Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting



Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, VIC 3052, Australia
*
Author to whom correspondence should be addressed.
Cells 2020, 9(11), 2413; https://doi.org/10.3390/cells9112413
Submission received: 5 October 2020
/
Revised: 30 October 2020
/
Accepted: 1 November 2020
/
Published: 4 November 2020
(This article belongs to the Special Issue Autophagy Meets Aging)
Abstract
:Over the past 20 years, there has been a drastically increased understanding of the genetic basis of Amyotrophic Lateral Sclerosis. Despite the identification of more than 40 different ALS-causing mutations, the accumulation of neurotoxic misfolded proteins, inclusions, and aggregates within motor neurons is the main pathological hallmark in all cases of ALS. These protein aggregates are proposed to disrupt cellular processes and ultimately result in neurodegeneration. One of the main reasons implicated in the accumulation of protein aggregates may be defective autophagy, a highly conserved intracellular “clearance” system delivering misfolded proteins, aggregates, and damaged organelles to lysosomes for degradation. Autophagy is one of the primary stress response mechanisms activated in highly sensitive and specialised neurons following insult to ensure their survival. The upregulation of autophagy through pharmacological autophagy-inducing agents has largely been shown to reduce intracellular protein aggregate levels and disease phenotypes in different in vitro and in vivo models of neurodegenerative diseases. In this review, we explore the intriguing interface between ALS and autophagy, provide a most comprehensive summary of autophagy-targeted drugs that have been examined or are being developed as potential treatments for ALS to date, and discuss potential therapeutic strategies for targeting autophagy in ALS.
1. Introduction
1.1. Amyotrophic Lateral Sclerosis
Motor neuron (MN) diseases are a group of neurodegenerative diseases where MNs selectively degenerate. Amyotrophic Lateral Sclerosis (ALS, known as motor neuron disease in United Kingdom and Australia, and Lou Gehrig’s disease in the USA) is the most common form [1,2], with approximately 1–2 newly diagnosed cases in every 100,000 people internationally every year [3].
In ALS, both the upper MNs in the motor cortex and the associated corticospinal tract, and the lower MNs in the brainstem and spinal cord selectively degenerate. As a result, neuromuscular function deteriorates, evoking weakness, muscle wasting, and paralysis [2,4,5,6]. In the majority of cases, the disease manifests itself between the ages of 50 and 60 [5]. Typically, 3–4 years after symptom onset, respiratory muscles also atrophy, culminating in death [2,4,5,6]. However, in approximately one-third of patients where symptoms begin at the bulbar level with dysarthria and dysphagia, survival is shortened to an average of 2 years [7,8,9].
ALS is a complex and heterogeneous disorder with likely multiple causes. More than 90% of cases are sporadic (SALS) with no obvious family history of disease, and 10% of cases are familial (FALS) with one or more identifiable genetic mutations (Figure 1). The first discovered ALS-associated gene was superoxide dismutase 1 (SOD1) [10]. However, more recently, an intronic hexanucleotide (GGGGCC) repeat expansion in the non-coding segment of the chromosome 9 opening reading frame 72 (C9orf72) gene was identified as the most prevalent cause of FALS [11,12]. After C9orf72 and SOD1, the two most common mutations implicated in FALS are in the genes TAR DNA binding partner 43 (TARDBP) [13] and fused in sarcoma (FUS) [14,15].
SALS and FALS are clinically indistinguishable, and the predominant cytoplasmic accumulation of ubiquitinated, hyaline, and skein-like aggregates within degenerating MNs and glial cells is a hallmark of both forms of ALS [17,18,19,20]. With the exception of SOD1- and FUS-linked ALS, the major pathological protein in all cases of ALS is TDP-43 and analysis of post-mortem tissues from ALS patients and mouse models has established that there is a direct correlation between MN loss and TDP-43 pathology [21,22].
Intracellular protein aggregates form when the level of misfolded proteins reaches a critical concentration, subsequently assembling into small soluble oligomers. Eventually, with time and increasing concentration of proteins, oligomers convert into the more metabolically stable insoluble aggregates [23,24,25]. According to the “seeding-nucleation” model, oligomerisation is a slow process as it is thermodynamically unfavourable. However, once an oligomeric seed is formed, it grows rapidly into high-molecular-weight protein complexes [26,27]. Consequently, the process of protein misfolding might begin years before the appearance of protein aggregates [28,29].
The exact role of these different aggregates in cells remains unknown. They can be initially cytoprotective by sequestering harmful protein species and burying the hydrophobic core of misfolded proteins, as opposed to having them exposed to the hydrophilic environment. Animal studies have supported this hypothesis, showing aggregates to be less toxic to cells than diffuse misfolded proteins [25]. Nonetheless, large intracellular aggregates disturb protein homeostasis, trigger cellular stress, and are closely associated with cell degeneration [17,24,25,30,31,32].
1.2. Cytoplasmic Protein Homeostasis and Degradation Pathways
Under normal physiological conditions, the protein quality control system circumvents aggregate-mediated toxicity by re-folding misfolded proteins using molecular chaperones or targeting them to one of two degradation pathways, the ubiquitin–proteasome system (UPS) or autophagy–lysosomal system [33].
The UPS degrades misfolded and short-lived soluble proteins through the protease complex [34]. The substrates have to be small enough to pass through the narrow pore of the proteasomal barrel. Studies in mouse models [35,36,37,38] and patients [39] indicate that the proteasome function is impaired in ALS spinal cord MNs.
In contrast to the UPS, autophagy does not have a size limitation for substrate clearance and can process large protein aggregates, not to mention organelles and intracellular bacteria [40,41,42,43]. The remainder of this review focuses on autophagy, its role in the pathogenesis of ALS, and its modulation as a possible therapeutic approach for the treatment of ALS.
There are three major sub-types of autophagy, namely, microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy [41]. The major degradative autophagy pathway is macroautophagy (hereafter referred to as autophagy).
Autophagy is an evolutionary conserved intracellular process that purges cells of insoluble proteins, entire organelles that are damaged or superfluous such as mitochondria via mitophagy, or endoplasmic reticulum via reticulophagy, as well as toxic metabolites; cancerous cells; and intact invading microorganisms such as bacteria, viruses, and protozoa [44,45]. Basal autophagy plays a pivotal role in protein and organelle quality control [46], innate and adaptive immunity [47], metabolism [48], development, and differentiation [49,50,51,52]. As well as its housekeeping functions, autophagy is vital under conditions of stress or starvation [44,53].
Neuronal autophagy is particularly crucial as neurons are terminally differentiated and non-proliferating cells. Their non-dividing feature does not allow their intracellular contents such as protein aggregates to become diluted by cell division. Hence, in a study of autophagy-deficient mice, protein aggregates accumulated predominantly in neurons [54,55]. Moreover, the high energetic demands of neurons necessitate the efficient turnover of proteins and organelles for proper cell functioning [56,57,58].
The autophagy dynamic process, referred to as autophagy flux, comprises multiple steps (Figure 2). Autophagy-inducing signals are recognised by AMP-activated protein kinase (AMPK), the central regulator of cellular energy balance. Subsequently, the autophagy-activating kinase Unc-51 like autophagy activating kinase 1 (ULK1) is upregulated, and the autophagy-inhibitory kinase mammalian target of rapamycin complex 1 (mTORC1) is downregulated [59,60,61]. The ULK1 complex recruits phosphatidylinositol 3-kinase class 3 (PI3KC3) comprising vacuolar protein sorting 34 (Vps34), P150, and the core protein Beclin 1 to produce phosphatidylinositol 3-phosphates (PI3Ps) [62,63,64]. Thus, a cup-shaped pre-autophagosomal structure forms where essential autophagy-related proteins (Atgs) are recruited [65]. One such protein is C-terminus of microtubule-associated protein 1A/1B-light chain 3 (LC3), which is cleaved, activated, and lipidated to LC3-II and transferred to the phospholipid bilayer of the pre-autophagosomal structure [66]. For selective autophagy, membrane-bound LC3-II recruits autophagy receptors such as p62, which recognise and target specific cargo to the forming autophagosome [67,68,69]. Thereupon, LC3-II hemi-fuses membranes to expand and surround cargo, forming the double-membrane vesicle called the autophagosome (Figure 2) [70,71,72,73,74].
Autophagosomes mature and are trafficked to the lysosome-rich microtubule-organising centre (the perinuclear region) where they dock and fuse with lysosomes for the formation of autolysosomes [75,76,77,78,79,80,81]. Autolysosomal contents, including entrapped LC3 and p62, are degraded, and generated micromolecules such as amino acids and fatty acids are released back into the cytosol for reuse (Figure 2). As autophagy products become abundant, mTORC1 signalling switches on anabolic pathways for protein synthesis and cell growth [82,83,84].
1.3. Autophagy in ALS
Increasing evidence suggests that ALS is a disease of protein dyshomeostasis, with autophagy dysfunction playing an important role in the pathogenesis of ALS. Autophagy appears to be highly induced in ALS, similar to other neurodegenerative diseases such as Parkinson’s, Huntington’s, and Alzheimer’s diseases [85]. Autophagy proteins, particularly those that are involved in the early stages of autophagy, such as LC3, Beclin 1, p62, and Atg5–Atg12 complex, are elevated in spinal MNs of SALS and FALS patients [86,87] and animal models [86,87,88,89,90]. As the disease progresses to the late symptomatic stages, accumulation of these autophagic factors is also observed in glial cells such as astrocytes and microglia [88]. The upregulation of these proteins is not accounted for by an increase of their transcripts [88]. There is also evidence from studies using electron microscopy showing the build-up of autophagosomes, but not of their matured form, autolysosomes, in spinal MNs of late-symptomatic ALS animal models and autopsied patients [89,91]. In vivo autophagy imaging of the spinal cord of ALS mice has also shown an elevated green fluorescent protein (GFP)-LC3 signal in MNs, indicative of autophagosome accumulation, from early to late symptomatic stages of disease [88].
In contrast, autophagy degradation appears to be reduced in ALS, as there is a build-up of protein aggregates in MNs and surrounding glial cells in the brain and spinal cord of ALS animal models [92] and patients [17,18,19,20]. The accumulation of morphologically altered mitochondria [93,94,95,96,97,98,99] and dysfunctional endoplasmic reticulum (ER) [86,100,101,102,103,104], which are typically cleared through autophagy [105], further reinforces this view.
Autophagy represents a stress adaptation pathway. Therefore, the presence of stressors that are prominent in diseased cells such as damaged DNA, abnormal protein and organelle accumulation, and ER and oxidative stress can over-induce autophagy and autophagosome formation and impair the degradation process [106]. It has been postulated that excessive autophagosome formation/accumulation, exceeding their clearance rate, causes stress and leads to type II autophagic cell death [107] and apoptosis [108]. In fact, inhibiting autophagy induction has beneficial effects in some neurodegeneration disease conditions [109]. Similarly, suppressing autophagosome formation genetically or by administering n-butylidenephthalide at the pre-symptomatic stage of disease in ALS mice attenuated their pathology and extended their lifespan [110,111,112].
Multiple ALS-linked genes, such as SQSTM1, OPTN, and TBK1, encode for core autophagy proteins and others, such as C9orf72, FUS, TDP-43, VAPB, UBQLN2, VCP, CHMP2B, ALS2, FIG4, TUBA4A, PFN1, and DCTN have a functional role in autophagy. Therefore, mutations of these genes can cause impairment in different stages of the autophagy pathway (Table 1, Figure 2). Moreover, the misfolded or aggregated protein products of some ALS-causing genes that are not directly involved in the autophagy process, such as SOD1, can abnormally interact with autophagy proteins to dysregulate their activity [90,113,114,115,116,117,118,119,120,121] (Table 1).
Moreover, any of the 40 mutant genes that have been associated with ALS to date [16,122,123] (Figure 1) can potentially overwhelm the autophagy pathway, as mutations can decrease the stability of their protein products and increase their misfolding and aggregation propensity [124,125,126]. Compensatory mechanisms that upregulate the translation of the aggregate-entrapped proteins to prevent their loss-of-function further exacerbate aggregate formation as the prion-like characteristic of these proteins corrupts and converts the newly synthesised proteins into misfolded replicates [127,128,129,130,131,132,133,134,135,136].
Furthermore, akin to other neurodegenerative diseases, ALS is an age-related disease. During normal ageing in the human brain, autophagy genes such as Atg5, Atg7, and Beclin 1 are transcriptionally downregulated [137,138]. In addition, in aged animal models and human MNs, there is a build-up of intra-lysosomal aggregates in the form of lipofuscin granules, extra-lysosomal aggregates, and defective mitochondria, which can overwhelm and impede autophagy [139]. Therefore, the age-related decline of autophagy proteins and lysosomal proteolytic activity [140,141] and the upsurge of autophagy substrates could reduce autophagy activity and potentially explain the late manifestation of ALS.
1.4. Autophagy-Targeted Treatments for ALS
Thus far, no effective treatment exists for ALS, despite the discovery of this disease nearly two centuries ago. Designing treatments that target specific ALS-causing genes to reduce the protein aggregation load is a potentially valid therapeutic approach. However, multifactorial diseases such as ALS with various aetiologies, from genetic to environmental and age-related, require treatments that target pathogenic processes and their progression in a timely and appropriate manner. The disappointingly modest effect of riluzole [168] and edaravone, the only two approved treatments available for patients that act to rectify excitotoxicity and oxidative stress, respectively, implies that a more central pathway needs to be targeted [169,170].
Dysregulation in autophagy is emerging as a significant contributor of pathogenesis in diverse neuropathologies, including ALS. Therefore, interest continues to grow in autophagy as a likely beneficial therapeutic target [171]. Different autophagy-modulating agents have been tested in ALS patients and disease models, and their findings are summarised below (Figure 3, Table 2).
1.5. Caloric Restriction and Its Mimetics
Caloric restriction through dietary intake reduction, starvation, or physical exercise can successfully induce autophagy by reducing glucose and insulin levels and thus inactivating mTORC1 [215,216] (Figure 3). However, this method is not considered a useful therapeutic approach because it can have harmful effects if not tightly regulated. In particular, males can be vulnerable to nutritional and metabolic stress, as caloric restriction or exercise reduces the lifespan of male, but not female ALS mice (Table 2) [172,173,217]. Moreover, the caloric restriction approach is time-inefficient and it fails to significantly affect brain autophagy, conceivably because of the stable nutrient supply to the brain [218]. Nonetheless, caloric restriction mimetics such as rapamycin, trehalose, spermidine, resveratrol, and metformin have been used to induce the beneficial effects of caloric restriction without its adverse consequences (Figure 3, Table 2).
Rapamycin is the most well-known pharmacological inducer of autophagy. Its administration has shown to be protective in several neurodegenerative disease animal models [219,220,221,222]. However, the effect of rapamycin on ALS differs depending on the animal model tested. In mice overexpressing human wild-type TDP-43 in the forebrain, rapamycin rescued motor neuron function [176]. However, in mice expressing mutant SOD1, rapamycin had no effect [174,178] or had detrimental effects [177]. The negative impact of rapamycin treatment on mutant SOD1 mice was partly attributed to immunosuppression of neuroprotective regulatory T cells [178], suggesting that global mTOR inhibition may not be useful for ALS.
1.6. Hormone Therapy
Female sex hormones can increase mitochondrial efficiency; reduce oxidative stress; and, more importantly, increase autophagy, as opposed to androgens, which decrease autophagy [223]. Hence, progesterone and oestrogen modulators such as raloxifene and tamoxifen have been used to induce autophagy in ALS (Figure 3, Table 2). In fact, female contraceptives may have protective roles in ALS [224] and ablating oestrogen production in female mutant SOD1 mice accelerates their rate of disease progression [225,226] (Table 1).
Therefore, differences in gonadal hormones could explain the slightly higher prevalence of ALS in men than in women [227]. Evidence to support this notion includes the equality of ALS ratio between the two genders at menopause, possibly a result of the age-related decline in oestrogen and progesterone levels in women [190], as well as the more infrequent susceptibility of ALS in pre-menopausal women compared to postmenopausal women [189].
1.7. Anti-Cancer Therapy
Cancer treatment drugs such as bosutinib and dasatinib suppress the phosphorylation of two closely related non-receptor tyrosine kinases, Src and c-Abl, to increase autophagy levels (Figure 3, Table 2).
The involvement of Src and c-Abl in the regulation of cell proliferation, apoptosis, and angiogenesis make them fundamental for neuron development [191]. However, in mature and healthy neurons, their kinase activity tends to be inactive, whereas, in sporadic and familial cases of ALS (and patients with other neurodegenerative diseases), their level of activity is significantly elevated. This inappropriate activation has been associated with neuroinflammation; cell cycle arrest; apoptosis; and, more intriguingly, autophagy inhibition [191] (Figure 3).
1.8. Psychotropic Therapy
Several mood-stabilising drugs such as lithium, valproate/valproic acid, and carbamazepine (Figure 3, Table 2) deplete inositol triphosphate (IP3) and thus have been used to stimulate autophagy.
IP3 interacts with its receptor (IP3R) on the ER to sequester the autophagy protein Beclin 1 and abolish its autophagy-inducing properties. IP3R activation also releases calcium from ER stores, which promotes calcium efflux to organelles such as mitochondria, leading to an increase in ATP production and subsequently autophagy inactivation [228]. An increase in intracellular calcium can also activate calpains to cleave and inactivate autophagy proteins [229] (Figure 3).
It is interesting to note that the majority of the proposed autophagy activators have mood-stabilising or antidepressant effects, raising the possibility for the involvement of protein dyshomeostasis in the pathogenesis of affective disorders [230].
1.9. Anti-Hypertensive Therapy
Although the effect of calcium flux on autophagy is ambiguous [231], calcium channel antagonists such as verapamil, and intracellular calcium modulating agents rilmenidine and berberine, used for the treatment of hypertension, have been used to promote autophagy (Figure 3, Table 2).
As mentioned in the previous section, increased mitochondrial calcium levels have an inhibitory effect on autophagy. However, under stressed conditions such as in ALS animal models, where there is an accumulation of misfolded proteins, cytosolic calcium can also positively regulate autophagy [232].
1.10. Anti-Histamine Therapy
2. Considerations for Effective Targeting Autophagy in ALS
The above-mentioned studies collectively suggest that a number of critical factors need to be taken into account when considering autophagy treatments for ALS. Firstly, the stage of the disease when treatment commences and duration of treatment. This is because genetic inhibition of autophagy in motor neurons of mutant SOD1 mice revealed that autophagy can be protective early in disease progression, but detrimental late in disease [110]. It is noteworthy to mention that some drugs such as clemastine were beneficial with acute treatment, while long-term treatment to the late phases of disease has no effect on disease progression or survival [214]. The stage of disease is likely to reflect differences in pathological substrates accumulating in affected tissues, such as soluble and insoluble misfolded proteins, oligomers, protofibrils, fibrils, aggregates, and inclusions, which could affect autophagy capacity and efficiency in cells.
Secondly, the pathway that the treatment targets to stimulate autophagy needs to be considered. Drugs such as rapamycin, metformin, progesterone, tamoxifen, bosutinib, dasatinib, latrepiradine, and clemastine act through the mTORC1 pathway to induce autophagy. However, mTORC1 is a master regulator of many autophagy-independent pathways in the cell such as protein synthesis, immunosuppression, cell cycle, and many more [236]. For example, the beneficial effects of rapamycin might be negated by its immunosuppressive effects, which might explain why SOD1 mice lacking mature lymphocytes show improved survival, compared to SOD1 mice with their immune system intact [178]. Moreover, upon prolonged treatment with rapamycin, the second mTOR kinase complex (mTORC2) becomes inhibited. This action can cause a reduction in the cell survival signalling pathway, which accounts for the upregulated levels of apoptotic proteins observed in long-term studies with rapamycin [237,238]. Therefore, autophagy-independent effects of mTOR inhibition must be taken into account, or drugs that modulate autophagy independently of the mTOR pathway should be considered [239,240]. Examples of these treatments include trehalose, spermidine, resveratrol, raloxifene, lithium, valproate, pimozide, methotrimeprazine, fluphenazine, verapamil, and rilmenidine. Despite this, treatment with mTOR-independent autophagy inducers such as trehalose [179,180,181,182] or rilmenidine [210] have yielded contrasting outcomes in mutant SOD1 mice, highlighting that bypassing mTOR signalling to invoke autophagy may not be straightforward.
Thirdly, it is critical to consider the stage of the autophagy pathway that the drug is acting on. Treatments that upregulated the autophagic flux/degradation such as trehalose [179,180,181], spermidine [176], tamoxifen [176], progesterone [190], bosutinib [191], lithium [193,196], carbamazepine [176], fluphenazine [206], and clemestine [213,214] showed beneficial effects in ALS mice. In contrast, rapamycin [177,207] and pimozide [177,207] induced autophagy but suppressed autophagic degradation, evidenced by accumulation of autophagosomes and autophagy substrates such as p62 and protein aggregates. These treatments exacerbated disease in ALS animal models. It is also worth noting that autophagic substrates, critically misfolded proteins and aggregates, are not always measured in tissues of ALS mice treated with autophagy-modulating drugs, which is essential to correlate with reported neuroprotective and clinical effects.
Fourthly, results of treatments may differ depending on the experimental model employed. In the absence of a mutation, such as in FTLD-U/TDP-25 cell culture and animal models where there is accumulation of substrates, autophagy can be successfully upregulated, irrespective of where it is targeted [175,176,189,201,211]. However, in animal models driven by mutant transgenes, autophagy may be impaired at different stages, and thus responses to treatments will differ. For example, loss of Beclin 1 in SOD1G93A and SOD1G127X mice exacerbates disease [86], whereas it improves the phenotype of SOD1G86R mice [86,121]. Similarly, the genotype of patients plays an important role in the outcome of clinical trials [241].
Lastly, these drugs are repurposed, and their original target might not be the brain (e.g., metformin). Therefore, they might not be able to sufficiently access the nervous system to affect neuronal autophagy [242]. For the treatment of ALS, it is crucial to be able to efficiently and adequately deliver therapeutics to the central nervous system, more specifically to MNs. This is one of the great challenges of treating neurological diseases; however, significant progress has been made in the development of various MN drug delivery systems [243,244,245].
Moreover, besides autophagy activation, some of these drugs target many other biological processes. Some of these off-target effects are desired and consistent with the multifunctional involvement of autophagy such as antioxidant, mitochondrial, and calcium effects (Figure 4), and some of which are harmful and unfavourable. For example, where researchers were vigilant enough to test the response of both male and female mice to a drug, it was shown that the effect of some treatments are sex-specific. Trehalose [180,181] and resveratrol [184,185,187], which can compete with oestrogen for binding with its receptor, and metformin [188] and latrepiradine [212], which inhibit oestrogen production, all demonstrated more protective effects in male mice compared to females.
3. Conclusions
In this review, we have summarised the main evidence for the enhancement of the autophagy pathway as a possible therapeutic strategy for the treatment of ALS. However, site of autophagy pathway dysfunction and the time at which autophagy becomes dysfunctional during the disease process needs to be elucidated before autophagy treatments can be used as a therapy for ALS. To date, there is no effective autophagy-inducing agent, and those currently being used are small molecules and drugs that often lack selectivity and possess undesirable side effects. Future studies should seek to develop more selective autophagy-enhancing drugs. Moreover, we speculate that autophagy is impaired, not at the autophagosome formation step, but at the autophagosome maturation/degradation step. Therefore, the late, degradative steps of autophagy may need to be targeted, rather than the early steps. In short, promotion of waste clearance and diminution of cytotoxicity is conferred by autophagosome accumulation.
Additionally, it is essential to note that currently there is no definitive diagnostic biomarker for ALS, and by the time the disease clinically manifests itself, more than 50% of neurons are lost [240]. However, the majority of the drugs discussed above were administered at pre-symptomatic or at the onset of symptomatic stage of the disease. To enhance the translational value of animal models, the potential drug needs to be administered to ALS mice at the post-symptomatic stage of the disease to better mimic the pathological conditions of when patients are likely to commence treatment. Meanwhile, reliable early diagnostic markers of disease onset need to be developed so that treatments can be initiated promptly. For example, a sensitive assay that can detect the presence of ALS-associated misfolded proteins early would allow their clearance before they accumulate into larger, degradation-resistant cargo, thus preventing the pathological cascade mediated by aggregate accumulation (Figure 4). Lastly, the effects of the drug should be studied in multiple models of ALS to account for heterogeneity of ALS and to better assess drug suitability. A successful autophagy-enhancing agent would have a potential therapeutic benefit to not only ALS patients but also patients with other proteinopathies that have protein aggregates as their cardinal feature.
Author Contributions
A.A., N.D.P. and F.S. have contributed to the preparation of the original draft of the manuscript. P.M.B. and B.J.T. have revised and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Health and Medical Research Council (project grants APP1138033 to F.S. and B.J.T. and APP1104299 to B.J.T.) and Stafford Fox Medical Research Foundation (2020). A.A. is a recipient of the Bethlehem Griffith Research Foundation scholarship (1808). N.D.P. is a recipient of the Motor Neurone Disease Research Institute of Australia Fellowship (1810). F.S. is a recipient of the FightMND Mid-career Fellowship (04MCR). B.J.T. is a recipient of the NHMRC-ARC Dementia Research Leadership Fellowship (1137024).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bäumer, D.; Talbot, K.; Turner, M.R. Advances in motor neurone disease. J. R. Soc. Med. 2014, 107, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salameh, J.S.; Brown, R.H., Jr.; Berry, J.D. Amyotrophic lateral sclerosis. Semin. Neurol. 2015, 35, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H. Amyotrophic Lateral Sclerosis: A Guide for Patients and Families, 3rd ed.; Demos Medical Publishing: New York, NY, USA, 2009. [Google Scholar]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Mulder, D.W.; Kurland, L.; Offord, K.; Beard, C. Familial adult motor neuron disease: Amyotrophic lateral sclerosis. Neurology 1986, 36, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Jokelainen, M. Amyotrophic lateral sclerosis in Finland: II: Clinical characteristics. Acta Neurol. Scand. 1977, 56, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.; Bede, P.; Montuschi, A.; Pender, N.; Chio, A.; Hardiman, O. Predicting prognosis in amyotrophic lateral sclerosis: A simple algorithm. J. Neurol. 2015, 262, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Consortium, E. Prognostic factors in ALS: A critical review. Amyotroph. Lateral. Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, T.J.; Bosco, D.; Leclerc, A.; Tamrazian, E.; Vanderburg, C.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.; Munsat, T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, M.; Brown, R.H. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef] [PubMed]
- Leigh, P.; Whitwell, H.; Garofalo, O.; Buller, J.; Swash, M.; Martin, J.; Gallo, J.-M.; Weller, R.; Anderton, B. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis: Morphology, distribution, and specificity. Brain 1991, 114, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Amari, M.; Takatama, M.; Aizawa, H.; Mihara, B.; Okamoto, K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006, 249, 13–18. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Brettschneider, J.; Arai, K.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Lee, E.B.; Kuwabara, S.; Shibuya, K.; Irwin, D.J.; Fang, L. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014, 128, 423–437. [Google Scholar] [CrossRef] [Green Version]
- Spiller, K.J.; Cheung, C.J.; Restrepo, C.R.; Kwong, L.K.; Stieber, A.M.; Trojanowski, J.Q.; Lee, V.M.-Y. Selective motor neuron resistance and recovery in a new inducible mouse model of TDP-43 proteinopathy. J. Neurosci. 2016, 36, 7707–7717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Ticozzi, N.; Ratti, A.; Silani, V. Protein aggregation and defective RNA metabolism as mechanisms for motor neuron damage. CNS Neurol. Disord. Drug Targets 2010, 9, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Soto, C.; Estrada, L.; Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2006, 31, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Schuster, J.; Funke, S.A. Methods for the specific detection and quantitation of amyloid-β oligomers in cerebrospinal fluid. J. Alzheimers Dis. 2016, 53, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemeyer, W.; Ramos, P.; Dohmen, R. Ubiquitin-proteasome system. Cell Mol. Life Sci. 2004, 61, 1562–1578. [Google Scholar] [CrossRef]
- Kabashi, E.; Agar, J.N.; Taylor, D.M.; Minotti, S.; Durham, H.D. Focal dysfunction of the proteasome: A pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2004, 89, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Peviani, M.; Cascio, P.; Debiasi, S.; Monti, C.; Bendotti, C. Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol. Dis. 2005, 18, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Marino, M.; Tortarolo, M.; Veglianese, P.; De Biasi, S.; Fontana, E.; Zuccarello, L.V.; Maynard, C.J.; Dantuma, N.P.; Bendotti, C. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashi, E.; Agar, J.N.; Hong, Y.; Taylor, D.M.; Minotti, S.; Figlewicz, D.A.; Durham, H.D. Proteasomes remain intact, but show early focal alteration in their composition in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2008, 105, 2353–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashi, E.; Agar, J.N.; Strong, M.J.; Durham, H.D. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2012, 13, 367–371. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar] [CrossRef]
- Carra, S.; Crippa, V.; Rusmini, P.; Boncoraglio, A.; Minoia, M.; Giorgetti, E.; Kampinga, H.H.; Poletti, A. Alteration of protein folding and degradation in motor neuron diseases: Implications and protective functions of small heat shock proteins. Prog. Neurobiol. 2012, 97, 83–100. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.-S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microbial Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.-M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L.; Iwata, J.-i.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.J.; Ding, Y.; Kohtz, S.; Mizushima, N.; Cristea, I.M.; Rout, M.P.; Chait, B.T.; Zhong, Y.; Heintz, N.; Yue, Z. Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 2006, 26, 8057–8068. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; DiFiglia, M.; Heintz, N.; Nixon, R.A.; Qin, Z.-H.; Ravikumar, B.; Stefanis, L.; Tolkovsky, A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005, 1, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome formation—The role of ULK1 and Beclin1–PI3KC3 complexes in setting the stage. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2013; Volume 23, pp. 301–309. [Google Scholar]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.; Ktistakis, N.T. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. 2013, 55, 17–27. [Google Scholar] [CrossRef]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Noda, N.N.; Kumeta, H.; Nakatogawa, H.; Satoo, K.; Adachi, W.; Ishii, J.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008, 13, 1211–1218. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schüchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.-Q.; Ni, T.; Hong, B.; Wang, H.-Y.; Jiang, F.-J.; Zou, S.; Chen, Y.; Zheng, X.-L.; Klionsky, D.J.; Liang, Y. Dual roles of Atg8− PE deconjugation by Atg4 in autophagy. Autophagy 2012, 8, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, D.; Isaacs, A.M. The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem. Soc. Trans. 2010, 38, 1469–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome–lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007–7021. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R.E.; Bucci, C.; Vieira, O.V.; Schroer, T.A.; Grinstein, S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: Role of Rab7 and RILP. Mol. Cell Biol. 2003, 23, 6494–6506. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end–directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.-L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, E.; Brännström, T.; Andersen, P.M.; Marklund, S.L. Low autophagy capacity implicated in motor system vulnerability to mutant superoxide dismutase. Acta Neuropathol. Commun. 2016, 4, 6–24. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Morimoto, N.; Liu, W.; Ohta, Y.; Deguchi, K.; Miyazaki, K.; Abe, K. In vivo optical imaging of motor neuron autophagy in a mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 985–992. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, X.; Le, W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008, 4, 290–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, N.; Nagai, M.; Ohta, Y.; Miyazaki, K.; Kurata, T.; Morimoto, M.; Murakami, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007, 1167, 112–117. [Google Scholar] [CrossRef]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Dykes-Hoberg, M.; Culotta, V.C.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef]
- Atsumi, T. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1981, 55, 193–198. [Google Scholar] [CrossRef]
- Napoli, L.; Crugnola, V.; Lamperti, C.; Silani, V.; Di Mauro, S.; Bresolin, N.; Moggio, M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1612–1613. [Google Scholar] [CrossRef] [Green Version]
- Siklós, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophc lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.J.; Suh, Y.-L. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct. Pathol. 2002, 26, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett 2019, 710, 132933. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef]
- Sasaki, S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilieva, E.V.; Ayala, V.; Jové, M.; Dalfó, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, L.; Roth, K.A. Autophagy: Cerebral home cooking. Am. J. Pathol. 2010, 176, 1065–1071. [Google Scholar] [CrossRef]
- Boya, P.; González-Polo, R.-A.; Casares, N.; Perfettini, J.-L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-A.; Gao, F.-B. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J. Neurosci. 2009, 29, 8506–8511. [Google Scholar] [CrossRef] [Green Version]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.M.; Zhang, J.J.; Li, S.; Chen, S.; Le, W.D. n-butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD 1G93A mouse model of amyotrophic lateral sclerosis. CNS Neurosci. Ther. 2017, 23, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, K.-W.; Chiou, T.-W.; Chiang, S.-F.; Yamashita, T.; Abe, K.; Borlongan, C.V.; Sanberg, P.R.; Lin, S.-Z.; Harn, H.-J. Autophagic down-regulation in motor neurons remarkably prolongs the survival of ALS mice. Neuropharmacology 2016, 108, 152–160. [Google Scholar] [CrossRef]
- Tak, Y.J.; Park, J.-H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef]
- Uversky, V.N. The roles of intrinsic disorder-based liquid-liquid phase transitions in the “Dr. Jekyll–Mr. Hyde” behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy 2017, 13, 2115–2162. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Ström, A.-L.; Fukada, K.; Lee, S.; Hayward, L.J.; Zhu, H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 2007, 282, 16691–16699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.K.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2018, 34–48. [Google Scholar] [CrossRef]
- Nassif, M.; Valenzuela, V.; Rojas-Rivera, D.; Vidal, R.; Matus, S.; Castillo, K.; Fuentealba, Y.; Kroemer, G.; Levine, B.; Hetz, C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 2014, 10, 1256–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragagnin, A.M.; Shadfar, S.; Vidal, M.; Jamali, S.; Atkin, J.D. Motor neuron susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Prudencio, M.; Hart, P.J.; Borchelt, D.R.; Andersen, P.M. Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. Hum. Mol. Genet. 2009, 18, 3217–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide andprotein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Camilloni, C.; Sala, B.M.; Sormanni, P.; Porcari, R.; Corazza, A.; De Rosa, M.; Zanini, S.; Barbiroli, A.; Esposito, G.; Bolognesi, M. Rational design of mutations that change the aggregation rate of a protein while maintaining its native structure and stability. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Grad, L.I.; Pokrishevsky, E.; Silverman, J.M.; Cashman, N.R. Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding. Prion 2014, 8, 331–335. [Google Scholar] [CrossRef]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymenidou, M.; Cleveland, D.W. The seeds of neurodegeneration: Prion-like spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Ravits, J. Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp. Neurol. 2014, 262, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and-independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Lu, T.; Furuya, T.; Degterev, A.; Mizushima, N.; Yoshimori, T.; MacDonald, M.; Yankner, B.; Yuan, J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006, 281, 14474–14485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Gustafsson, B.; Brunk, U. Autophagy, organelles and ageing. J. Pathol. 2007, 211, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef]
- Fernando, R.; Castro, J.P.; Flore, T.; Deubel, S.; Grune, T.; Ott, C. Age-Related Maintenance of the Autophagy-Lysosomal System Is Dependent on Skeletal Muscle Type. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Soo, K.Y.; Halloran, M.; Sundaramoorthy, V.; Parakh, S.; Toth, R.P.; Southam, K.A.; McLean, C.A.; Lock, P.; King, A.; Farg, M.A. Rab1-dependent ER–Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 2015, 130, 679–697. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996–4007. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.K.; Huang, C.C.; Shen, C.J. Regulation of autophagy by the neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z. TDP-43 loss of function increases TFEB activity and blocks autophagosome–lysosome fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef] [Green Version]
- Fiesel, F.C.; Voigt, A.; Weber, S.S.; Van den Haute, C.; Waldenmaier, A.; Görner, K.; Walter, M.; Anderson, M.L.; Kern, J.V.; Rasse, T.M. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 2010, 29, 209–221. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Suzuki, H.; Kanekura, K.; Levine, T.P.; Kohno, K.; Olkkonen, V.M.; Aiso, S.; Matsuoka, M. ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-expressed wild-type VAPB. J. Neurochem. 2009, 108, 973–985. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C. ER-mitochondria signaling regulates autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jantrapirom, S.; Piccolo, L.L.; Yoshida, H.; Yamaguchi, M. Depletion of Ubiquilin induces an augmentation in soluble ubiquitinated Drosophila TDP-43 to drive neurotoxicity in the fly. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3038–3049. [Google Scholar] [CrossRef]
- Rothenberg, C.; Srinivasan, D.; Mah, L.; Kaushik, S.; Peterhoff, C.M.; Ugolino, J.; Fang, S.; Cuervo, A.M.; Nixon, R.A.; Monteiro, M.J. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 2010, 19, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, J.R.; Kolaitis, R.-M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Fukushi, J.; Hori, S.; Matsuda, N.; Okatsu, K.; Kakiyama, Y.; Kawawaki, J.; Kakizuka, A.; Tanaka, K. Different dynamic movements of wild-type and pathogenic VCP s and their cofactors to damaged mitochondria in a P arkin-mediated mitochondrial quality control system. Genes Cells 2013, 18, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.C.; Tresse, E.; Kolaitis, R.-M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, A.M.; Johannsen, P.; Holm, I.; Nielsen, J.E. Frontotemporal dementia caused by CHMP2B mutations. Curr. Alzheimer Res. 2011, 8, 246–251. [Google Scholar] [CrossRef]
- Urwin, H.; Ghazi-Noori, S.; Collinge, J.; Isaacs, A. The role of CHMP2B in frontotemporal dementia. Biochem. Soc. Trans. 2009, 37, 208–212. [Google Scholar] [CrossRef]
- Rusten, T.E.; Filimonenko, M.; Rodahl, L.M.; Stenmark, H.; Simonsen, A. ESCRTing autophagic clearance of aggregating proteins. Autophagy 2008, 4, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Hadano, S.; Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Akatsuka, A.; Koike, M.; Aoki, M.; Uchiyama, Y.; Itoyama, Y.; Ikeda, J.-E. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1H46R-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS ONE 2010, 5, e9805. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rademakers, R.; van Blitterswijk, M. Excess of rare damaging TUBA4A variants suggests cytoskeletal defects in ALS. Neuron 2014, 84, 241–243. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Liao, Q.; Lu, K.; Zhou, J.; Huang, C.; Bi, F. Riluzole exhibits no therapeutic efficacy on a transgenic rat model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2020, 17, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Rodríguez, J.J.; Verkhratsky, A. Glial calcium and diseases of the nervous system. Cell Calcium 2010, 47, 140–149. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Corti, O.; Blomgren, K.; Poletti, A.; Beart, P.M. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J. Neurochem. 2020, 154, 354–371. [Google Scholar] [CrossRef] [Green Version]
- Hamadeh, M.J.; Rodriguez, M.C.; Kaczor, J.J.; Tarnopolsky, M.A. Caloric restriction transiently improves motor performance but hastens clinical onset of disease in the Cu/Zn-superoxide dismutase mutant G93A mouse. Muscle Nerve 2005, 31, 214–220. [Google Scholar] [CrossRef]
- Hamadeh, M.J.; Tarnopolsky, M.A. Transient caloric restriction in early adulthood hastens disease endpoint in male, but not female, Cu/Zn-SOD mutant G93A mice. Muscle Nerve 2006, 34, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Bokov, A.; Muller, F.L.; Jernigan, A.L.; Maslin, K.; Diaz, V.; Richardson, A.; Van Remmen, H. Dietary restriction but not rapamycin extends disease onset and survival of the H46R/H48Q mouse model of ALS. Neurobiol. Aging 2012, 33, 1829–1832. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem. 2009, 284, 27416–27424. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.-F.; Guo, B.-S.; Liu, Y.-C.; Wu, C.-C.; Yang, C.-H.; Tsai, K.-J.; Shen, C.-K.J. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Staats, K.A.; Hernandez, S.; Schönefeldt, S.; Bento-Abreu, A.; Dooley, J.; Van Damme, P.; Liston, A.; Robberecht, W.; Van Den Bosch, L. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol. Neurodegener. 2013, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Escrevente, C.; Costa, J. Mutant superoxide dismutase 1 overexpression in NSC-34 cells: Effect of trehalose on aggregation, TDP-43 localization and levels of co-expressed glycoproteins. Neurosci. Lett. 2010, 475, 145–149. [Google Scholar] [CrossRef]
- Mancuso, R.; Del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1 G93A ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Choi, J.-R.; Shin, K.S.; Kang, S.J. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012, 1483, 112–117. [Google Scholar] [CrossRef]
- Markert, C.D.; Kim, E.; Gifondorwa, D.J.; Childers, M.K.; Milligan, C.E. A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J. Med. Food 2010, 13, 1081–1085. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Tang, L.; Zhang, N.; Fan, D. Protective effects of resveratrol through the up-regulation of SIRT1 expression in the mutant hSOD1-G93A-bearing motor neuron-like cell culture model of amyotrophic lateral sclerosis. Neurosci. Lett. 2011, 503, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chen, L.; Zhang, X.; Li, J.; Le, W. Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1G93A mouse model of amyotrophic lateral sclerosis. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Kaneb, H.M.; Sharp, P.S.; Rahmani-Kondori, N.; Wells, D.J. Metformin treatment has no beneficial effect in a dose-response survival study in the sod1g93a mouse model of ALS and is harmful in female mice. PLoS ONE 2011, 6, e24189. [Google Scholar] [CrossRef]
- Zhou, F.; Dong, H.; Liu, Y.; Yan, L.; Sun, C.; Hao, P.; Liu, Y.; Zhai, J.; Liu, Y. Raloxifene, a promising estrogen replacement, limits TDP-25 cell death by enhancing autophagy and suppressing apoptosis. Brain Res. Bull. 2018, 140, 281–290. [Google Scholar] [CrossRef]
- Kim, J.; Kim, T.-Y.; Cho, K.-S.; Kim, H.N.; Koh, J.-Y. Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 59, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsumata, R.; Ishigaki, S.; Katsuno, M.; Kawai, K.; Sone, J.; Huang, Z.; Adachi, H.; Tanaka, F.; Urano, F.; Sobue, G. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e46185. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, U.-L.S. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): A phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013, 12, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.P.; Zinman, L.; Simpson, E.; McKinley, J.; Jackson, K.E.; Pinto, H.; Kaufman, P.; Conwit, R.A.; Schoenfeld, D.; Shefner, J. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010, 9, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Cho, S.I.; Lim, H.R.; Lee, J.K.; Lee, Y.A.; Noh, J.S.; Joo, I.S.; Kim, K.-W.; Gwag, B.J. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Pharmacol. 2007, 71, 965–975. [Google Scholar] [CrossRef]
- Chio, A.; Borghero, G.; Calvo, A.; Capasso, M.; Caponnetto, C.; Corbo, M.; Giannini, F.; Logroscino, G.; Mandrioli, J.; Marcello, N. Lithium carbonate in amyotrophic lateral sclerosis: Lack of efficacy in a dose-finding trial. Neurology 2010, 75, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, E.; Veldink, J.H.; Huisman, M.H.; Draak, T.; Uijtendaal, E.V.; van der Kooi, A.J.; Schelhaas, H.J.; de Visser, M.; van der Tweel, I.; van den Berg, L.H. Lithium lacks effect on survival in amyotrophic lateral sclerosis: A phase IIb randomised sequential trial. J. Neurol. Neurosurg. Psychiatry 2012, 83, 557–564. [Google Scholar] [CrossRef]
- Miller, R.; Moore, D.; Forshew, D.; Katz, J.; Barohn, R.; Valan, M.; Bromberg, M.; Goslin, K.; Graves, M.; McCluskey, L. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis: Examining a more efficient trial design. Neurology 2011, 77, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Sugai, F.; Yamamoto, Y.; Miyaguchi, K.; Zhou, Z.; Sumi, H.; Hamasaki, T.; Goto, M.; Sakoda, S. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur. J. Neurosci. 2004, 20, 3179–3183. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, M.; Teng, J.; Che, X.; Zhang, W.; Feng, S.; Zhou, S.; Zhang, Y.; Wu, E.; Ding, X. Valproate attenuates 25-kDa C-terminal fragment of TDP-43-induced neuronal toxicity via suppressing endoplasmic reticulum stress and activating autophagy. Int. J. Biol. Sci. 2015, 11, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Rouaux, C.; Panteleeva, I.; René, F.; de Aguilar, J.-L.G.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.-L.; Loeffler, J.-P. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Piepers, S.; Veldink, J.H.; De Jong, S.W.; Van Der Tweel, I.; Van Der Pol, W.L.; Uijtendaal, E.V.; Schelhaas, H.J.; Scheffer, H.; De Visser, M.; De Jong, J.V. Randomized sequential trial of valproic acid in amyotrophic lateral sclerosis. Ann. Neurol. 2009, 66, 227–234. [Google Scholar] [CrossRef]
- Boll, M.-C.; Bayliss, L.; Vargas-Cañas, S.; Burgos, J.; Montes, S.; Peñaloza-Solano, G.; Rios, C.; Alcaraz-Zubeldia, M. Clinical and biological changes under treatment with lithium carbonate and valproic acid in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 340, 103–108. [Google Scholar] [CrossRef]
- Feng, H.-L.; Leng, Y.; Ma, C.-H.; Zhang, J.; Ren, M.; Chuang, D.-M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, S.; Thammisetty, S.S.; Julien, J.-P. Chronic Administration of Pimozide Fails to Attenuate Motor and Pathological Deficits in Two Mouse Models of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2018, 15, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Szczudlik, A.; Tomik, B.; Słowik, A.; Kasprzyk, K. Assessment of the efficacy of treatment with pimozide in patients with amyotrophic lateral sclerosis. Introductory notes. Neurol. Neurochir. Pol. 1998, 32, 821–829. [Google Scholar] [PubMed]
- Miller, R.; Smith, S.; Murphy, J.; Brinkamann, J.R.; Graves, J.; Mendoza, M.; Sands, M.; Ringel, S. A clinical trial of verapamil in amyotrophic lateral sclerosis. Muscle Nerve 1996, 19, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-F.; Lee, Y.-C.; Lee, K.-H.; Lin, H.-C.; Chen, C.-L.; Shen, C.-K.J.; Huang, C.-C. Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J. Biomed. Sci. 2016, 23, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, K.S.; Mitchem, M.R.; Hogg, M.C.; Prehn, J.H. “Preconditioning” with latrepirdine, an adenosine 5′-monophosphate-activated protein kinase activator, delays amyotrophic lateral sclerosis progression in SOD1G93A mice. Neurobiol. Aging 2015, 36, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Fabbrizio, P.; Parisi, C.; Amadio, S.; Volonté, C. Clemastine confers neuroprotection and induces an anti-inflammatory phenotype in SOD1 G93A mouse model of amyotrophic lateral sclerosis. Mol. Neurobiol. 2016, 53, 518–531. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonté, C. Actions of the antihistaminergic clemastine on presymptomatic SOD1-G93A mice ameliorate ALS disease progression. J. Neuroinflamm. 2016, 13, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Rodriguez, C.; Devries, M.; Yasuda, N.; Tarnopolsky, M.A. Effects of high-intensity endurance exercise training in the G93A mouse model of amyotrophic lateral sclerosis. Muscle Nerve 2004, 29, 656–662. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’kane, C.J. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2005, 15, 433–442. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, K.; Brown, K.; Hunger, N.; Simpson, E. Regulation of LKB1 expression by sex hormones in adipocytes. Int. J. Obes. 2012, 36, 982–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, J.P.; Visser, A.E.; D’ovidio, F.; Vermeulen, R.; Beghi, E.; Chio, A.; Veldink, J.H.; Logroscino, G.; van den Berg, L.H.; Hardiman, O. A case-control study of hormonal exposures as etiologic factors for ALS in women: Euro-MOTOR. Neurology 2017, 89, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Trieu, V.N.; Uckun, F.M. Genistein is neuroprotective in murine models of familial amyotrophic lateral sclerosis and stroke. Biochem. Biophys. Res. Commun. 1999, 258, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, G.; Van Muiswinkel, F.; Sturkenboom, J.; Wokke, J.; Bär, P.; Van den Berg, L. Ovariectomy and 17β-estradiol modulate disease progression of a mouse model of ALS. Brain Res. 2004, 1021, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Aktekin, M.R.; Uysal, H. Epidemiology of Amyotrophic Lateral Sclerosis. Turk. Noroloji Dergisi 2020, 26, 187–196. [Google Scholar] [CrossRef]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2012, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Mangan, D. Autophagy activation as a mechanism of action of psychoactive drugs, revised and expanded version. Biol. Sci. 2015. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Sun, F.; Xu, X.; Wang, X.; Zhang, B. Regulation of autophagy by Ca 2+. Tumour Biol. 2016, 37, 15467–15476. [Google Scholar] [CrossRef] [Green Version]
- Baronio, D.; Gonchoroski, T.; Castro, K.; Zanatta, G.; Gottfried, C.; Riesgo, R. Histaminergic system in brain disorders: Lessons from the translational approach and future perspectives. Ann. Gen. Psychiatry 2014, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thors, B.; Halldórsson, H.; Thorgeirsson, G. eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisová, P.; Alvarez, S.; Kilbride, S.M.; Anilkumar, U.; Baumann, B.; Jordán, J.; Bernas, T.; Huber, H.J.; Düssmann, H.; Prehn, J.H. Latrepirdine is a potent activator of AMP-activated protein kinase and reduces neuronal excitability. Transl. Psychiatry 2013, 3, e317. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.G.; Liu, F.; Song, X.F.; Wang, Z.H.; Dong, Z.Q.; Hu, Z.Q.; Lan, R.Z.; Guan, W.; Zhou, T.G.; Xu, X.M. Rapamycin regulates Akt and ERK phosphorylation through mTORC1 and mTORC2 signaling pathways. Mol. Carcinog. 2010, 49, 603–610. [Google Scholar] [CrossRef]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.A.; Hands, S.; Hafiz, F.; Mizushima, N.; Tolkovsky, A.M.; Wyttenbach, A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol. Pharmacol. 2008, 73, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetkov, A.S.; Mitra, S.; Finkbeiner, S. Protein turnover differences between neurons and other cells. Autophagy 2009, 5, 1037–1038. [Google Scholar]
- Van Eijk, R.P.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Shaw, P.J.; Leigh, P.N.; Young, C.A.; Shaw, C.E.; Mora, G.; Mandrioli, J. Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology 2017, 89, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, J.; Xi, M.; Jia, Y.; Wong, Y.; Zhao, J.; Ding, L.; Zhang, J.; Wen, A. Pharmacogenetic variation and metformin response. Curr. Drug Metab. 2013, 14, 1070–1082. [Google Scholar] [CrossRef]
- O’Leary, V.B.; Ovsepian, S.V.; Bodeker, M.; Dolly, J.O. Improved lentiviral transduction of ALS motoneurons in vivo via dual targeting. Mol. Pharm. 2013, 10, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Ovsepian, S.V.; O’Leary, V.B.; Ntziachristos, V.; Dolly, J.O. Circumventing brain barriers: Nanovehicles for retroaxonal therapeutic delivery. Trends Mol. Med. 2016, 22, 983–993. [Google Scholar] [CrossRef]
- Ovespian, S.V.; Bodeker, M.; O’Leary, V.B.; Lawrence, G.W.; Dolly, J.O. Internalization and retrograde axonal trafficking of tetanus toxin in motor neurons and trans-synaptic propagation at central synapses exceed those of its C-terminal-binding fragments. Brain Struct. Funct. 2015, 220, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The prevalence of the most commonly known genetic causes of Amyotrophic Lateral Sclerosis (ALS). Other genes that are more rarely associated with ALS are not included in the diagram above are: sequestosome 1 (SQSTM1), dynactin subunit 1 (DCTN1), VAMP associated protein B and C (VAPB), D-amino acid oxidase (DAO), TATA-box binding protein associated factor 15 (TAF15), ubiquilin 2 (UBQLN2), heterogenous nuclear ribonucleoprotein A1 (hnRNPA1), heterogenous nuclear ribonucleoproteins A2/B1 (hnRNPA2B1), matrin 3 (MATR3), tubulin alpha 4a (TUBA4A), sec1 family domain containing 1 (SCFD1), myelin associated oligodendrocyte basic protein (MOBP), chromosome 21 open reading frame 2 (C21orf2), cyclin F (CCNF), NIMA related kinase 1 (NEK1), neurofilament heavy (NEFH), dnaJ heat shock protein family (DNAJ), EWS RNA binding protein 1 (EWSR1), senataxin (SETX), calcium- responsive transactivator (CREST), elongator acetyltransferase complex subunit 3 (ELP3), charged multivesicular body protein 2B (CHMP2B), alsin rho nucleotide exchange factor ALS2 (ALS2), sigma non-opioid intracellular receptor 1 (SIGMARI), FIG4 phosphoinositide 5-phosphatase (FIG4), spastic paraplegia 11 (SPG11), peripherin (PRPH), neuropathy target esterase (NTE), serum paraoxonase and arylesterase 1-3 (PON1-3), cholinergic receptor nicotinic alpha 3 (CHRNA3), cholinergic receptor nicotinic alpha 4 (CHRNA4), cholinergic receptor nicotinic beta 4 (CHRNB4), erb-b2 receptor tyrosine kinase 4 (ERBB4), coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10), amyotrophic lateral sclerosis 3 (ALS3), amyotrophic lateral sclerosis 7 (ALS7), amyotrophic lateral sclerosis 6-21 (ALS6-21), amyotrophic lateral sclerosis-frontotemporal dementia (ALS-FTD) [16].
Figure 1.
The prevalence of the most commonly known genetic causes of Amyotrophic Lateral Sclerosis (ALS). Other genes that are more rarely associated with ALS are not included in the diagram above are: sequestosome 1 (SQSTM1), dynactin subunit 1 (DCTN1), VAMP associated protein B and C (VAPB), D-amino acid oxidase (DAO), TATA-box binding protein associated factor 15 (TAF15), ubiquilin 2 (UBQLN2), heterogenous nuclear ribonucleoprotein A1 (hnRNPA1), heterogenous nuclear ribonucleoproteins A2/B1 (hnRNPA2B1), matrin 3 (MATR3), tubulin alpha 4a (TUBA4A), sec1 family domain containing 1 (SCFD1), myelin associated oligodendrocyte basic protein (MOBP), chromosome 21 open reading frame 2 (C21orf2), cyclin F (CCNF), NIMA related kinase 1 (NEK1), neurofilament heavy (NEFH), dnaJ heat shock protein family (DNAJ), EWS RNA binding protein 1 (EWSR1), senataxin (SETX), calcium- responsive transactivator (CREST), elongator acetyltransferase complex subunit 3 (ELP3), charged multivesicular body protein 2B (CHMP2B), alsin rho nucleotide exchange factor ALS2 (ALS2), sigma non-opioid intracellular receptor 1 (SIGMARI), FIG4 phosphoinositide 5-phosphatase (FIG4), spastic paraplegia 11 (SPG11), peripherin (PRPH), neuropathy target esterase (NTE), serum paraoxonase and arylesterase 1-3 (PON1-3), cholinergic receptor nicotinic alpha 3 (CHRNA3), cholinergic receptor nicotinic alpha 4 (CHRNA4), cholinergic receptor nicotinic beta 4 (CHRNB4), erb-b2 receptor tyrosine kinase 4 (ERBB4), coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10), amyotrophic lateral sclerosis 3 (ALS3), amyotrophic lateral sclerosis 7 (ALS7), amyotrophic lateral sclerosis 6-21 (ALS6-21), amyotrophic lateral sclerosis-frontotemporal dementia (ALS-FTD) [16].

Figure 2.
The two main stages of the autophagy pathway. Stage 1 involves autophagy induction where autophagy signals result in the formation of the Unc-51 like autophagy activating kinase 1 (ULK1) complex followed by the phosphatidylinositol 3-kinase class 3 (PI3KC3) complex. This leads to pre-autophagosomal structure formation and autophagosome formation and expansion. Stage 2 involves autophagosome transport for fusion with lysosomes. This leads to autolysosome formation and degradation of its contents. ALS-associated proteins that impair autophagy are indicated beside each step of the pathway. FIP200, FAK family kinase-interacting protein of 200 kDa; mTORC1, mammalian target of rapamycin complex I; ULK1, Unc-51 like autophagy activating kinase 1; p150/Vps15, vacuolar protein sorting 15; Vps34, vacuolar protein sorting 34; LC3-II, C-termini of microtubule-associated protein 1A/1B-light chain 3B; UVRAG, UV radiation resistance associated; Atg13, autophagy-related protein 13; Atg14L, autophagy-related protein 14.
Figure 2.
The two main stages of the autophagy pathway. Stage 1 involves autophagy induction where autophagy signals result in the formation of the Unc-51 like autophagy activating kinase 1 (ULK1) complex followed by the phosphatidylinositol 3-kinase class 3 (PI3KC3) complex. This leads to pre-autophagosomal structure formation and autophagosome formation and expansion. Stage 2 involves autophagosome transport for fusion with lysosomes. This leads to autolysosome formation and degradation of its contents. ALS-associated proteins that impair autophagy are indicated beside each step of the pathway. FIP200, FAK family kinase-interacting protein of 200 kDa; mTORC1, mammalian target of rapamycin complex I; ULK1, Unc-51 like autophagy activating kinase 1; p150/Vps15, vacuolar protein sorting 15; Vps34, vacuolar protein sorting 34; LC3-II, C-termini of microtubule-associated protein 1A/1B-light chain 3B; UVRAG, UV radiation resistance associated; Atg13, autophagy-related protein 13; Atg14L, autophagy-related protein 14.

Figure 3.
Autophagy-targeted treatments that have been studied in ALS. For autophagy to be activated, pathways depicted in red need to be upregulated and pathways depicted in blue need to be downregulated. The pathway that is targeted by the treatment is depicted in green (arrow if its effect is stimulatory and flathead if its effect is inhibitory). InsP, inositol phosphate; IMPase, inositol monophosphatase; PIP2, phosphatidylinositol 4,5- biphosphate; DAG, diacylglycerol; PKC, protein kinase C; MAPK, mitogen-activated protein kinase; cAMP, cyclic adenosine monophosphate; PLC, phospholipase C; IP3, inositol triphosphate; IP3R, inositol triphosphate receptor; CaMKKβ, calcium/calmodulin-dependent protein kinase 2; TAK-1, transforming growth factor beta-activated kinase 1; NAD+, nicotinamide adenine dinucleotide; SIRT1, NAD-dependent deacetylate sirtuin-1; LKB1, liver kinase B1; PKA, protein kinase A; AKT/PKB, protein kinase B; GF, growth factor; FOXO4, forkhead box 4, FOXO1, forkhead box 1; GF, growth factors; RTK, receptor tyrosine kinase; PI3KI, class I phosphoinositol 3-kinase; PIP3, phosphatidylinositol-3,4,5-triphosphate; GSK3, glycogen synthase kinase 3; TIP60, tat-interactive protein 60 kDa; TSC1–TSC2, tuberous sclerosis complex; Rheb, Ras homolog enriched in brain; mTORC1, mammalian target of rapamycin complex I; ULK1, Unc-51 like autophagy activating kinase 1; PI3KIII, class III phosphoinositol 3-kinase; S6K, ribosomal protein S6 kinase beta-1; 4E-BP, eukaryotic translation initiation factor 4E-binding protein 1; eIF4E, eukaryotic translation initiation factor 4E; EP300, E1A binding protein P300.
Figure 3.
Autophagy-targeted treatments that have been studied in ALS. For autophagy to be activated, pathways depicted in red need to be upregulated and pathways depicted in blue need to be downregulated. The pathway that is targeted by the treatment is depicted in green (arrow if its effect is stimulatory and flathead if its effect is inhibitory). InsP, inositol phosphate; IMPase, inositol monophosphatase; PIP2, phosphatidylinositol 4,5- biphosphate; DAG, diacylglycerol; PKC, protein kinase C; MAPK, mitogen-activated protein kinase; cAMP, cyclic adenosine monophosphate; PLC, phospholipase C; IP3, inositol triphosphate; IP3R, inositol triphosphate receptor; CaMKKβ, calcium/calmodulin-dependent protein kinase 2; TAK-1, transforming growth factor beta-activated kinase 1; NAD+, nicotinamide adenine dinucleotide; SIRT1, NAD-dependent deacetylate sirtuin-1; LKB1, liver kinase B1; PKA, protein kinase A; AKT/PKB, protein kinase B; GF, growth factor; FOXO4, forkhead box 4, FOXO1, forkhead box 1; GF, growth factors; RTK, receptor tyrosine kinase; PI3KI, class I phosphoinositol 3-kinase; PIP3, phosphatidylinositol-3,4,5-triphosphate; GSK3, glycogen synthase kinase 3; TIP60, tat-interactive protein 60 kDa; TSC1–TSC2, tuberous sclerosis complex; Rheb, Ras homolog enriched in brain; mTORC1, mammalian target of rapamycin complex I; ULK1, Unc-51 like autophagy activating kinase 1; PI3KIII, class III phosphoinositol 3-kinase; S6K, ribosomal protein S6 kinase beta-1; 4E-BP, eukaryotic translation initiation factor 4E-binding protein 1; eIF4E, eukaryotic translation initiation factor 4E; EP300, E1A binding protein P300.

Figure 4.
The interrelationship between autophagy and other pathogenic mechanisms in ALS. Dysfunction of autophagy, specifically, aggrephagy (aggregate-specific autophagy), mitophagy (aggregate-specific autophagy), and reticulophagy (ER-specific autophagy) leads to the build-up of protein aggregates, damaged mitochondria, and ER stress, respectively. Thus, inducing reactive oxygen species (ROS) production, which persistently builds up in ALS to inhibit autophagy, stabilises aggregates and induces inflammation and excitotoxicity. Excitotoxicity over-activates neurons, resulting in cytoplasmic and mitochondrial calcium augmentation and hence further mitochondrial damage. As a result of these impairments, cells undergo apoptosis and death.
Figure 4.
The interrelationship between autophagy and other pathogenic mechanisms in ALS. Dysfunction of autophagy, specifically, aggrephagy (aggregate-specific autophagy), mitophagy (aggregate-specific autophagy), and reticulophagy (ER-specific autophagy) leads to the build-up of protein aggregates, damaged mitochondria, and ER stress, respectively. Thus, inducing reactive oxygen species (ROS) production, which persistently builds up in ALS to inhibit autophagy, stabilises aggregates and induces inflammation and excitotoxicity. Excitotoxicity over-activates neurons, resulting in cytoplasmic and mitochondrial calcium augmentation and hence further mitochondrial damage. As a result of these impairments, cells undergo apoptosis and death.


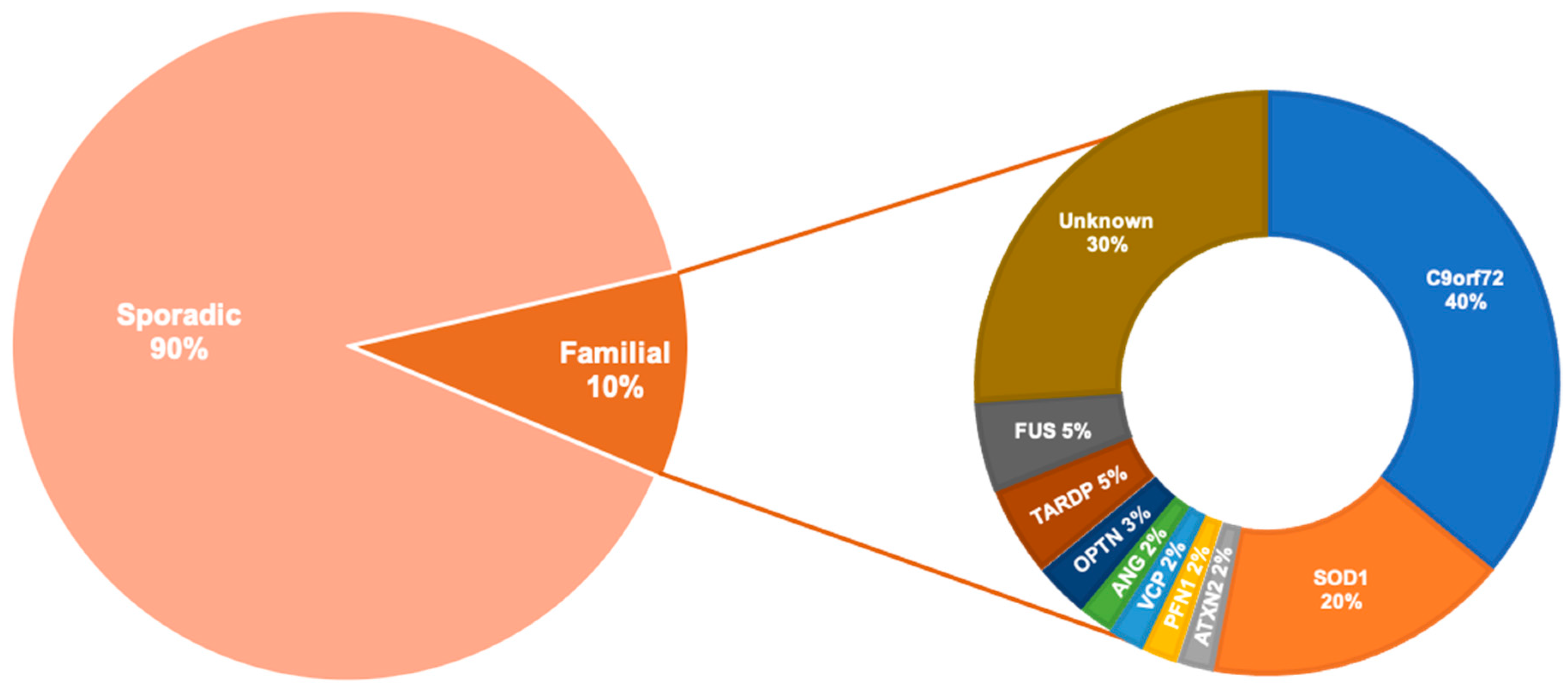{kind=link}
{kind=link}
{kind=link}
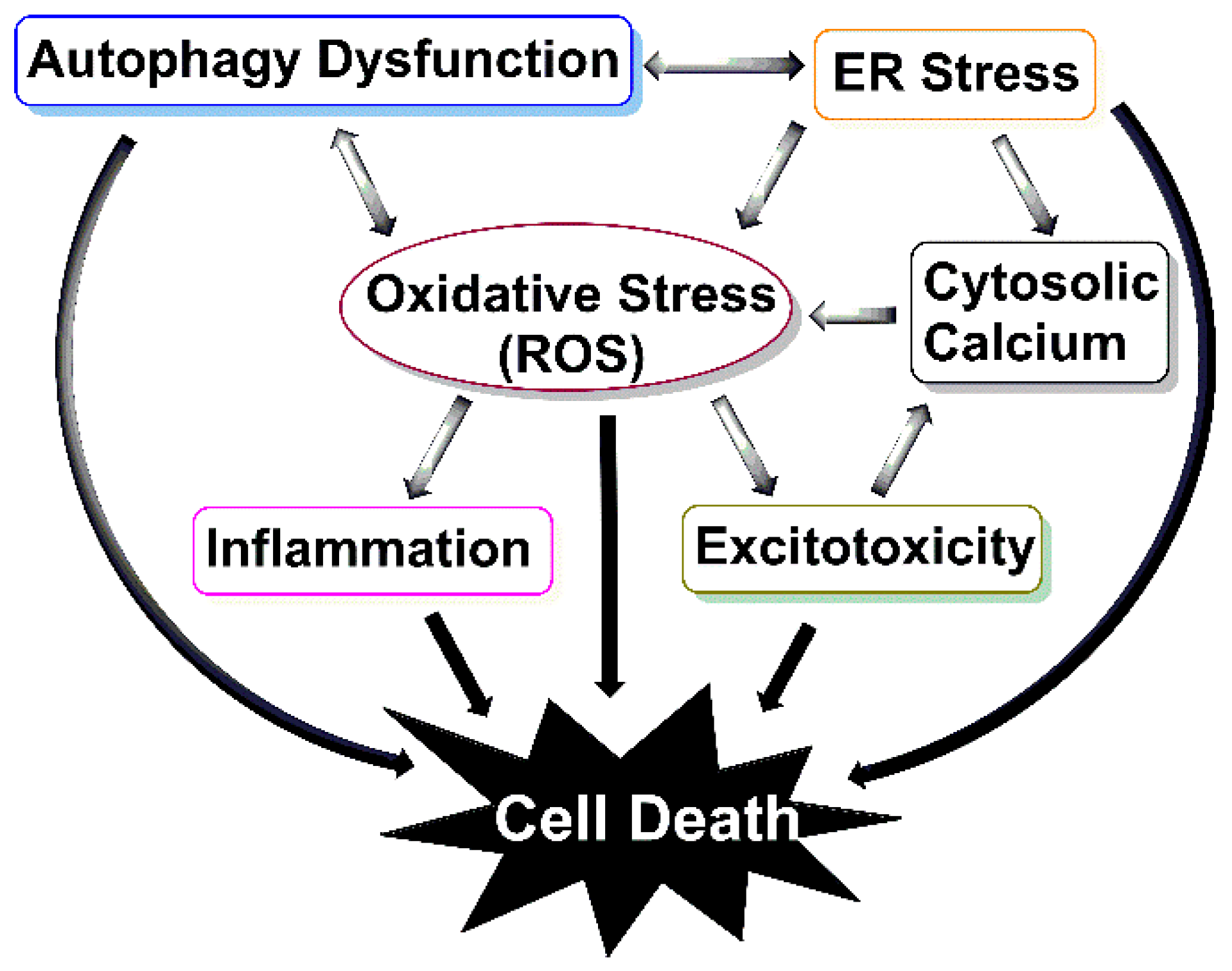{kind=link}
Table 1.
The association between ALS genes and autophagy.
| Gene | Protein | Function | Effect on Autophagy | Reference |
|---|---|---|---|---|
| SOD1 | Superoxide dismutase 1 | Antioxidant enzyme: convert superoxide radical anions to oxygen/hydrogen peroxide. | Mutant associates with Beclin1–B cell lymphoma 2 (Bcl2) complex to disrupt the activity of Beclin1 → impairing autophagosome formation/expansion. | [121] |
| Mutant and misfolded wild-type sequesters dynein proteins into inclusions → impairing retrograde transport and autolysosome formation. | [90,119,120] | |||
| Mutant binds voltage-dependent anion-selective channel protein 1 (VDAC1) and Bcl2 → impairing mitophagy. | [114,115,116,117,118] | |||
| Mutant binds to optineurin → impairing mitophagy. | [113] | |||
| C9ORF72 | Chromosome 9 opening reading frame 72 | Guanine nucleotide exchange factor (GEFs) for Rab GTPases—allows trafficking via actin, regulates lysosome biogenesis and maturation. | Deficiency disrupts ULK1 complex trafficking → impairing autophagosome formation. | [120] |
| Deficiency disrupts transfer of lysosomal proteins to lysosomes → impairing autolysosome formation. | [142] | |||
| Deficiency disrupts its association with early endosomes for lysosome maturation → impairing autolysosome formation. | [142] | |||
| FUS | Fused in sarcoma | RNA-binding protein—regulates RNA splicing, processing, transport, and translation. | Mutant disrupts ER–Golgi transport and ER–mitochondrial interactions → impairing autophagosome formation. | [143,144] |
| Deficiency decreases Parkin → impairing mitophagy. | [145,146] | |||
| TDP-43 | TAR DNA binding partner 43 | RNA-binding protein—regulates RNA splicing, processing, transport, and translation. | Mutant disrupts ER–Golgi transport and ER–mitochondrial interactions → impairing autophagosome formation. | [143,144] |
| Deficiency decreases Parkin → impairing mitophagy. | [145,146] | |||
| Deficiency destabilises Atg7 mRNA and proteins → impairing autophagosome expansion. | [147] | |||
| Mutant inhibits dynactin and HDAC6 → impairing retrograde transport for autolysosome formation. | [148,149] | |||
| SQSTM1 | Sequestosome 1/p62 | Autophagy receptor—recruit cargo to autophagy. | Mutant disrupts recognition of ubiquitinated cargo and their delivery to autophagosomes → impairing cargo degradation. | [120] |
| OPTN | Optineurin | Autophagy receptor—recruits cargo to autophagy. | Mutant disrupts recognition of ubiquitinated cargo/mitochondria and their delivery to autophagosomes → impairing cargo degradation and mitophagy. | [120] |
| Deficiency disrupts its interaction with myosin VI → impairing retrograde transport for autolysosome formation. | [120] | |||
| TBK1 | Tank-binding kinase 1 | Autophagy receptor activator—phosphorylates p62, OPTN, etc. for selective autophagy. | Mutant disrupts its kinase activity of autophagy receptors → impairing cargo/mitochondria degradation. | [150,151] |
| Mutant disrupts its kinase activity of microtubule-binding proteins → impairing retrograde transport for autolysosome formation. | [120] | |||
| VAPB and VAPC | VAMP-associated protein B and C | ER proteins—activate endoplasmic reticulum-associated protein degradation (ERAD) and autophagy. | Mutant disrupts its interaction with PTPIP51 to allow ER–mitochondrial contact → impairing autophagosome formation. | [152,153] |
| UBQLN2 | Ubiquilin | ER protein—recognises ubiquitinated misfolded proteins. | Mutant disrupts recognition of ubiquitinated cargo and their delivery to autophagosomes → impairing cargo degradation. | [154,155] |
| VCP | Valosin-containing protein | ER protein—forms aggresomes and translocates them out of ER for degradation. | Mutant disrupts formation of aggresomes and translocates out of ER → impairing cargo degradation | [120,156] |
| Mutant disrupts granulophagy (degradation of stress granules). | [157,158] | |||
| Mutant disrupts its recruitment to damaged mitochondria → impairing mitophagy. | [159,160] | |||
| CHMP2B | Charged multivesicular body protein 2 B | Endosomal protein—generates multi-vesicular bodies (MVBs). | Mutant disrupts Rab5 conversion to Rab7 → impairing retrograde transport for autolysosome formation. | [161,162,163] |
| Mutant disrupts dissociation of its ESCRT-III complex from the endosomal membrane MVB generation → impairing autophagosome maturation. | [161,162,163] | |||
| Mutant disrupts biogenesis and maintenance of lysosomes → impairing autolysosome formation. | [161,162,163] | |||
| ALS2 | Alsin | Endosomal protein—generates MVBs. | Mutant disrupts its GEF activity of Rab5 → impairing autophagosome maturation. | [164] |
| FIG4 | Polyphosphoinositide 5-phosphatase | Endosomal protein—generates MVBs. | Mutant disrupts PI3P production → impairing autophagosome maturation. | [165] |
| TUBA4A | Tubulin alpha 4A | Cytoskeleton protein—allows transport within cells. | Mutant disrupts the dynamic and stability of microtubules → impairing retrograde transport for autolysosome formation. | [120,166,167] |
| PFN1 | Profilin1 | Cytoskeleton protein— allows transport within cells. | Mutant disrupts its interaction with actin → impairing retrograde transport for autolysosome formation. | [120,166,167] |
| DCTN | Dynactin | Cytoskeleton protein—allows transport within cells. | Mutant disrupts its complex formation with dynein to bind to microtubules → impairing retrograde transport for autolysosome formation. | [120,166,167] |
Table 2.
The effect of autophagy-targeted treatments on ALS models. Sur., survival; Ref., reference; F, female mice; M, male; ↑, increased/restoration; ↓, decreased; ↔, unaffected; co, combination of treatments; *, p62 remained unchanged.
Table 2.
The effect of autophagy-targeted treatments on ALS models. Sur., survival; Ref., reference; F, female mice; M, male; ↑, increased/restoration; ↓, decreased; ↔, unaffected; co, combination of treatments; *, p62 remained unchanged.
| Treatment |
Treatment Dosing | Model | Treatment Duration | Autophagy | MNs | Sur. | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Caloric Restriction | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑function | ↓ | [172] | |||
| SOD1-G93A mice | Short-term (pre-symptomatic) | ↓ (M) | [173] | |||||
| SOD1-H46R/H48Q mice | Long-term (pre-symptomatic) | ↑function | ↑ | [174] | ||||
| Caloric Restriction Mimetics | Rapamycin | 0.5 µg/mL | N2A and SH-SYSY cells TDP-25 | 24 h | ↑flux | [175] | ||
| I.P. injection 2.24 mg/kg/day | SOD1-G93A mice | Long-term (pre-symptomatic) | ↔ | [174] | ||||
| I.P. injection 2.24 mg/kg/day | SOD1-H46R/H48Q mice | Long-term (pre-symptomatic) | ↔ | [174] | ||||
| I.P. injection 10 mg/kg 3×/week | FTLD-U mice (M) | Short-term (symptomatic) | ↑flux | ↑function | [176] | |||
| I.P. injected 2 mg/kg/day | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑induction ↓degradation | ↓number | ↓ | [177] | ||
| Oral 2.33 mg/kg/day | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑induction | ↔ | [178] | |||
| Oral 2.33 mg/kg/day | RAG1(-/-) x SOD1-G93A mice | Long-term (pre-symptomatic) | ↑degradation | ↑ | [178] | |||
| Trehalose | I.P. injection 2 g/kg 3×/week | SOD1-G86R mice | Long-term (pre-symptomatic) | ↑flux | ↑function | ↑ | [179] | |
| 100 mM | NSC-34 cells SOD1-G86R | 24 h | ↑flux | [179] | ||||
| Oral 2% w/v/day | SOD1-G93A mice (F) | Short-term (pre-symptomatic) | ↑flux | ↑function | [180] | |||
| Oral 2% w/v/day | SOD1-G93A mice (F) | Long-term (pre-symptomatic) | ↑induction | ↔ | [180] | |||
| 100 mM | NSC-34 cells SOD1-G93A | 24 h | ↑flux | [180] | ||||
| Oral 2% w/v/day | SOD1-G93A mice (M) | Long-term (pre-symptomatic) | ↑degradation | ↑number | ↑ | [181] | ||
| 10 mM | NSC-34 cells SOD1-G93A | 72 h | ↑degradation | [182] | ||||
| Spermidine | I.P. injection 50 mg/kg 3×/week | FTLD-U mice (M) | Short-term (symptomatic) | ↑flux | ↑function ↑number | [176] | ||
| Resveratrol | Oral 160 mg/kg/day | SOD1-G93A mice | Short-term (pre-symptomatic and symptomatic) | ↑induction | ↑function | ↑ | [183] | |
| I.P. injection 20 mg/kg 2×/week | SOD1-G93A mice (M) | Long-term (pre-symptomatic) | ↑function | ↑ | [184] | |||
| Oral 25 mg/kg/day | SOD1-G93A mice (F) | Long-term (pre-symptomatic) | ↔ | [185] | ||||
| 10 µM | VSC4.1 cells SOD1-G93A | 24 h | [186] | |||||
| I.P. injection 25 mg/kg/day | SOD1-G93A mice (M) | Long-term (pre-symptomatic) | ↑number | ↑ | [187] | |||
| Metformin | Oral 2 mg/mL | SOD1-G93A mice | Short-term (pre-symptomatic) | ↑number | ↔ (M) ↓ (F) | [188] | ||
| Hormone Therapy | Raloxifene | 0.1 µM | NSC-34 cells TDP-25 | 24 h | ↑degradation | [189] | ||
| Tamoxifen | S.C. injection 50 mg/kg 3×/week | FTLD-U mice (M) | Short-term (symptomatic) | ↑flux | ↑function ↑number | [176] | ||
| Progesterone | I.P. injection 4 mg/kg/day | SOD1-G93A mice (M) | Long-term (pre-symptomatic) | ↑degradation | ↑ | [190] | ||
| Cancer Therapy | Bosutinib | 10 µM | Patient iPSC derived MNs SOD1-L144FVX, SOD1-G93S, TDP-43 M337V, TDP-43 Q343R, TDP-43 G298S, C9orf72, SALS | 7 days | ↑degradation | ↑number | [191] | |
| I.P. injection 5 mg/kg/day | SOD1-G93A mice | Short-term | ↑degradation | ↑number | ↑ | [191] | ||
| Dasatinib | Oral gavage 25 mg/kg/day | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑function | ↑ | [192] | ||
| Oral gavage 5 mg/kg/day | SOD1-G93A mice | Long-term (pre-symptomatic) | ↔ | [192] | ||||
| Psychotropic Therapy | Lithium | I.P. injection 1 mEq/kg/day | SOD1-G93A mice (M) | Long-term (symptomatic) | ↑flux | ↑function | ↑ | [193] |
| Plasma range of 0.4–0.8 mmol/L | Patients | 15 months | ↑ | [193] | ||||
| Plasma range of 0.4–0.8 mmol/L | Patients | 18 months | ↔ | [194] | ||||
| Plasma range of 0.4–0.8 mmol/L | Patients | 12 months | ↔ | [195] | ||||
| Lithium carbonate 200 mg/kg/day OR Neu2000 30 mg/kg/day OR combination | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑function ↑number ↑↑co | ↑ ↑↑co | [196] | |||
| Plasma range of 0.4–0.8 mmol/L | Patients | 15 months | ↔ | [197] | ||||
| Plasma range of 0.4–0.8 mmol/L | Patients | 16 months | ↔ | [198] | ||||
| Plasma range of 0.3–0.8 mmol/L | Patients | 13 months | ↔ | [199] | ||||
| Valproate or valproic acid | Oral 0.26% w/v 530 mg/kg/day | SOD1-G93A mice (M) | Long-term (pre-symptomatic) | ↑ | [200] | |||
| 1–2 mM | SH-SY5Y cells TDP-25 | 48 h | ↑ induction | [201] | ||||
| I.P. injection 250 mg/kg/day | SOD1-G86R mice | Long-term (pre-symptomatic) | ↑number | ↔ | [202] | |||
| 1500 mg | Patients | 12 months | ↔ | [203] | ||||
| Valproic acid + lithium carbonate | 0.3–0.75 mmol/L | Patients | 18 months | ↑ | [204] | |||
| I.P. injection lithium OR R valproate OR combination 60 mg/kg/2× day | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑function ↑↑co | ↑ ↑↑co | [205] | |||
| Carbamazepine | 50 mg/kg 3×/week | FTLD-U mice (M) | Short-term (symptomatic) | ↑flux | ↑function ↑number | [176] | ||
| Fluphenazine | 5 µM | Primary neurons TDP43-A315T | 48 h | ↑flux | ↑ | [206] | ||
| Methotrimeprazine | 5 µM | Primary neurons TDP43-A315T | 48 h | ↑flux | ↑ | [206] | ||
| Pimozide | I.P. injection 1 mg/kg/every 2 days | SOD1-G93A mice | Long-term (pre-symptomatic) | ↓degradation | ↓function ↔number | ↓ | [207] | |
| I.P. injection 1 mg/kg/every 2 days | SOD1-G93A mice | Long-term (symptomatic) | ↔degradation | ↓function ↔number | ↓ | [207] | ||
| I.P. injection 1 mg/kg/every 2 days | TDP43-A315T mice | Short-term | ↓degradation | ↓function | ↓ | [207] | ||
| 1 mg/day | Patients | 3–12 months | ↑ | [208] | ||||
| Anti- hypertensive | Verapamil | 240 mg/day | Patients | 6 months | ↔ | [209] | ||
| Rilmenidine | I.P. injection 10 mg/kg 4×/week | SOD1-G93A mice | Long-term (pre-symptomatic) | ↑induction | ↓number | ↓ | [210] | |
| 10 µM | NSC-34 cells SOD1- A4V | 24 h | ↑degradation * | [210] | ||||
| 10 µM | Stem cells SOD1-WT | 24 h | ↑flux | [210] | ||||
| Berberine | 10–30 µg/ml | N2a cells TDP-25 | 6 h, 24 h | ↑flux | [211] | |||
| Anti-histamine | Latrepiradine | I.P. injection I µg/kg/day | SOD1-G93A mice | Short-term (pre-symptomatic) | ↑induction | ↑function (M) | ↑ (M) | [212] |
| Clemastine | I.P. injection 10 mg/kg/5×/week | SOD1- G93A mice (F) | Long-term (pre-symptomatic) | ↑number | ↔ | [213] | ||
| I.P. injection 50 mg/kg/5×/week | SOD1-G93A mice (F) | Short-term (pre-symptomatic) | ↑flux | ↑function | ↑ | [214] | ||
| I.P. injection 50 mg/kg 5×/week | SOD1-G93A mice (F) | Long-term (pre-symptomatic) | ↔ | ↔ | [214] | |||
| 30 µM | NSC-34 cells SOD1-G93A | 6 h, 24 h | ↑flux (6 hrs) ↓flux (24 hrs) | [214] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. https://doi.org/10.3390/cells9112413
AMA Style
Amin A, Perera ND, Beart PM, Turner BJ, Shabanpoor F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells. 2020; 9(11):2413. https://doi.org/10.3390/cells9112413
Chicago/Turabian StyleAmin, Azin, Nirma D. Perera, Philip M. Beart, Bradley J. Turner, and Fazel Shabanpoor. 2020. "Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting" Cells 9, no. 11: 2413. https://doi.org/10.3390/cells9112413
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.