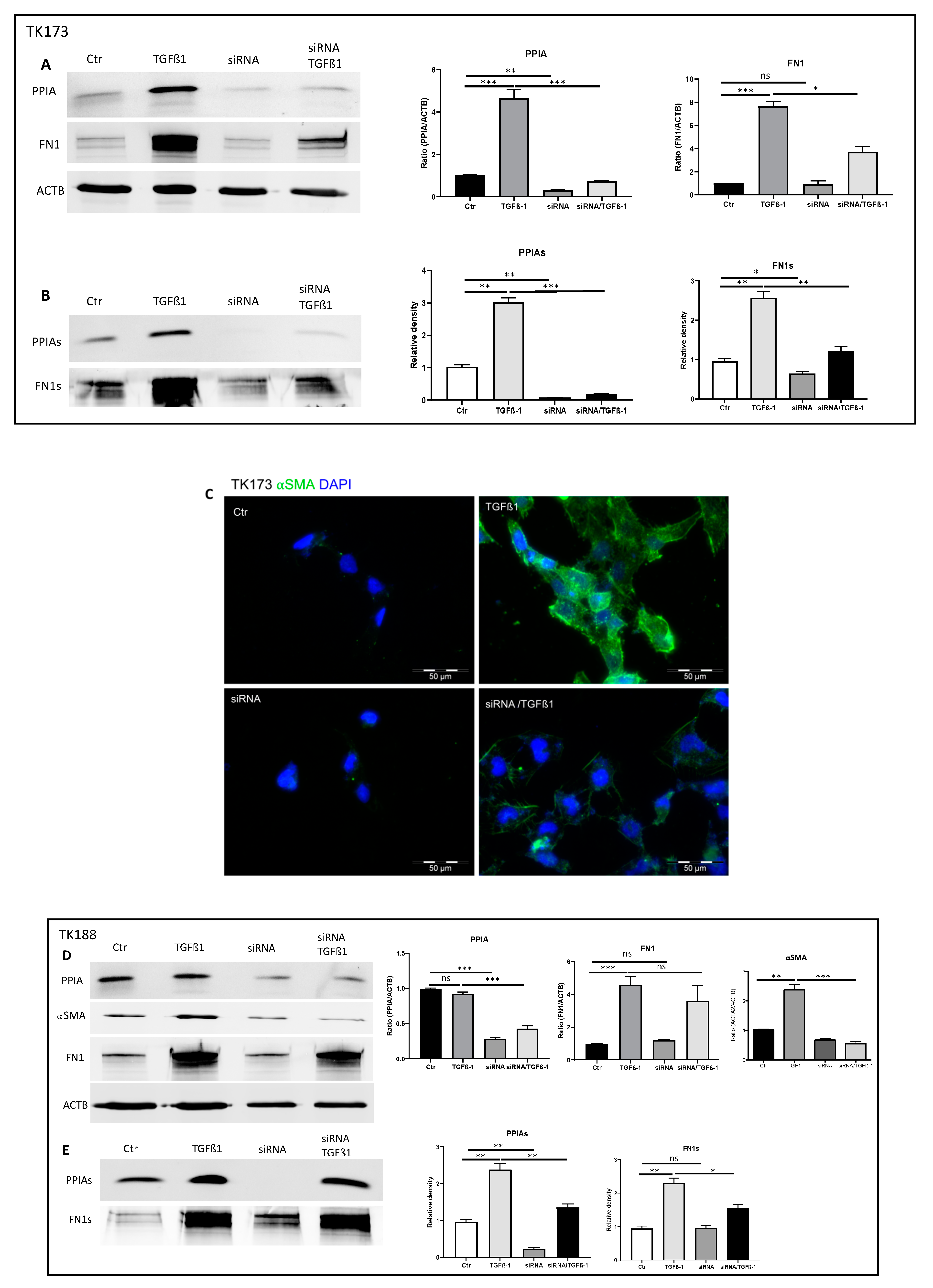
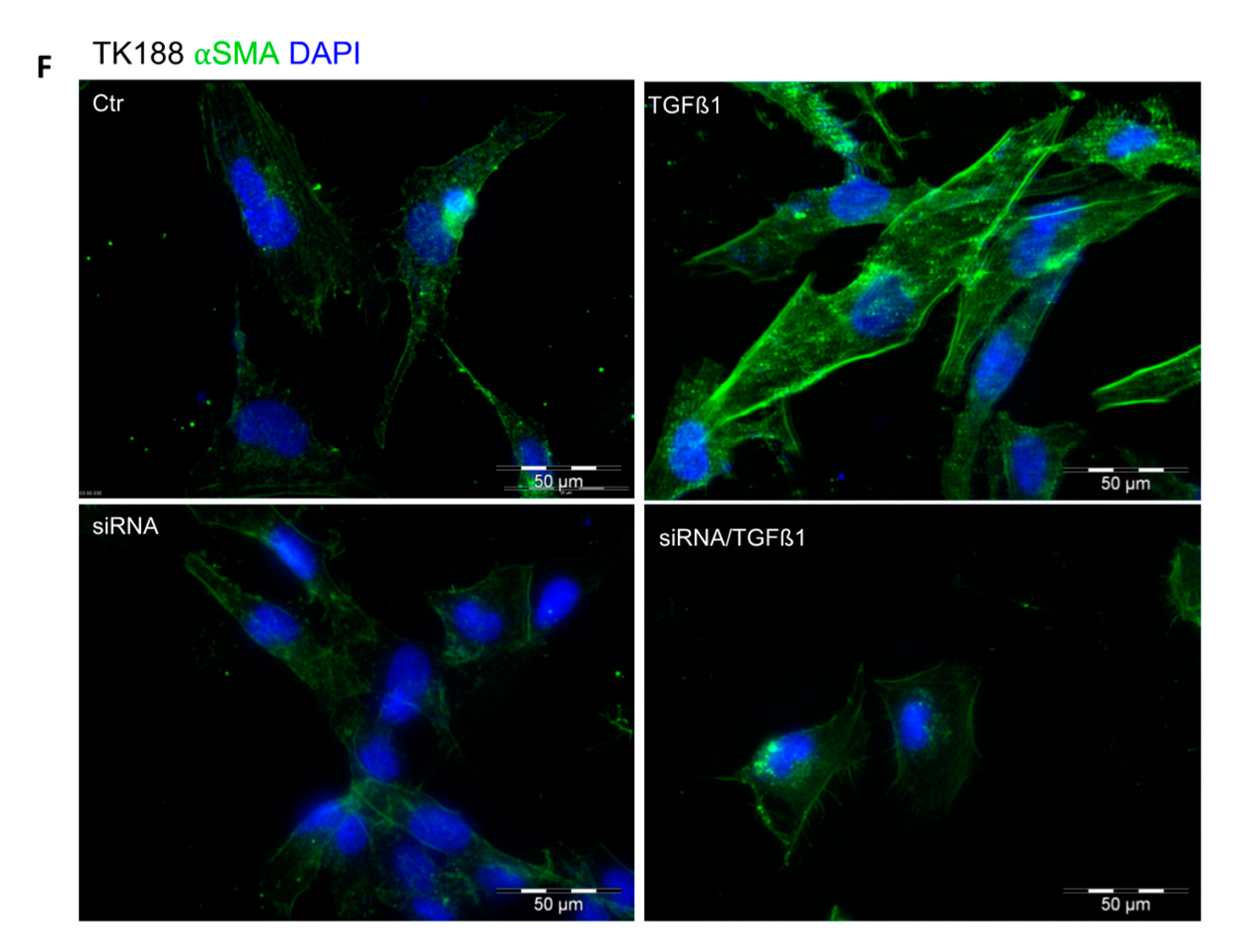
3.9. siRNA Mediated Downregulation of PPIA and Its Impact on Cell Transformation and EMC Expression and Accumulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, B.-B.; Stuart, L.; Feener, E.P. Label-free quantitative analysis of one-dimensional PAGE LC/MS/MS proteome: Application on angiotensin II-stimulated smooth muscle cells secretome. Mol. Cell. Proteom. 2008, 7, 2399–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundacker, N.C.; Haudek, V.J.; Wimmer, H.; Slany, A.; Griss, J.; Bochkov, V.; Zielinski, C.; Wagner, O.; Stöckl, J.; Gerner, C. Cytoplasmic proteome and secretome profiles of differently stimulated human dendritic cells. J. Proteome Res. 2009, 8, 2799–2811. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Song, J.W.; Matsumoto, K.; Dadafarin, S.; Dihazi, H.; Müller, G.; Goligorsky, M.S. The third path of tubulointerstitial fibrosis: Aberrant endothelial secretome. Kidney Int. 2017, 92, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Caccia, D.; Zanetti Domingues, L.; Miccichè, F.; De Bortoli, M.; Carniti, C.; Mondellini, P.; Bongarzone, I. Secretome compartment is a valuable source of biomarkers for cancer-relevant pathways. J. Proteome Res. 2011, 10, 4196–4207. [Google Scholar] [CrossRef]
- Doroudgar, S.; Glembotski, C.C. The cardiokine story unfolds: Ischemic stress-induced protein secretion in the heart. Trends Mol. Med. 2011, 17, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Marzoq, A.J.; Mustafa, S.A.; Heidrich, L.; Hoheisel, J.D.; Alhamdani, M.S.S. Impact of the secretome of activated pancreatic stellate cells on growth and differentiation of pancreatic tumour cells. Sci. Rep. 2019, 9, 5303. [Google Scholar] [CrossRef] [Green Version]
- Pocock, J.M.; Storisteanu, D.M.L.; Reeves, M.B.; Juss, J.K.; Wills, M.R.; Cowburn, A.S.; Chilvers, E.R. Human Cytomegalovirus Delays Neutrophil Apoptosis and Stimulates the Release of a Prosurvival Secretome. Front. Immunol. 2017, 8, 1185. [Google Scholar] [CrossRef] [Green Version]
- Hendrata, M.; Sudiono, J. A Computational Model for Investigating Tumor Apoptosis Induced by Mesenchymal Stem Cell-Derived Secretome. Comput. Math. Methods Med. 2016, 2016, 4910603. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Risdon, R.A.; Sloper, J.C.; De Wardener, H.E. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet Lond. Engl. 1968, 2, 363–366. [Google Scholar] [CrossRef]
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- Katz, A.; Caramori, M.L.A.; Sisson-Ross, S.; Groppoli, T.; Basgen, J.M.; Mauer, M. An increase in the cell component of the cortical interstitium antedates interstitial fibrosis in type 1 diabetic patients. Kidney Int. 2002, 61, 2058–2066. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.-Y.; Chen, S.S.-L. Hepatitis C virus and cellular stress response: Implications to molecular pathogenesis of liver diseases. Viruses 2012, 4, 2251–2290. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Frank, J.; Rodemann, H.P.; Engler-Blum, G. Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis. Exp. Nephrol. 1995, 3, 127–133. [Google Scholar]
- Dihazi, H.; Dihazi, G.H.; Jahn, O.; Meyer, S.; Nolte, J.; Asif, A.R.; Mueller, G.A.; Engel, W. Multipotent adult germline stem cells and embryonic stem cells functional proteomics revealed an important role of eukaryotic initiation factor 5A (Eif5a) in stem cell differentiation. J. Proteome Res. 2011, 10, 1962–1973. [Google Scholar] [CrossRef]
- Dihazi, H.; Dihazi, G.H.; Bibi, A.; Eltoweissy, M.; Mueller, C.A.; Asif, A.R.; Rubel, D.; Vasko, R.; Mueller, G.A. Secretion of ERP57 is important for extracellular matrix accumulation and progression of renal fibrosis, and is an early sign of disease onset. J. Cell Sci. 2013, 126, 3649–3663. [Google Scholar] [CrossRef] [Green Version]
- Wessel, D.; Flügge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Jahn, O.; Hesse, D.; Reinelt, M.; Kratzin, H.D. Technical innovations for the automated identification of gel-separated proteins by MALDI-TOF mass spectrometry. Anal. Bioanal. Chem. 2006, 386, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Immunoblotting in the clinical laboratory. J. Clin. Chem. Clin. Biochem. 1989, 27, 495–501. [Google Scholar] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. 1979. Biotechnology 1992, 24, 145–149. [Google Scholar]
- Schmidt, C.; Hesse, D.; Raabe, M.; Urlaub, H.; Jahn, O. An automated in-gel digestion/iTRAQ-labeling workflow for robust quantification of gel-separated proteins. Proteomics 2013, 13, 1417–1422. [Google Scholar] [CrossRef]
- Sondermann, J.R.; Barry, A.M.; Jahn, O.; Michel, N.; Abdelaziz, R.; Kügler, S.; Gomez-Varela, D.; Schmidt, M. Vti1b promotes TRPV1 sensitization during inflammatory pain. Pain 2019, 160, 508–527. [Google Scholar] [CrossRef]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipphardt, M.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Fibrogenic Secretome of Sirtuin 1-Deficient Endothelial Cells: Wnt, Notch and Glycocalyx Rheostat. Front. Physiol. 2018, 9, 1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipphardt, M.; Dihazi, H.; Jeon, N.L.; Dadafarin, S.; Ratliff, B.B.; Rowe, D.W.; Müller, G.A.; Goligorsky, M.S. Dickkopf-3 in aberrant endothelial secretome triggers renal fibroblast activation and endothelial-mesenchymal transition. Nephrol. Dial. Transplant. 2019, 34, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Endothelial dysfunction is a superinducer of syndecan-4: Fibrogenic role of its ectodomain. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Smith, T.; Ferreira, L.R.; Hebert, C.; Norris, K.; Sauk, J.J. Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum. J. Biol. Chem. 1995, 270, 18323–18328. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Vijayakumar, S.; Schiene-Fischer, C.; Li, H.; Purkerson, J.M.; Malesevic, M.; Liebscher, J.; Al-Awqati, Q.; Schwartz, G.J. Secreted cyclophilin A, a peptidylprolyl cis-trans isomerase, mediates matrix assembly of hensin, a protein implicated in epithelial differentiation. J. Biol. Chem. 2009, 284, 6465–6475. [Google Scholar] [CrossRef] [Green Version]
- Gewin, L.; Zent, R. How does TGF-β mediate tubulointerstitial fibrosis? Semin. Nephrol. 2012, 32, 228–235. [Google Scholar] [CrossRef] [Green Version]
- García-Sánchez, O.; López-Hernández, F.J.; López-Novoa, J.M. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 2010, 77, 950–955. [Google Scholar] [CrossRef] [Green Version]
- López-Hernández, F.J.; López-Novoa, J.M. Role of TGF-β in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012, 347, 141–154. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Meissner, F.; Scheltema, R.A.; Mollenkopf, H.-J.; Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 2013, 340, 475–478. [Google Scholar] [CrossRef]
- Mukherjee, P.; Mani, S. Methodologies to decipher the cell secretome. Biochim. Biophys. Acta 2013, 1834, 2226–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccia, D.; Dugo, M.; Callari, M.; Bongarzone, I. Bioinformatics tools for secretome analysis. Biochim. Biophys. Acta 2013, 1834, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Hayano, T.; Suzuki, M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 1989, 337, 473–475. [Google Scholar] [CrossRef]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef]
- Hoffmann, H.; Schiene-Fischer, C. Functional aspects of extracellular cyclophilins. Biol. Chem. 2014, 395, 721–735. [Google Scholar] [CrossRef]
- Seizer, P.; Klingel, K.; Sauter, M.; Westermann, D.; Ochmann, C.; Schönberger, T.; Schleicher, R.; Stellos, K.; Schmidt, E.-M.; Borst, O.; et al. Cyclophilin A affects inflammation, virus elimination and myocardial fibrosis in coxsackievirus B3-induced myocarditis. J. Mol. Cell. Cardiol. 2012, 53, 6–14. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Wang, T.; You, H.; Jia, J. N-methyl-4-isoleucine cyclosporine attenuates CCl -induced liver fibrosis in rats by interacting with cyclophilin B and D. J. Gastroenterol. Hepatol. 2011, 26, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, V.; Zybarth, G.; O’Connor, M.; Dai, W.W.; Franchin, G.; Hao, T.; Guo, H.; Hung, H.-C.; Toole, B.; Gallay, P.; et al. Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J. Biol. Chem. 2002, 277, 22959–22965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malesevic, M.; Gutknecht, D.; Prell, E.; Klein, C.; Schumann, M.; Nowak, R.A.; Simon, J.C.; Schiene-Fischer, C.; Saalbach, A. Anti-inflammatory effects of extracellular cyclosporins are exclusively mediated by CD147. J. Med. Chem. 2013, 56, 7302–7311. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Gene Name | Accession | Entry | IEP | MW | Ctr (fmol) | TGFβ1 (fmol) |
|---|---|---|---|---|---|---|---|
| Peptidyl-prolyl cis-trans isomerase A | PPIA | P62937 | PPIA_HUMAN | 7.85 | 18,240.64 | 411.95 | 568.08 |
| Heterogeneous nuclear ribonucleoprotein U | HNRNPU | Q00839 | HNRPU_HUMAN | 5.66 | 91,325.94 | 329.21 | |
| Ubiquitin-40S ribosomal protein S27a | RPS27A | P62979 | RS27A_HUMAN | 10.24 | 18,307.10 | 135.32 | |
| Galectin-1 | LGALS1 | P09382 | LEG1_HUMAN | 5.15 | 15,057.89 | 61.14 | 103.95 |
| E3 SUMO-protein ligase RanBP2 | RANBP2 | P49792 | RBP2_HUMAN | 5.81 | 362,590.78 | 78.65 | |
| Heat shock protein 75 kDa_ mitochondrial | TRAP1 | Q12931 | TRAP1_HUMAN | 8.31 | 80,395.28 | 11.33 | 69.58 |
| Fibronectin | FN1 | P02751 | FINC_HUMAN | 5.36 | 266,217.54 | 64.91 | |
| Plasminogen activator inhibitor 1 | SERPINE1 | P05121 | PAI1_HUMAN | 6.79 | 45,117.13 | 5.66 | 56.30 |
| Bifunctional purine biosynthesis protein PURH | ATIC | P31939 | PUR9_HUMAN | 6.30 | 65,129.21 | 10.66 | 43.70 |
| Intercellular adhesion molecule 1 | ICAM1 | P05362 | ICAM1_HUMAN | 7.92 | 58,623.66 | 18.39 | 40.33 |
| Splicing factor 3B subunit 1 | SF3B1 | O75533 | SF3B1_HUMAN | 6.69 | 146,572.02 | 2.43 | 36.54 |
| Myosin-10 | MYH10 | P35580 | MYH10_HUMAN | 5.28 | 229,968.92 | 7.87 | 31.49 |
| Peptidyl-prolyl cis-trans isomerase B | PPIB | P23284 | PPIB_HUMAN | 10.06 | 23,799.61 | 14.31 | 29.33 |
| Ras-related protein Rap-1A | RAP1A | P62834 | RAP1A_HUMAN | 6.53 | 21,329.38 | 28.94 | |
| 40S ribosomal protein S20 | RPS20 | P60866 | RS20_HUMAN | 10.71 | 13,486.79 | 27.24 | |
| Aldo-keto reductase family 1 member B1 | AKR1B1 | P15121 | ALDR_HUMAN | 6.59 | 36,252.64 | 8.73 | 27.09 |
| Brain acid soluble protein 1 | BASP1 | P80723 | BASP1_HUMAN | 4.42 | 22,693.42 | 6.91 | 21.55 |
| Myosin-11 | MYH11 | P35749 | MYH11_HUMAN | 5.25 | 228,195.03 | 4.50 | 21.10 |
| Ras-related C3 botulinum toxin substrate 1 | RAC1 | P63000 | RAC1_HUMAN | 8.58 | 21,849.32 | 20.56 | |
| Tumor protein D54 | TPD52L2 | O43399 | TPD54_HUMAN | 5.08 | 22,294.78 | 4.45 | 13.04 |
| Integrin alpha-5 | ITGA5 | P08648 | ITA5_HUMAN | 5.41 | 115,677.14 | 4.45 | 11.68 |
| Cytoplasmic dynein 1 heavy chain 1 | DYNC1H1 | Q14204 | DYHC1_HUMAN | 5.99 | 535,145.98 | 10.62 | |
| Bifunctional glutamate/proline--tRNA ligase | EPRS | P07814 | SYEP_HUMAN | 7.00 | 172,187.91 | 9.30 | |
| CD166 antigen | ALCAM | Q13740 | CD166_HUMAN | 5.87 | 65,786.70 | 4.70 | 9.04 |
| Serpin B12 | SERPINB12 | Q96P63 | SPB12_HUMAN | 5.22 | 46,675.69 | 8.14 | |
| Small nuclear ribonucleoprotein F | SNRPF | P62306 | RUXF_HUMAN | 4.43 | 9782.26 | 8.03 | |
| Integrin alpha-V OS=Homo sapiens | ITGAV | P06756 | ITAV_HUMAN | 5.32 | 117,121.56 | 7.89 | |
| Thrombospondin-1 | THBS1 | P07996 | TSP1_HUMAN | 4.53 | 133,374.73 | 7.83 | |
| Proteasome subunit beta type-1 | PSMB1 | P20618 | PSB1_HUMAN | 8.22 | 26,717.54 | 7.69 | |
| Protein S100-A10 | S100A10 | P60903 | S10AA_HUMAN | 7.27 | 11,317.21 | 7.46 | |
| S-phase kinase-associated protein 1 | SKP1 | P63208 | SKP1_HUMAN | 4.20 | 18,829.12 | 7.42 | |
| Splicing factor_ proline- and glutamine-rich | SEPQ | P23246 | SFPQ_HUMAN | 9.95 | 76,263.60 | 7.39 | |
| LIM and SH3 domain protein 1 | LASP1 | Q14847 | LASP1_HUMAN | 6.70 | 30,116.41 | 7.30 | |
| Coatomer subunit beta | COPB1 | P53618 | COPB_HUMAN | 5.66 | 108,282.98 | 6.84 | |
| Isoleucine--tRNA ligase_ cytoplasmic | IARS | P41252 | SYIC_HUMAN | 5.77 | 145,809.96 | 6.35 | |
| Heterogeneous nuclear ribonucleoprotein D-like | HNRNPDL | O14979 | HNRDL_HUMAN | 9.96 | 46,608.67 | 6.26 | |
| Tropomyosin beta chain | TPM2 | P07951 | TPM2_HUMAN | 4.46 | 32,964.85 | 5.84 | |
| GTPase NRas | NRAS | P01111 | RASN_HUMAN | 4.82 | 21,514.35 | 5.82 | |
| Cystatin-A | CSTA | P01040 | CYTA_HUMAN | 5.22 | 11,006.51 | 5.36 | |
| Protein Niban | FAM129A | Q9BZQ8 | NIBAN_HUMAN | 4.54 | 104,104.30 | 5.10 | |
| ELAV-like protein 1 | ELAVL1 | Q15717 | ELAV1_HUMAN | 9.57 | 36,263.04 | 5.07 | |
| Podocalyxin | PODXL | O00592 | PODXL_HUMAN | 5.16 | 59,091.56 | 4.99 | |
| Putative heat shock protein HSP 90-beta 2 | HSP90AB2P | Q58FF8 | H90B2_HUMAN | 4.59 | 44,520.09 | 4.97 | |
| Ephrin type-A receptor 2 | EPHA2 | P29317 | EPHA2_HUMAN | 5.83 | 109,749.33 | 4.64 | |
| Collagen alpha-1(VI) chain | COL6A1 | P12109 | CO6A1_HUMAN | 5.09 | 109,670.06 | 4.58 | |
| Transportin-1 | TNPO1 | Q92973 | TNPO1_HUMAN | 4.65 | 103,837.95 | 4.46 | |
| Phosphatidylinositol transfer protein beta isoform | PITPNB | P48739 | PIPNB_HUMAN | 6.47 | 31,825.26 | 4.46 | |
| Thioredoxin-dependent peroxide reductase_ mitochondrial | PRDX3 | P30048 | PRDX3_HUMAN | 7.70 | 28,034.84 | 4.13 | |
| Putative heat shock protein HSP 90-beta 4 | HSP90AB4P | Q58FF6 | H90B4_HUMAN | 4.45 | 58,891.78 | 4.00 | |
| Phosphoribosylformylglycinamidine synthase | PFAS | O15067 | PUR4_HUMAN | 5.41 | 146,388.37 | 3.94 | |
| Epidermal growth factor receptor | EGFR | P00533 | EGFR_HUMAN | 6.27 | 137,699.25 | 3.85 | |
| Glucosidase 2 subunit beta | PRKCSH | P14314 | GLU2B_HUMAN | 4.13 | 60,394.97 | 3.83 | |
| Peripherin | PRPH | P41219 | PERI_HUMAN | 5.21 | 53,765.00 | 3.74 | |
| Ras GTPase-activating-like protein IQGAP3 | IQGAP3 | Q86VI3 | IQGA3_HUMAN | 7.37 | 185,383.40 | 3.64 | |
| Hippocalcin-like protein 1 | HPCAL1 | P37235 | HPCL1_HUMAN | 5.03 | 22,427.29 | 3.54 | |
| Glial fibrillary acidic protein | GFAP | P14136 | GFAP_HUMAN | 5.26 | 49,937.33 | 3.41 | |
| Ras-related protein Ral-B | RALB | P11234 | RALB_HUMAN | 6.26 | 23,522.64 | 3.38 | |
| Structural maintenance of chromosomes protein 4 | SMC4 | Q9NTJ3 | SMC4_HUMAN | 6.38 | 147,866.56 | 3.34 | |
| Aminoacyl tRNA synthase complex-interacting multifunctional protein 2 | AIMP2 | Q13155 | AIMP2_HUMAN | 8.27 | 35,691.02 | 3.31 | |
| Ras suppressor protein 1 | RSU1 | Q15404 | RSU1_HUMAN | 9.23 | 31,540.35 | 3.31 | |
| Ras-related protein Rab-23 | RAB23 | Q9ULC3 | RAB23_HUMAN | 6.24 | 26,887.39 | 3.21 | |
| Collagen alpha-2(VI) chain | COL6A2 | P12110 | CO6A2_HUMAN | 5.81 | 109,777.13 | 2.90 | |
| Structural maintenance of chromosomes protein 2 | SMC2 | O95347 | SMC2_HUMAN | 8.73 | 136,169.56 | 2.75 | |
| FACT complex subunit SPT16 | SUPT16H | Q9Y5B9 | SP16H_HUMAN | 5.37 | 120,484.25 | 2.71 | |
| Aminopeptidase N | ANPEP | P15144 | AMPN_HUMAN | 5.16 | 109,938.95 | 2.62 | |
| Glycogen phosphorylase_ brain form | PYGB | P11216 | PYGB_HUMAN | 6.43 | 97,380.45 | 2.54 | |
| CAD protein | CAD | P27708 | PYR1_HUMAN | 6.03 | 245,322.25 | 2.49 | |
| DNA replication licensing factor MCM2 | MCM2 | P49736 | MCM2_HUMAN | 5.20 | 102,580.63 | 2.45 | |
| Signal transducer and activator of transcription 2 | STAT2 | P52630 | STAT2_HUMAN | 5.20 | 98,657.73 | 2.44 | |
| Valine--tRNA ligase | VARS | P26640 | SYVC_HUMAN | 7.37 | 141,730.79 | 2.30 | |
| Peroxidasin homolog | PXDN | Q92626 | PXDN_HUMAN | 6.79 | 167,898.08 | 2.23 | |
| Microtubule-associated protein 4 | MAP4 | P27816 | MAP4_HUMAN | 5.14 | 121,518.45 | 2.15 | |
| Myoferlin | MYOF | Q9NZM1 | MYOF_HUMAN | 5.79 | 236,248.81 | 2.13 | |
| Kinesin-like protein KIF23 | KIF23 | Q02241 | KIF23_HUMAN | 8.59 | 111,085.51 | 2.03 | |
| Procollagen-lysine_2-oxoglutarate 5-dioxygenase 2 | PLOD2 | O00469 | PLOD2_HUMAN | 6.27 | 85,427.10 | 1.45 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dihazi, G.H.; Eltoweissy, M.; Jahn, O.; Tampe, B.; Zeisberg, M.; Wülfrath, H.S.; Müller, G.A.; Dihazi, H. The Secretome Analysis of Activated Human Renal Fibroblasts Revealed Beneficial Effect of the Modulation of the Secreted Peptidyl-Prolyl Cis-Trans Isomerase A in Kidney Fibrosis. Cells 2020, 9, 1724. https://doi.org/10.3390/cells9071724
Dihazi GH, Eltoweissy M, Jahn O, Tampe B, Zeisberg M, Wülfrath HS, Müller GA, Dihazi H. The Secretome Analysis of Activated Human Renal Fibroblasts Revealed Beneficial Effect of the Modulation of the Secreted Peptidyl-Prolyl Cis-Trans Isomerase A in Kidney Fibrosis. Cells. 2020; 9(7):1724. https://doi.org/10.3390/cells9071724
Chicago/Turabian StyleDihazi, Gry H., Marwa Eltoweissy, Olaf Jahn, Björn Tampe, Michael Zeisberg, Hauke S. Wülfrath, Gerhard A. Müller, and Hassan Dihazi. 2020. "The Secretome Analysis of Activated Human Renal Fibroblasts Revealed Beneficial Effect of the Modulation of the Secreted Peptidyl-Prolyl Cis-Trans Isomerase A in Kidney Fibrosis" Cells 9, no. 7: 1724. https://doi.org/10.3390/cells9071724