
Cystathionine Beta-Synthase Deficiency: Three Consecutive Cases Detected in 40 Days by Newborn Screening in Emilia Romagna (Italy) and a Comprehensive Review of the Literature
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Literature Review
3.1. Epidemiology
3.2. Pathogenesis
3.3. Clinical Presentation
3.3.1. Pyridoxine-Responsive
3.3.2. Pyridoxine Non-Responsive
3.4. Diagnosis
3.5. Expanded Newborn Screening
3.6. Treatment
4. Results
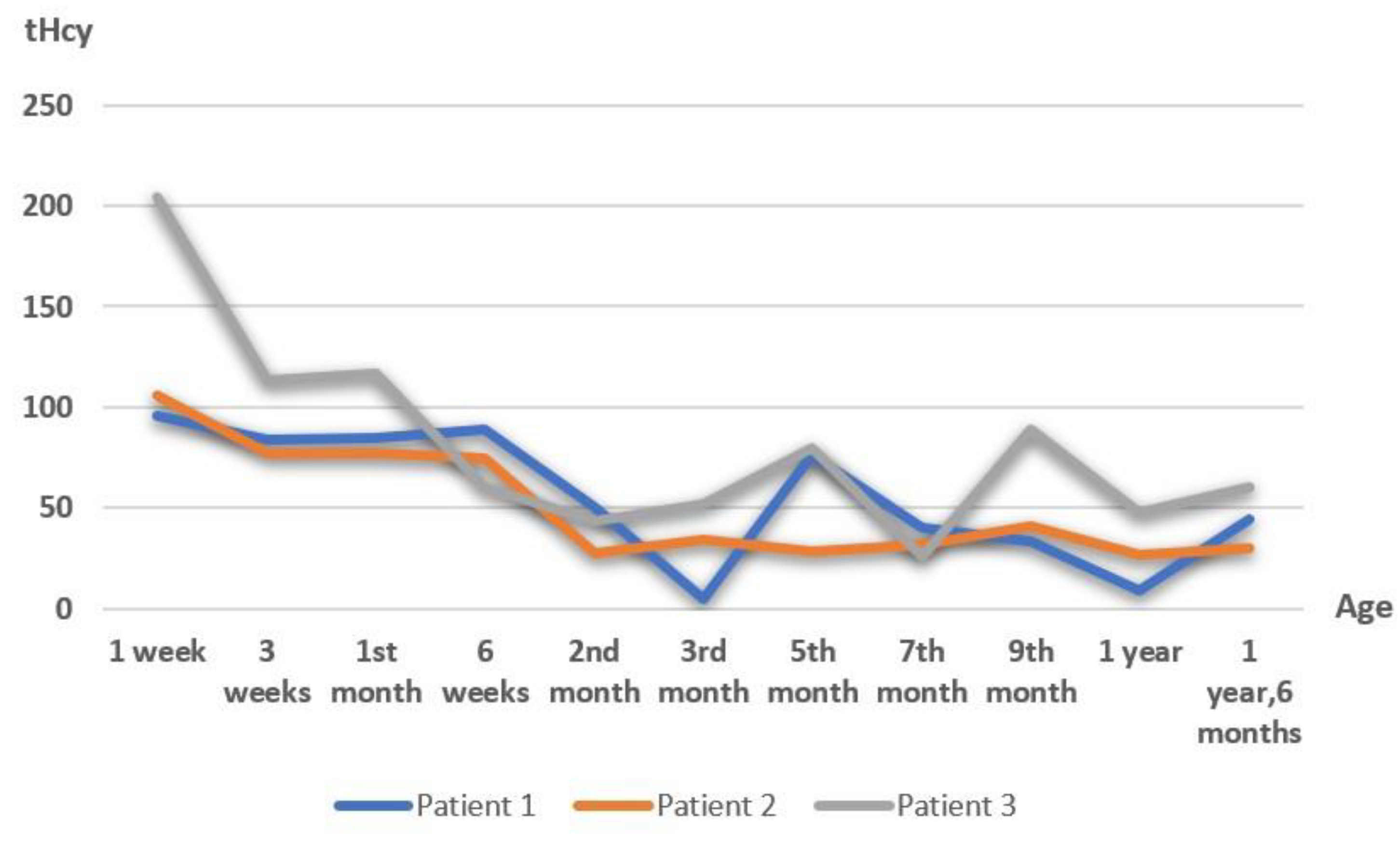
4.1. Our Experience
4.2. Proposal Flow-Chart
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morris, A.M.; Kožich, V. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H. Hypermethioninemias of genetic and non-genetic origin: A review. Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Jepchumba, V.K. Mechanisms of homocysteine-induced damage to the endothelial, medial and adventitial layers of the arterial wall. Biochimie 2020, 173, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Yap, S. Classical homocystinuria: Newborn screening with early treatment effectively prevents complications. Hamdan Med. J. 2012, 5, 351–362. [Google Scholar] [CrossRef]
- Weisfeld-Adams, J.D.; Bender, H.A. Neurologic and neurodevelopmental phenotypes in young children with early-treated combined methylmalonic acidemia and homocystinuria, cobalamin C type. Mol. Genet. Metab. 2013, 110, 241–247. [Google Scholar] [CrossRef]
- Gerrard, A.; Dawson, C.J. Homocystinuria diagnosis and management: It is not all classical. Clin. Pathol. 2022, 75, 744–750. [Google Scholar] [CrossRef]
- Huemer, M.; Kožich, V. Newborn screening for homocystinurias and methylation disorders: Systematic review and proposed guidelines. J. Inherit. Metab. Dis. 2015, 38, 1007–1019. [Google Scholar] [CrossRef] [Green Version]
- Diekman, E.F.; de Koning, T.J. Survival and psychomotor development with early betaine treatment in patients with severe methylenetetrahydrofolate reductase deficiency. JAMA Neurol. 2014, 71, 188–194. [Google Scholar] [CrossRef]
- Ombrone, D.; Giocaliere, E. Expanded newborn screening by mass spectrometry: New tests, future perspectives. Mass Spectrom. Rev. 2016, 35, 71–84. [Google Scholar] [CrossRef]
- Ruoppolo, M.; Malvagia, S. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience. Int. J. Neonatal Screen. 2022, 8, 47. [Google Scholar] [CrossRef]
- Emilia-Romagna-Ufficio di Statistica. Available online: https://statistica.regione.emilia-romagna.it/documentazione/pubblicazioni/documenti_catalogati/rapporto-popolazione-residente-emilia-romagna-2022 (accessed on 1 January 2022).
- Technical Reports of the ENS on the Italian Territory. Available online: https://www.simmesn.it/it/documenti/rapporti-tecnici-screening-neonatale.htm (accessed on 20 December 2022).
- Riley, S.D.; Barber, S.M. CARE guidelines for case reports: Explanation and elaboration document. J. Clin. Epidemiol. 2017, 89, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Gan-Schreier, H.; Kebbewar, M. Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. J. Pediatr. 2010, 156, 427–432. [Google Scholar] [CrossRef]
- Villani, G.R.; Albano, L. Hypermethioninemia in Campania: Results from 10 years of newborn screening. Mol. Genet. Metab. Rep. 2019, 21, 100520. [Google Scholar] [CrossRef]
- Hermann, A.; Sitdikova, G. Homocysteine: Biochemistry, Molecular Biology and Role in Disease. Biomolecules 2021, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine and the pathogenesis of atherosclerosis. Expert. Rev. Clin. Pharmacol. 2015, 8, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Skovby, F.; Gaustadnes, M. A revisit to the natural history of homocystinuria due to cystathionine β-synthase deficiency. Mol. Genet. Metab. 2010, 99, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.H.; Burke, J. Ophthalmic abnormalities in homocystinuria: The value of screening. Eye 1998, 12 Pt 3a, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Gus, P.I.; Donis, K.C. Ocular manifestations in classic homocystinuria. Ophthalmic Genet. 2021, 42, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.R.; Coughlin, C. Low bone mineral density is a common finding in patients with homocystinuria. Mol. Genet. Metab. 2016, 117, 351–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saboul, C.; Darteyre, S. Inaugural cerebral sinovenous thrombosis revealing homocystinuria in a 2-year-old boy. J. Child Neurol. 2015, 30, 107–112. [Google Scholar] [CrossRef]
- Kalil, M.A.; Donis, K.C. Cardiovascular findings in classic homocystinuria. Mol. Genet. Metab. Rep. 2020, 25, 100693. [Google Scholar] [CrossRef] [PubMed]
- Magner, M.; Krupková, L. Vascular presentation of cystathionine β-synthase deficiency in adulthood. J. Inherit. Metab. Dis. 2011, 34, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, S.; Rushe, H. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine β-synthase deficiency. J. Inherit. Metab. Dis. 2001, 24, 437–447. [Google Scholar] [CrossRef] [PubMed]
- El Bashir, H.; Dekair, L. Neurodevelopmental and Cognitive Outcomes of Classical Homocystinuria: Experience from Qatar. JIMD Rep. 2015, 21, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Mazzei, D.H.; Rodriguez, S.M. A forgotten lethal psychosis: A case report. Eur. Child Adolesc. Psychiatry 2014, 23, 235–238. [Google Scholar] [CrossRef]
- Harker, L.A.; Slichter, S.J. Homocystinuria: Vascular injury and arterial thrombosis. N. Engl. J. Med. 1974, 291, 537–543. [Google Scholar] [CrossRef]
- Cochran, F.B.; Sweetman, L. Pyridoxine-unresponsive homocystinuria with an unusual clinical course. Am. J. Med. Genet. 1990, 35, 519–522. [Google Scholar] [CrossRef]
- Bass, H.N.; LaGrave, D. Spontaneous pneumothorax in association with pyridoxine-responsive homocystinuria. J. Inherit. Metab. Dis. 1997, 20, 831–832. [Google Scholar] [CrossRef]
- Ilan, Y.; Eid, A. Gastrointestinal involvement in homocystinuria. J. Gastroenterol. Hepatol. 1993, 8, 60–62. [Google Scholar] [CrossRef]
- Yuan, J.; Wei, Z. Vitamin B12 Attenuates Acute Pancreatitis by Suppressing Oxidative Stress and Improving Mitochondria Dysfunction via CBS/SIRT1 Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 7936316. [Google Scholar] [CrossRef]
- Levy, H.L.; Vargas, J.E. Reproductive fitness in maternal homocystinuria due to cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2002, 25, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Yokoyama, K. Long-Term Outcomes of Adult Patients with Homocystinuria before and after Newborn Screening. Int. J. Neonatal. Screen. 2020, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P.; Korson, M. Metabolic profiling of total homocysteine and related compounds in hyperhomocysteinemia: Utility and limitations in diagnosing the cause of puzzling thrombophilia in a family. JIMD Rep. 2013, 11, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Refsum, H.; David Smith, A. Facts and recommendations about total homocysteine determinations: An expert opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alodaib, A.N.; Carpenter, K. Homocysteine measurement in dried blood spot for neonatal detection of homocystinurias. JIMD Rep. 2012, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bártl, J.; Chrastina, P. Simultaneous determination of cystathionine, total homocysteine, and methionine in dried blood spots by liquid chromatography/tandem mass spectrometry and its utility for the management of patients with homocystinuria. Clin. Chim. Acta 2014, 437, 211–217. [Google Scholar] [CrossRef]
- Mendes, M.I.; Colaco, H.G. Reduced response of cystathionine beta-synthase (CBS) to S-adenosylmethionine (SAM): Identification and functional analysis of CBS gene mutations in Homocystinuria patients. J. Inherit. Metab. Dis. 2013, 37, 245. [Google Scholar] [CrossRef]
- Fowler, B.; Giles, L. Chorionic villus sampling: Diagnostic uses and limitations of enzyme assays. J. Inherit. Metab. Dis. 1989, 12 (Suppl. S1), 105–117. [Google Scholar] [CrossRef]
- Kožich, V.; Sokolovà, J. Cystathionine β-synthase deficiency in the E-HOD registry-part I: Pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis. J. Inherit. Metab. Dis. 2021, 44, 677–692. [Google Scholar] [CrossRef]
- Levy, H.L. Early Development of Newborn Screening for HCU and Current Challenges. Int. J. Neonatal. Screen. 2021, 7, 67. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y. Analysis of five cases of hypermethioninemia diagnosed by neonatal screening. J. Pediatr. Endocrinol. Metab. 2020, 33, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.; Abdenur, J.E. Mudd’s disease (MAT I/III deficiency): A survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet. J. Rare Dis. 2015, 10, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweinberger, B.M.; Wyse, A.T. Mechanistic basis of hypermethioninemia. Amino Acids 2016, 48, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Skovby, F. The natural history of homocystinuria due to cystathionine ß-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar] [PubMed]
- Maase, R.; Skrinska, V. A Rapid Screening Method for the Measurement of Neonatal Total Homocysteine in Dried Blood Spots by Liquid Chromatography-Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2017, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Yap, S.; Barry-Kinsella, C. Maternal pyridoxine non-responsive homocystinuria: The role of dietary treatment and anticoagulation. BJOG 2001, 108, 425–428. [Google Scholar] [CrossRef]
- Sacharow, S.J.; Picker, J.D. Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency; 2004; [updated 2017 May 18]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews® [Internet]; Seattle (WA), University of Washington: Seattle, DC, USA, 1993–2023. [Google Scholar] [PubMed]
- Varlibas, F.; Cobanoglu, O.; Ergin, B.; Tireli, H. Different phenotypy in three siblings with homocystinuria. Neurologist 2009, 15, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Mourot-Cottet, R. Regressive pyridoxine-induced sensory neuronopathy in a patient with homocystinuria. BMJ Case Rep. 2018, 2018, bcr2018225059. [Google Scholar] [CrossRef]
- Saudubray, J.M. Inborn Metabolic Diseases: Diagnosis and Treatment, 7th ed.; Springer: Berlin, Germany, 2022. [Google Scholar]
- Hemminger, A.; Wills, B.K. Vitamin B6 Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Adam, S.; Almeida, M.F. Dietary practices in pyridoxine non-responsive homocystinuria: A European survey. Mol. Genet. Metab. 2013, 110, 454–459. [Google Scholar] [CrossRef]
- Yap, S.; Naughten, E. Homocystinuria due to cystathionine beta-synthase deficiency in Ireland-25 years experience of a newborn screened and treated population with reference to clinical outcome and biochemical control. J. Inherit. Metab. Dis. 1998, 21, 738–747. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Mar, M.H. Concentrations of choline-containing compounds and betaine in common foods. J. Nutr. 2003, 133, 1302–1307. [Google Scholar] [CrossRef] [Green Version]
- Diamant, S.; Rosenthal, D. Dicarboxylic amino acids and glycine-betaine regulate chaperone-mediated protein-disaggregation under stress. Mol. Microbiol. 2003, 49, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Krijt, J. Restoring assembly and activity of cystathionine beta-synthase mutants by ligands and chemical chaperones. J. Inherit. Metab. Dis. 2011, 34, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, A.; Sakura, N. Limited effectiveness of betaine therapy for cystathionine beta synthase deficiency. Pediatr. Int. 2003, 45, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.H.; Faiyaz-Ul-Haque, M. Clinical and molecular findings of 13 families from Saudi Arabia and a family from Sudan with homocystinuria. Clin. Genet. 2012, 81, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Sebastio, G.; Sperandeo, M.P. The molecular basis of homocystinuria due to cystathionine beta-synthase deficiency in Italian families, and report of four novel mutations. Am. J. Hum. Genet. 1995, 56, 1324–1333. [Google Scholar]
- Kluijtmans, L.A.; van den Heuvel, L.P. Molecular genetic analysis in mild hyperhomocysteinemia: A common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am. J. Hum. Genet. 1996, 58, 35–41. [Google Scholar]
- Gazzetta Ufficiale. 167/2016. Available online: https://www.gazzettaufficiale.it/eli/id/2016/08/31/16G00180/sg (accessed on 10 December 2022).
- Dondi, A.; Candela, E. Parents’ Perception of Food Insecurity and of Its Effects on Their Children in Italy Six Months after the COVID-19 Pandemic Outbreak. Nutrients 2020, 13, 121. [Google Scholar] [CrossRef]
- Cruysberg, J.R.; Boers, G.H. Delay in diagnosis of homocystinuria: Retrospective study of consecutive patients. BMJ. 1996, 313, 1037–1040. [Google Scholar] [CrossRef] [Green Version]
- Loeber, J.G.; Burgard, P. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J. Inherit. Metab. Dis. 2012, 35, 603–611. [Google Scholar] [CrossRef]
- Okun, J.G.; Gan-Schreier, H. Newborn Screening for Vitamin B6 Non-responsive Classical Homocystinuria: Systematical Evaluation of a Two-Tier Strategy. JIMD Rep. 2017, 32, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, E.G.; Scott, A.J. A cautionary tale of pyridoxine toxicity in cystathionine beta-synthase deficiency detected by two-tier newborn screening highlights the need for clear pyridoxine dosing guidelines. Am. J. Med. Genet. A 2020, 182, 2704–2708. [Google Scholar] [CrossRef] [PubMed]
- Homocystinuria (HCU) Dietetic Management Pathway. Available online: https://www.bimdg.org.uk/store/enbs/HCU_Dietetic_Management_Pathway_V1_April_2015_215380_12052015.pdf (accessed on 30 December 2022).
- Imbard, A.; Toumazi, A. Efficacy and pharmacokinetics of betaine in CBS and cblC deficiencies: A cross-over randomized controlled trial. Orphanet. J. Rare Dis. 2022, 17, 417. [Google Scholar] [CrossRef] [PubMed]
- Schwahn, B.C.; Schefner, T. Cystathionine beta synthase defciency and brain edema associated with methionine excess under betaine supplementation: Four new cases and a review of the evidence. JIMD Rep. 2020, 52, 3–10. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Gestational Age Birth Weight | I DBS MET μmol/L | 2TT DBS Hcy | Plasma tHcy (μmol/L) | Plasma MET (μmol/L) | |
|---|---|---|---|---|---|
| Patient 1 | 38 weeks | ||||
| 102.51 | Positive | 95.6 | 727.15 | ||
| 3840 g | |||||
| Patient 2 | 40 weeks 3895 g | 45.02 | Positive | 105.8 | 93.46 |
| Patient 3 | 39 weeks | 599.99 | |||
| 3030 g | 79.12 | Positive | 204.3 |
| Ethnic Group | Variant Allele V1 | Variant Allele V2 | HGMD | ACMG | Reference | |
|---|---|---|---|---|---|---|
| Patient 1 | North Africa | |||||
| c.969 G > A | c.969 G > A | P | P | [60] | ||
| Patient 2 | Caucasian | c.770 C > T | c.1330 G > A | V1: P V2: P | V1: LP V2: LP | V1: [61] V2: [62] |
| Patient 3 | Caucasian | |||||
| A | V1: LP V2: LP | A | ||||
| c.1333 C > T | c.1219_1223 + 5del |
| Plasma tHcy after 14 days Pyridoxine Trial μmol/L | Current Treatment | |
|---|---|---|
| Patient 1 | 83.5 (−12.6%, NR) | Pyridoxine (10 mg/kg/day) + OH-B12(1000 mcg) + Folate (5 mg) from the onset. Diet therapy from 1 month of life (Met 9 mg/kg, Natural Protein Intake 0.57 g/kg/die, Total Protein Intake 2.2 g/kg/die). Betaine (80 mg/kg) from 45 days of life |
| Patient 2 | 76.9 (−27%, PR) | Pyridoxine (10 mg/kg/day) + OH-B12(1000 mcg) + Folate (5 mg) from the onset. Diet therapy from 2 months of life (Met 20 mg/kg, Natural Protein Intake 1.2 g/kg/die, Total Protein Intake 2.6 g/kg/die). Betaine (90 mg/kg) from 75 days of life |
| Patient 3 | 113.3 (−44%, PR) | Pyridoxine (10 mg/kg/day) + OH-B12 (1000 mcg) + Folate (5 mg) from the onset. Diet therapy from 1 month of life (Met 10 mg/kg, Natural Protein Intake 0.7 g/kg/die, Total Protein Intake 2.7 g/kg/die). Betaine (105 mg/kg) from 60 days of life |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candela, E.; Zagariello, M.; Di Natale, V.; Ortolano, R.; Righetti, F.; Assirelli, V.; Biasucci, G.; Cassio, A.; Pession, A.; Baronio, F. Cystathionine Beta-Synthase Deficiency: Three Consecutive Cases Detected in 40 Days by Newborn Screening in Emilia Romagna (Italy) and a Comprehensive Review of the Literature. Children 2023, 10, 396. https://doi.org/10.3390/children10020396
Candela E, Zagariello M, Di Natale V, Ortolano R, Righetti F, Assirelli V, Biasucci G, Cassio A, Pession A, Baronio F. Cystathionine Beta-Synthase Deficiency: Three Consecutive Cases Detected in 40 Days by Newborn Screening in Emilia Romagna (Italy) and a Comprehensive Review of the Literature. Children. 2023; 10(2):396. https://doi.org/10.3390/children10020396
Chicago/Turabian StyleCandela, Egidio, Michele Zagariello, Valeria Di Natale, Rita Ortolano, Francesca Righetti, Valentina Assirelli, Giacomo Biasucci, Alessandra Cassio, Andrea Pession, and Federico Baronio. 2023. "Cystathionine Beta-Synthase Deficiency: Three Consecutive Cases Detected in 40 Days by Newborn Screening in Emilia Romagna (Italy) and a Comprehensive Review of the Literature" Children 10, no. 2: 396. https://doi.org/10.3390/children10020396