
Chemical Vapor Deposition of Zirconium Compounds: A Review

Oak Ridge National Laboratory, Materials Science and Technology Division, Oak Ridge, TN 37830, USA
*
Author to whom correspondence should be addressed.
Coatings 2023, 13(2), 266; https://doi.org/10.3390/coatings13020266
Submission received: 2 December 2022
/
Revised: 13 January 2023
/
Accepted: 17 January 2023
/
Published: 23 January 2023
(This article belongs to the Special Issue Chemical Vapor Deposition (CVD) of Coatings and Films)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Coatings of zirconium compounds are used in a wide variety of fields, yet an understanding and descriptions of deposition mechanisms are scant in the public literature. The mechanisms of deposition for metallic zirconium, ZrC, ZrN, ZrO2, ZrB2, and zirconium silicides are discussed based on the direct vapor deposition research of those compounds where possible or compared to complementary titanium systems when direct research is lacking. Both inorganic and organometallic deposition systems are discussed. As a class of compounds, an understanding of the vapor deposition mechanisms can be significantly improved by investigations on metallic zirconium deposition by zirconium halides and hydrogen and by in situ analysis techniques such as Fourier-transform infrared (FTIR) spectroscopy or x-ray photoelectron spectroscopy (XPS).
1. Introduction
Coatings of zirconium compounds, such as ZrC, ZrN, ZrO2, ZrSi2, and ZrB2, have been studied for use in a variety of applications in the fields of aerospace, nuclear, and microelectronics. Despite the interest in Zr-based coatings over the past seventy years [1], there is very little published on the kinetics and mechanisms of deposition for many of these materials. Understanding the deposition chemistry of the target material and byproducts is fundamental to controlling the reaction.
In nuclear science, zirconium metal alloys and compounds are used in a wide array of applications due to the very low thermal neutron capture cross-section of zirconium [2]. Zirconium and zirconium alloys have been used in fuel and fuel cladding [3,4,5,6,7], as well as for structural applications [2], ZrH2 in hydride fuels and as a neutron reflector [8,9,10,11], ZrC as cladding for TRISO fuel particles [12,13], ZrN as the matrix in nitride fuels [14,15], zirconium silicides as neutron reflectors and cladding [16,17,18], and ZrO2 in matrix material in fast reactors and as fuel cladding [11,19,20,21]. Zirconium compounds have also been used outside of nuclear science. The refractory nature of many zirconium compounds combined with favorable mechanical and electrical properties lends them to a variety of specialty applications. Tool tips and coatings have been made from ZrB2 and ZrN [22,23,24,25,26], and optical coatings from ZrN and ZrO2 [27,28,29]. In microelectronics, zirconium silicides can be used as ohmic contacts on silicon [30,31]. Diffusion barriers have been made from ZrB2 and ZrN [32,33,34], and ZrN can also be used in diodes, transistors, and low-resistance contacts [35,36]. The low electrical conductivity and chemical inertness of ZrO2 make it advantageous for transistor insulation and in gate dielectrics [37,38]. Lastly, ZrO2, ZrB2, and ZrC have all been investigated or used as thermal barrier coatings in aerospace [39,40,41,42,43]. All of these applications are specifically for zirconium compounds, and ignore the usage of equivalent compounds of hafnium. It should be noted that zirconium and hafnium are chemically identical [44], so many if not all, of the chemical behaviors identified in the processing and handling of zirconium, also apply to hafnium and its compounds.
Following a description of chemical vapor deposition (CVD) and the chemical behavior of zirconium compounds, this review discusses the vapor deposition of some binary Zr-based coatings. Where possible, the current understanding of deposition kinetics and reaction mechanisms for zirconium compounds are discussed. When knowledge of a zirconium system is lacking, comparisons to complementary Group IV compounds (i.e., titanium and hafnium systems) are made to predict behavior for zirconium deposition. The surface chemistry and production of zirconium have previously been reviewed [45,46], and several sources cover the chemical behavior and historical metallurgy of zirconium and zirconium compounds [47,48]. To the best of the authors’ knowledge, this is the first review of the CVD of zirconium compounds.
1.1. Chemical Vapor Deposition
CVD has been defined as the deposition of a solid on a heated surface from a chemical reaction in the vapor phase and belongs to the class of atomistic vapor-transfer processes (i.e., deposition of species as atoms or molecules, or in a combination of these) [49,50]. The design and components of CVD reactors, the chemistry of general CVD reactions, the history of CVD developments, and reviews of the CVD of several classes of materials are discussed thoroughly elsewhere [51,52,53]; a brief description is included here.
As defined, CVD requires the following few general components: a heated substrate, a controlled vapor environment, and a supply of volatile precursors [50,54,55]. Precursors—the chemical reactants or starting materials—can be inorganic, organometallic, or a combination of these. The liquid and solid precursors can be used by heating them above room temperature and transporting them to the reactor by carrier gases, such as argon, nitrogen, or hydrogen. The vapor environment is typically an air-tight vessel, such as a quartz or metal tube with vacuum flanges connected to a vacuum pump to control the system pressure, but it can be a completely sealed vessel. Finally, the heating of substrates is achieved typically by either “hot-wall” or “cold-wall” systems. Hot-wall systems use a heating source to heat the pressure vessel, or a section of the vessel, to radiantly heat the substrate; for example, a clam-shell tube furnace. Cold-wall systems selectively heat just the substrate, which can be achieved by resistive or RF induction heating of the substrate directly or by mounting the substrate to a heater plate.
The fundamental kinetics and thermodynamics of CVD are covered in more detail elsewhere, but the general CVD reaction can be divided into five stages [56]. (1) Reactant gases first enter the reactor by forced flow before (2) diffusing through the boundary layer (the layer between the bulk gas flow and the zero-velocity gas at the substrate surface). (3) Gases come into contact with the substrate surface, then (4) deposition reactions take place on the surface. Finally, (5) gaseous byproducts diffuse away from the substrate through the boundary layer. The slowest step in the sequence determines the reaction rate and is generally either surface reaction kinetics (step 4) or mass transport (step 2). The parameters that affect the rate-limiting step in this sequence—temperature and pressure—also affect the microstructure of the deposit.
Deposit microstructure is relevant in any coating application, as the microstructure directly affects the thermomechanical properties of the film. Based on the results of sputtering studies for a variety of metals, [57,58] the microstructure formed can be related to both the temperature of the substrate and the partial pressure of reactants. Generally, higher temperatures increase surface and/or bulk diffusion, thereby improving the organization of surface species and increasing grain structure. Likewise, the reactant partial pressure affects microstructure. Low partial pressures can produce voids at grain boundaries, while high partial pressures (especially at higher temperatures) promote gas phase reactions and soot formation. Additional information about the control of CVD microstructure can be found elsewhere [56,59]; as microstructure development is largely independent of the chemical mechanism of deposit formation, it is beyond the scope of this review.
CVD is adaptable to many process variations [49], allowing for changes in the elemental composition of the deposit, and does not normally require an ultrahigh vacuum. It is not restricted to line-of-sight deposition, as is typical of physical vapor deposition processes such as sputtering. However, CVD equipment can be costly to operate and maintain; precursors are inherently reactive, often flammable, and sometimes extremely toxic; many CVD processes are most versatile above 600 °C, which excludes many substrates. Some of these limitations are overcome by using lower-temperature techniques, such as plasma-enhanced CVD (PE-CVD) [54,60,61], laser-assisted CVD (LA-CVD) [55,60,61], or metal-organic CVD (MO-CVD) [62]. As discussed further in later sections, MO-CVD behaves chemically much the same way as CVD with inorganic precursors—via thermal excitation to decompose or otherwise activate species for reaction in either the gas phase or on the substrate surface. The key difference between organometallic and inorganic precursors is the temperatures needed for a reaction to occur; organometallic compounds generally are more reactive than, for example, metal halides, and can form deposits on lower-temperature capable substrates. PE- and LA-CVD use plasma or lasers to form highly reactive species to deposit on low-temperature or even room-temperature substrates. The chemical reactions and kinetics of these processes are significantly different than what are found in conventional thermal CVD and are beyond the scope of this review.
1.2. Chemistry of Zirconium Compounds
Covalent materials such as SiC have very narrow stoichiometric ranges before becoming biphasic as follows: silicon-rich compositions produce Si-SiC mixtures and carbon-rich compositions produce C-SiC mixtures. In contrast, zirconium intermetallic compounds generally exist as solid solutions of, for example, carbon in metallic zirconium to form zirconium carbide (ZrC) [63]. Small elements such as boron, nitrogen, carbon, and oxygen can all readily dissolve into zirconium to occupy octahedral sites either singly (ZrB, ZrN, ZrC, etc.) or as mixtures such as zirconium cyanonitride, Zr(O,C,N) [63]. In some cases, the additional filling can occupy tetrahedral sites by a rhombohedral distortion of the original rock-salt crystal structure [63,64]. Stoichiometric compositions exist solely on geometry and not from balancing of valences—pure ZrC exists in a C/Zr range of 0.58-0.98 [65]—while the N/Zr ratio in zirconium nitride can range from 1.03 (ZrN) to 1.35 (Zr3N4) [64]. Historically, researchers attempted to characterize the composition of intermetallic zirconium compounds by X-ray diffraction (XRD), using the lattice parameters to estimate stoichiometry. However, samples are easily contaminated—for example, carbon vacancies in ZrC being filled by oxygen or nitrogen from the air. Since each species (carbon, oxygen, nitrogen, or vacancy) affects the lattice parameters differently, it is unreliable to use only XRD to determine stoichiometry. Recent efforts have largely overcome this limitation by augmenting XRD with other analytical methods [66,67].
Larger elements from the second row or higher in the periodic table, such as silicon, are too large to occupy interstitial sites and form semimetallic compositions, combining metallic and covalent bond characteristics and maintaining most of the original metallic zirconium structure. Semimetallic compositions can also have a range of compositions, as in the case of the zirconium silicides with Si/Zr ranging from 0.25 (Zr4Si) to 2 (ZrSi2) [63].
Covalent zirconium compounds can be formed from the halogens [63], and direct halogenation of zirconium is one of the most common methods of introducing zirconium halides to vapor deposition processes [68]. Zirconium halides are also a key intermediate product in the refining of zirconium metal and the separation of zirconium from hafnium [46]. The purification of zirconium is primarily important for nuclear applications, where the high neutron-capture cross section of hafnium is detrimental [46]. Otherwise, the chemical similarities between zirconium and hafnium are close enough that the naturally occurring Zr-Hf (1–3% Hf) can be used directly after refining from ore [44,46].
2. Metallic Zirconium
Early reports on the vapor deposition of metallic zirconium started in 1896, initially as studies on plating metallic filaments prior to evolving into methods for zirconium purification [69,70,71,72,73]. The van Arkel-de Boer iodide process (1920s) [72,73], discussed in more detail elsewhere [46,74], was scaled up by the 1950s as part of the development of nuclear reactors [1,75]. Improvements to the Kroll process [76] led to the replacement of the iodide process for the purification of bulk zirconium, but metallic zirconium coatings by zirconium iodides, chlorides, and bromides have continued [46].
In the van Arkel-de Boer iodide process, a sealed, evacuated bulb is charged with ZrI4 or I2 and crude zirconium. The iodine or iodide is heated along with a zirconium or tungsten filament; ZrI4 vapor is generated either by evaporation or by reaction of iodine with the crude metal source. This was originally accomplished with filament temperatures of 2000 °C [72,73], but this high temperature was decreased to 1200-1300 °C with further process development [44]. The zirconium iodide vapor deposits purified metallic Zr on the hot filament and (re)generates I2 as follows [74,77]:
As the process was scaled up, glass bulbs were replaced with metallic containers, and the process was adopted to be dynamic instead of static [74].
Deposition from Zirconium Halides
Studies on the deposition of zirconium from its halides have found that describing the reactions generally as decomposition (Equation (2)) or reduction (Equation (3)) reactions [1] does not fully represent the chemistry of this system as follows [77]:
Instead, the iodides [77,78], chlorides [79], and bromides [80] of zirconium are interconnected with their respective subhalides by disproportionation reactions, as described for ZrI4 (Equation (4)) as follows [77]:
These equilibria for the disproportionation of zirconium halides contribute significantly to low-temperature (<800 °C) deposition of zirconium, and to a lesser extent at higher temperatures, as decomposition or reduction mechanisms become preferential. Regardless of temperature, the formation of subhalides is a critical step in zirconium deposition.
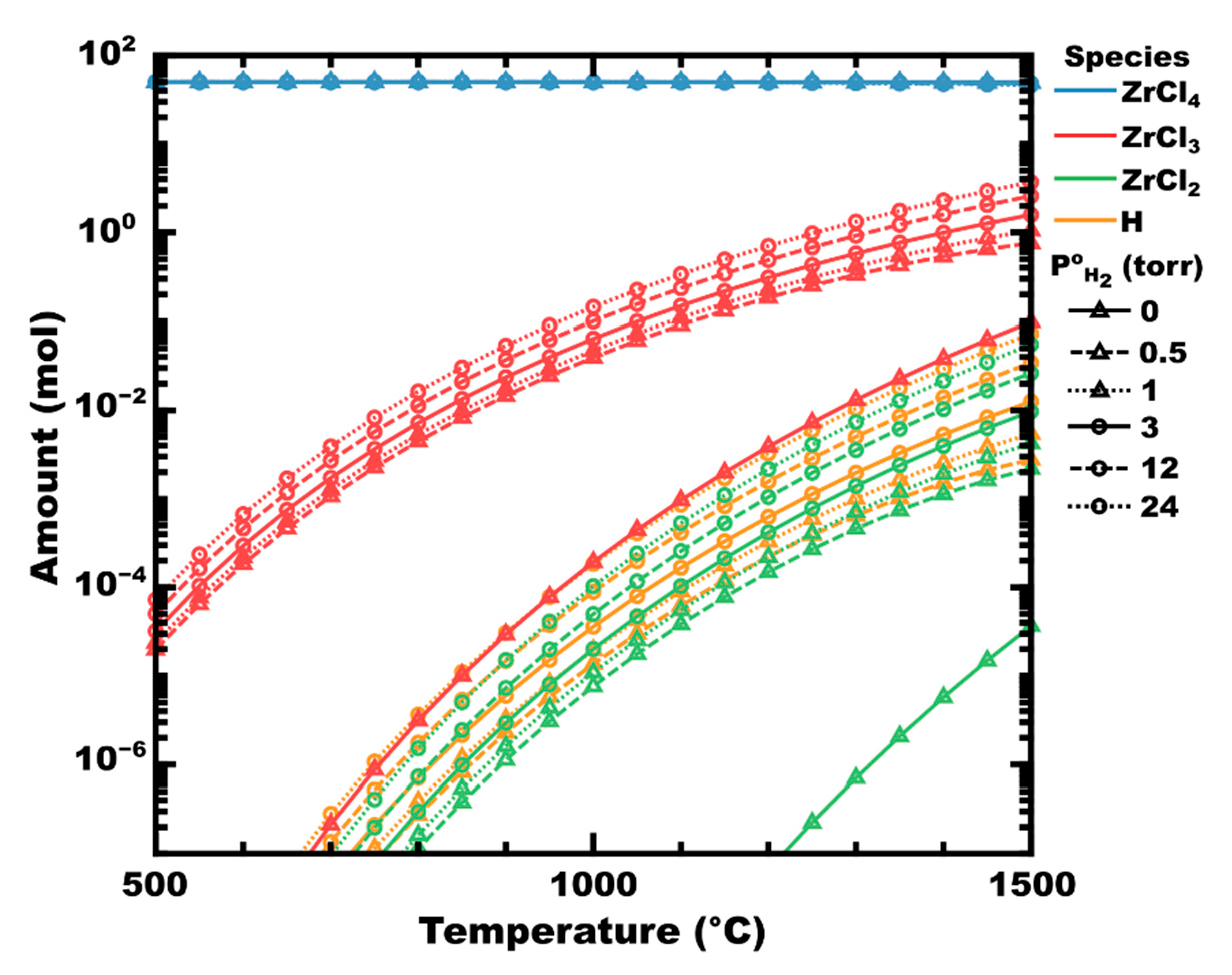
Examining the decomposition of zirconium tetrachloride, two paths are expected to occur depending on the absence or presence of hydrogen. In the absence of hydrogen, ZrCl4 can be expected to dissociate into subchlorides of zirconium and chlorine, as shown in Figure 1 (initial hydrogen partial pressure, P°H2 = 0, shown as solid lines with triangles). At P°H2 = 0.5 torr (dashes with triangles), corresponding to a hydrogen mole fraction (χH2) of 0.01, the partial pressure of zirconium chlorides is significantly higher than in the absence of hydrogen. For example, at 700 °C, there is approximately 0.1 µmol of ZrCl3 at equilibrium in the absence of hydrogen (red solid line with triangles). However, at P°H2 0.5 torr, the equilibrium amount of ZrCl3 has increased to 107 µmol, an increase of three orders of magnitude. As temperature increases, thermal decomposition (vs. chemical decomposition) increases, but even at 1500 °C, there is still nearly an order of magnitude increase in the equilibrium amount of ZrCl3 as P°H2 increases from 0 to 0.5. Beyond this initial spike, the increase in subchloride partial pressure is less significant. In the absence of a substrate (e.g., Zr metal) to drive the reaction forward by disproportionation, no condensed phases are predicted to form thermodynamically (calculated in FactSage 8.1).
Atomic hydrogen radicals have been shown to significantly reduce the activation energy of dissociation reactions, for example, in thermal CVD of CH4 or plasma-enhanced CVD of BCl3 [81,82,83,84,85]. Accordingly, the addition of H2 to ZrCl4 might improve the dissociation rate of chlorides via the abstraction of chlorine by atomic hydrogen (Equation (5)). Indeed, the equilibrium amount of atomic hydrogen in Figure 1 is calculated to increase to a similar degree as the zirconium chlorides as P°H2 increases (i.e., ×10 to ×100, depending on temperature).
With the formation of zirconium subhalides and the subsequent adsorption to a substrate, the further reduction could proceed by either a continued disproportionation (Equation (4)) or, in the presence of hydrogen, a hydrogen-mediated surface reaction could occur similar to the formation of silicon from SiCl2-H2 [86,87]. However, there appears to be no information in the published literature on heterogeneous reactions in the deposition of zirconium halides in the presence of hydrogen.
3. Zirconium Carbide
The carbide of zirconium has been of particular interest to the nuclear community as a particle fuel coating [68,88,89,90,91,92,93], as well as a thermal barrier coating for nuclear thermal propulsion [65,94,95,96]. Accordingly, the deposition of ZrC has been the subject of both experimental and theoretical studies.
Studies have primarily focused on ZrX4-CxHy-H2 systems. The most popular system by far has been ZrCl4-CH4-H2 [68,89,90,94,97,98,99,100,101,102,103,104]; however, investigations have included zirconium bromide or iodide [91,105,106,107,108], as well as other hydrocarbons such as propane and hexane [68,89,90,103]. Typical deposition conditions are temperatures of 1000–1400 °C; total reaction pressures are reported up to 760 torr; however, published partial pressures of zirconium halides are consistently below 50 torr and are typically around 25 torr.
Despite its use as a critical material in the nuclear industry that has garnered international research interest for over half a century, the understanding of the mechanisms for the deposition of ZrC remains inadequate. There is currently no published research analyzing the surface chemistry—the chemical reactions occurring on the coating surface—of this system. Currently, the mechanisms proposed are as follows: (i) a solid-state diffusion mechanism and (ii) a “droplet” mechanism.
3.1. Diffusion Mechanism
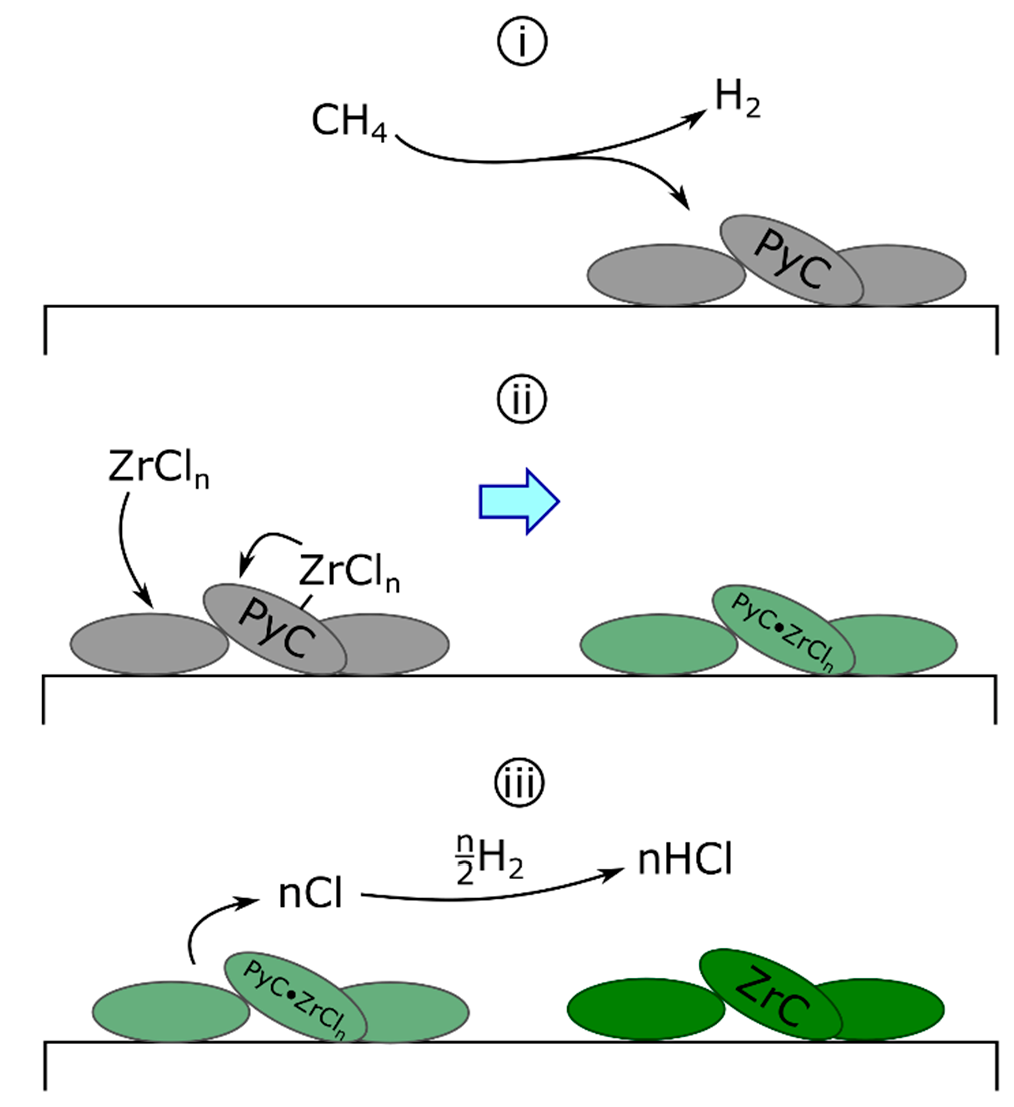
The solid-state diffusion mechanism was first proposed for the deposition of ZrC on graphite tubes by ZrCl4-H2-HCl [94]. Initially, the carbon source was the substrate itself, diffusing through the building zirconium layer to form the carbide. This concept was later altered for the codeposition of zirconium and carbon (Scheme 1) as follows: the separate deposition of carbon and zirconium, where pyrolytic carbon (PyC) deposition is described generically (Equation 6a) and hydrogen reduces zirconium chlorides homogeneously to deposit fully reduced zirconium (Equations (6b) and (6c)) as follows [68,88,89]:
Following the initial deposition, zirconium is proposed to diffuse into the previously deposited graphite to form the carbide.
Studying the effect of gas flow rates and bed area in the fluidized bed CVD of oxide particles [88], Wagner and colleagues observed that increasing the partial pressure of hydrogen (PH2) improved the “metallic luster” of the deposit while increasing PCH4 dulled the appearance of the coating. Coating appearance was a qualitative representation of the amount of free carbon in the C-ZrC deposits formed. From these results, Wagner and colleagues concluded that the improvement in deposit luster was due to an increase in zirconium deposition, described generally in Equation (6c) and that hydrogen could not be influencing the reaction by any other mechanism [88].
The diffusion mechanism proposed by Wagner and colleagues ignores the effect of hydrogen on carbon deposition. In a series of works investigating the CVD of PyC [81,109], the Hüttinger group observed significant inhibition of PyC deposition by hydrogen. From their analysis, Hüttinger and co-workers propose that small, stable hydrocarbons form progressively heavier species via unsaturated or radical intermediates. Increasing PH2 works to inhibit this mechanism by pushing reactants towards stable species such as CH4 or C2H6. Additionally, surface-adsorbed hydrogen can inhibit the deposition of heavier hydrocarbons by blocking surface sites [110]. The diffusion mechanism also ignores the significant difference in vapor pressure of ZrCl4 compared to ZrCl3 or ZrCl2. Even under reduced pressure, both subchlorides are much more likely to condense (i.e., vapor deposit) compared to the tetrachloride [79], suggesting a heterogeneous adsorption process. Finally, carbon is more likely to diffuse into zirconium than vice versa [63], as was originally suggested by Wallace [94], as demonstrated by tracer diffusion studies in β-Zr and ZrCx [111].
3.2. Droplet Mechanism
First proposed in 1979 for ZrC [107], the droplet mechanism continues to be referenced in the literature [100,103,112,113,114,115]. The defining feature of this mechanism (Scheme 2) is the formation of polyunsaturated (low hydrogenation) liquid or plastic carbonaceous droplets on the surface of the substrate via Equation (7a). Zirconium chlorides from the surface or reaction gas then diffuse into the droplet [100] (Equation (7b)) prior to forming ZrC and releasing atomic chlorine (Equation (7c)). Finally, evolved atomic chlorine reacts with hydrogen to form HCl (Equation (7d)) [103].
This mechanism is essentially the diffusion mechanism modified by including a mechanism for carbon deposition proposed by Grisdale and colleagues in the early 1950s [116,117]. In Grisdale’s work at Bell Labs, a tumbling bed of ceramic substrates (e.g., beads or rods) were heated to a range of 975–1300 °C; once at temperature, a hydrocarbon source diluted by nitrogen was introduced to deposit carbon black. At a “suitably high” hydrocarbon partial pressure, a sooty fog developed in the furnace [116]. The sooty droplets were thought to form from homogeneous nucleation of heavy hydrocarbons, eventually condensing onto the substrates as complex hydrocarbon plastic droplets. Dehydrogenation continued to form a carbonized coating [117].
The PyC droplet mechanism, as proposed for the work at Bell Labs, is consistent with more contemporary studies on PyC deposition. As the residence time increases, progressively heavier hydrocarbons form, including aromatics such as benzene, naphthalene, and indene within a 1-s residence time from a methane source gas [81]. Thus, a combination of high hydrocarbon partial pressures and long residence times could be expected to form sooty products in the gas phase. However, the CVD ZrC studies proposing the droplet theory [100,103,107,112,113] have sufficiently low hydrocarbon partial pressures that soot formation is unlikely. Additionally, Zr-Cl bonds are quite stable, with standard enthalpies of formation around −250 kJ/mol [118]. Compared to the standard enthalpy of formation for atomic chlorine (121.2 kJ/mol) [118], Zr-Cl bonds are unlikely to decompose to atomic chlorine, requiring around 370 kJ/mol per Zr-Cl bond. In comparison, the experimental enthalpy of formation for ZrCx ranges from −146 to −204 kJ/mol as ‘x’ increases from 0.63 to 0.98 [119], implying a more exothermic mechanism than the droplet mechanism. Lastly, the arguments against the diffusion mechanism—e.g., carbon diffusing into zirconium versus zirconium into carbon—apply to the droplet mechanism as well.
3.3. The Role of Hydrogen
Some of the earliest studies on CVD ZrC have suggested that zirconium and carbon deposit independently [90,91,105], supporting a separate treatment of zirconium and carbon deposition mechanisms. The effect of hydrogen on ZrCl4 in the deposition of zirconium has been described in a previous section, and a discussion of the effect of hydrogen on PyC deposition follows.
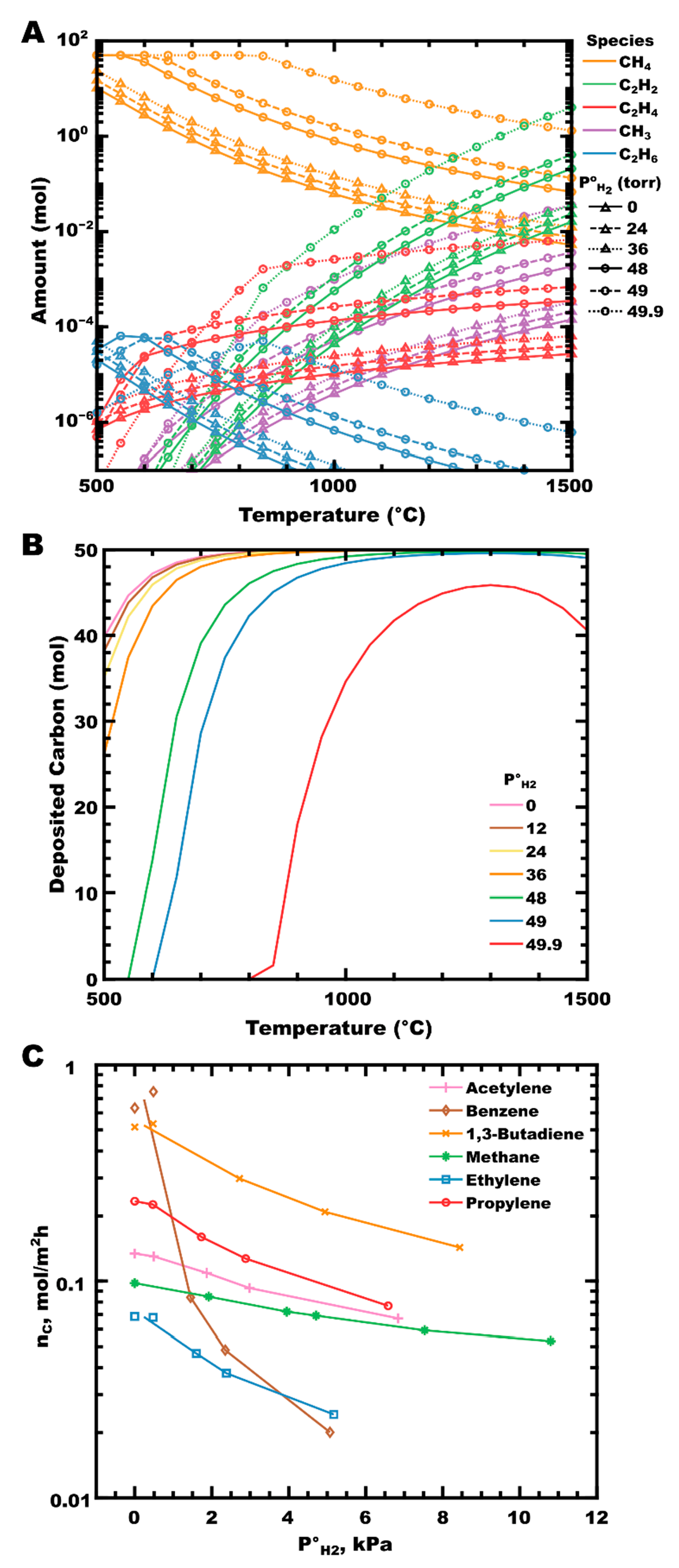
An examination of CH4-H2 mixtures by calculating thermodynamic equilibrium (Figure 2A) shows the increase in the moles of hydrocarbon at equilibrium as the P°H2 increases. In the absence of hydrogen (solid lines with triangles), methane (gold curves) dissociates primarily into a methyl radical (purple curve), with further homogeneous reactions forming acetylene, ethene, and ethane as the majority gaseous reaction products (green, red, and blue curves, respectively). Very little change in the equilibrium partial pressures is observed, even up to P°H2 of 24 torr (dashed lines with triangles; equivalent to a hydrogen mole fraction of χH2 = 0.48). The calculated amount of deposited PyC has decreased slightly over this range (Figure 2B, pink vs. yellow curves). As the initial partial pressure of hydrogen increases further (P°H2 ≥ 48 torr, χH2 ≥ 0.96), the amount of deposited PyC decreases significantly with a corresponding increase in the partial pressures of all hydrocarbon species. Hydrogen participates in the equilibrium reactions occurring in the gas phase, shifting the equilibrium towards (less reactive) saturated hydrocarbons, and can impact the reaction kinetically by inhibiting surface deposition by blocking surface sites, as observed experimentally by the Hüttinger group in the deposition of PyC from several hydrocarbons (Figure 2C) [81,110,120]. In their work, PyC deposition rates decrease in proportion to increasing P°H2 for all hydrocarbon sources reported. Benzene is particularly affected. This feature was attributed by Becker, and colleagues to the preferential adsorption of benzene parallel to a surface (compared to adsorption end-on or perpendicular to the surface), resulting in increased contact surface area. As the coverage of atomic hydrogen on the surface increases, more surface sites are blocked per unit surface area, reducing the area available for benzene to adsorb in the lowest energy orientation [110]. Additional experimental support can be found when comparing the results of ZrC deposition to thermodynamic predictions, where a greater suppression of carbon deposition is observed with increasing hydrogen concentration than is predicted from thermodynamic equilibrium alone [88,91,104,106,121].
Once zirconium and carbon species are bound to the surface, several possible reaction paths can be envisioned. Zirconium subchlorides and methyl groups, for example, could alternate binding (Zr to C, C to Zr), reacting to produce hydrogen chloride and thus form new surface sites via a Langmuir-Hinshelwood mechanism, as is seen in silicon carbide deposition [122]. Alternatively, carbon and zirconium could nucleate independently, followed by ZrC formation via solid-state diffusion of carbon into zirconium grains, consistent with the deposition of ZrN from ZrCl4 in the presence of nitrogen or ammonia and with the deposition of ZrB2 from ZrCl4-BCl3-H2 (vide infra). However, without a systematic study of surface chemistries and growth mechanisms, the discussion of CVD ZrC mechanics is necessarily limited.
4. Zirconium Nitride
Group IV nitrides can form as cubic MN or rhombohedral M3N4, where M = Ti, Zr, Hf [64,123]. The cubic phases are metallic, extremely hard, high melting (~3000 °C), conductive, and chemically stable materials [123]. These properties make them attractive for such applications as cutting tools, optical coatings, microelectronic contacts, and diffusion barriers. When deposited by CVD, ZrN has been grown by either inorganic (ZrCl4-N2-H2 or ZrCl4-NH3) [124,125] or organometallic [64,123,126,127,128,129,130,131] (e.g., Zr(N(CH3)2)4-NH3) routes.
4.1. Inorganic CVD
Zirconium nitride can be deposited from ZrCl4 in the presence of N2-H2 mixtures or NH3 alone. Similar to the deposition of ZrC discussed above, little analysis has been published on the surface chemistry of ZrN deposition from these systems.
In the deposition of ZrCl4-NH3 [125], the formation of ZrN powder was possible at >1000 °C. Lower temperatures produced Zr3N4, ZrClN, and/or ZrCl4•2NH3 adducts depending on the reaction temperature. Earlier studies exposing ZrX4 powders (X = Cl, Br, I) to ammonia also identified an initial formation of ZrCl4•4NH3 adduct followed by decomposition to Zr3N4 [63]. It was proposed that ZrCl4 first reacts with ammonia to form ZrCl4•2NH3 [125]. The adduct reacts with additional ammonia to form ZrClN and NH4Cl. ZrClN, in turn, reacts with ammonia to form ZrNx, Zr3N4, and NH4Cl. At approximately 900–1000 °C, the ratio of N/Zr in the deposit begins to decrease. The Zr3N4-ZrN1.28 mixture observed at ~700 °C shifted to phase-pure ZrN1.28 by 1100 °C. Further increases in temperature continued to alter the N/Zr ratio, decreasing to ZrN1.12 by 1400 °C. A stable ZrN1.03 phase could be isolated from ZrN1.28 deposits by annealing in argon at ≥1100 °C [125]. The stoichiometry of compounds formed in this study was evaluated by both XRD and gravimetric chemical analyses, with an estimated accuracy for the atomic ratio N/Zr of ±0.01.
The addition of hydrogen to the ammonia-based system (i.e., ZrCl4-NH3-H2) should improve the efficiency of ZrN deposition. Replacing the argon carrier gas [125] with hydrogen would promote the formation of zirconium subchlorides and improve the efficiency of zirconium deposition, as discussed in the section on zirconium metal deposition. Mixtures of NH3-H2 are also used in gas nitriding. Gas nitriding is treated theoretically by the sum of Equations (8a) and (8b) (giving overall Equation (8c)) as follows [132]:
where [N] represents dissolved nitrogen. These reactions can be used to derive the chemical potential of nitrogen in the system, which is directly related to the partial pressures of ammonia and hydrogen [132,133]. Thus, by controlling the ratio of ammonia to hydrogen in the gas stream, the nitriding potential of the system can be manipulated. It is worth noting that thermodynamically, Equations (8a) and (8b) are accurate—at equilibrium, ammonia is predicted to be completely dissociated at >450 °C. However, reaction kinetics dominate the nitriding process under typical conditions [134], and experimental values of ammonia dissociation have been shown to be <20% at 600 °C [134]. Additionally, the fugacity of the hypothetical N2 gas in Equation (8b) has been calculated as several gigapascals for the equilibrium between Fe and NH3-H2, indicating that nitrogen gas is less suitable than ammonia for producing nitrides from metals [134]. Therefore, representing nitriding by Equation (8c) is appropriate in dynamic systems [134,135].
While Group IV nitride formation in the presence of nitrogen is less efficient than ammonia, examples of the growth of ZrN, HfN, and TiN from N2/H2 gas mixtures have been published. In the work of van Arkel and de Boer to form high-purity metals [72], the authors reported that metal nitride coatings grew to a limited extent on resistively heated tungsten filaments in the presence of trace nitrogen in hydrogen—e.g., ZrN growth from ZrCl4 + H2/N2. Unfortunately, no further details of the reaction (temperature, pressures, gas ratios, etc.) were included in the manuscript. Subsequent work by Moers targeting the growth of refractory nitrides is more informative [136]. Here, nitrides of zirconium, hafnium, and titanium were grown from metal chlorides in the presence of ammonia, N2/H2 mixtures, or pure N2—the order of gases corresponding to ease of deposition. While the temperature of the resistively heated tungsten filament was not stated for the ZrCl4-NH3 reaction, ZrN deposition proceeds from 2000 to 2400 °C in N2/H2 and at >3000 °C with N2 alone. Similar results were also reported for HfN and TiN depositions. The deposition temperatures reported are significantly over the melting point of zirconium metal (1854 °C) [137]; while this feature was highlighted by Moers, its impact on the deposition mechanism remains undetermined.
Additionally, ZrN and HfN whiskers have been grown by MCl4-N2-H2 mixtures (M = Zr or Hf) [124,138]. The growth of whiskers by CVD has been proposed to occur via a vapor-liquid-solid mechanism [139], where gaseous reagents dissolve into a liquid bead (e.g., Pd, Mn, Fe, Ni, or Sb) [124,138] isolated at the top of a growing whisker. Whiskers up to 100 µm long with a 5 µm diameter have been reported [138]. The process also appears to be temperature-sensitive as follows: whiskers can be grown from 1000 to 1200 °C, but higher temperatures only grow flat coatings [124,138]. Once in solution, whisker-growth reactions occur more rapidly than in the gas phase, promoting a deposition front and whisker growth under proper conditions. In whisker growth systems, the relatively rapid solution reactions were originally attributed to liquid droplets acting as the preferential site for vapor precursors, resulting in supersaturation and precipitation [139]. Alternatively, it could be expected that solvent cage effects are increasing the probability of chemisorption and reaction at the droplet-solid interface [140]. Briefly, the frequency of collision of a gas-phase reaction is not increased by an equivalent reaction in solution if a reaction can occur in both systems [141]. However, the distribution of collisions is significantly altered [142], increasing the collision rate by 2–3 fold in the solution compared to the gas phase [140]. Increasing the number of potential collisions prior to a reactant diffuses away from a surface is particularly important for molecules with low sticking coefficients, such as nitrogen.
While surface chemistry of ZrN growth by ZrCl4 + N2/(H2) has not been reported, the similarities to metallic Zr deposition (i.e., ZrCl4-H2) [1] and nitriding of zirconium (i.e., Zr(m) + N2(g)) [63,143] suggests a mechanism where metallic zirconium deposits and is simultaneously nitrided. This is supported by studies on the surface chemistry of N2 on pure zirconium, where several groups have found evidence that N2 dissociates upon chemisorption and diffuses to octahedral sites between the first and second zirconium layers [45]. Further, zirconium nitride can be prepared by forming zirconium metal (by reducing ZrO2 with magnesium, for instance) in the presence of nitrogen [63,144]. Atomic nitrogen is attributed as the reactive nitrogen species when nitriding via N2-H2 [145,146], additionally with charged species (e.g., N+, N2+, NH+, NH2+) in plasma nitriding [146,147,148]. Atomic nitrogen is also the chemisorbed species on other metals such as tungsten and ruthenium [149,150].
4.2. Organometallic CVD
With applications in such diverse fields as radiological coatings, optics, and microelectronics, deposition of zirconium nitrides (ZrN, Zr3N4) at low temperatures (<500 °C) and with a low halide contamination have been of interest [123]. To that end, research has been reported for the deposition of ZrN from zirconium amido complexes, such as tetrakis(diethylamino)zirconium (Zr(NEt2)4) [64,123,127,128,129,151,152], tetrakis(dimethylamino)zirconium (Zr(NMe2)4) [128,129,130,131,152,153], and tetrakis(ethylmethylamino)zirconium (Zr(NEtMe)4) [129,152]. Deposition of ZrN from these organometallic precursors can be accomplished as a single-source precursor (e.g., Zr(NMe2)4 alone) [127,128,129,130,131,151] or in the presence of ammonia [123,129,152,153], nitrogen, or hydrogen [128].
When used as single-source precursors, amido-containing zirconium complexes usually suffer from high carbon contamination [129,130], sometimes to such an extent that resulting deposits are described as carbonitrides [128]. Growth rates can also be very slow, for example, as low as 1.1 µm/day at 280 °C [131].
In contrast, depositing ZrN from the same organozirconium compounds in the presence of ammonia can be much more successful. Early work on this system (specifically, Zr(NEt2)4 + NH3) [64,123] conceived the system based on the result of solution reactivity studies [154,155,156], proposing that transamination and amine elimination reactions between Zr(NEt2)4 and NH3 occur to produce zirconium nitrides (Equation (9)) [123,157].
However, the reactivity of this reaction is high enough that premature deposition and consumption of reactants can occur [129]. Dividing the reaction into sequential pulses via atomic layer deposition (ALD) of Zr(NMe2)4 + NH3 was recently demonstrated to produce ZrN coatings with minimal carbon and oxygen contamination, albeit at 0.3 um/day growth rate in a non-optimized system [153].
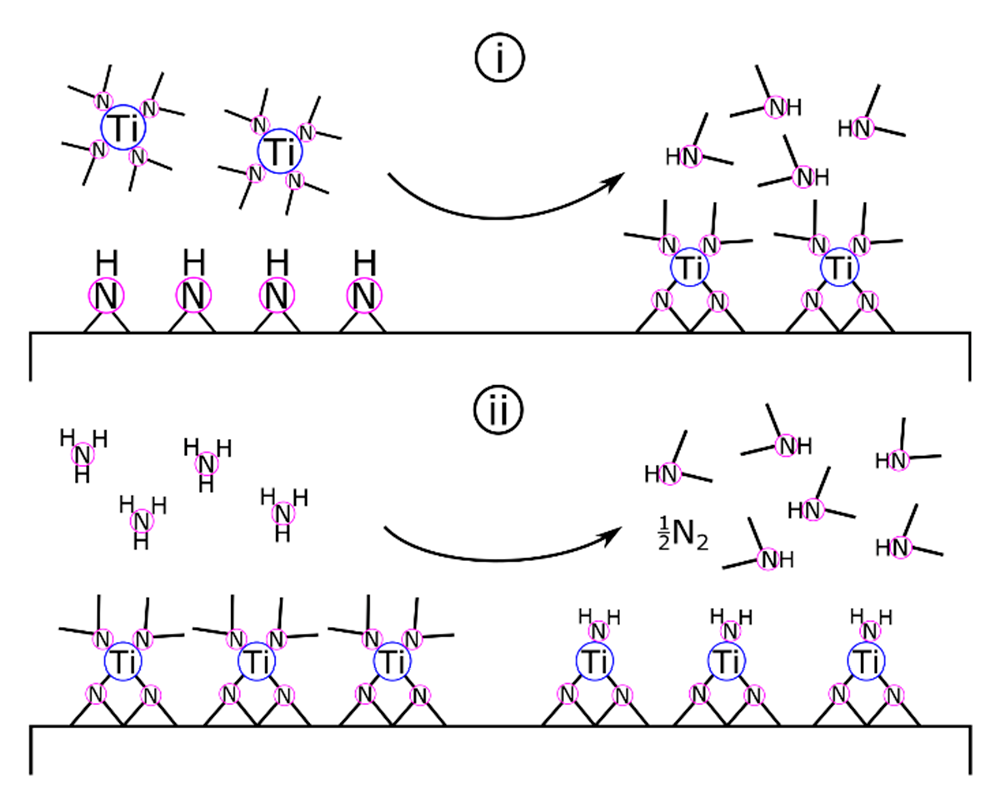
Surface chemistry and film growth studies on ALD of TiN [157] from Ti(NMe2)4 + NH3 support the transamination and amine elimination mechanisms proposed for organometallic ZrN deposition [123]. Elam and colleagues proposed that the TiN-equivalent to Equation (9) occurred as two binary reactions (Equations (10a) and (10b)) as follows [157]:
where asterisks designate surface-bound species. As shown in Scheme 3, Elam and colleagues proposed that Ti(NMe2)4 and surface-bound amides and imides react by transamination exchange to form Ti(IV) surface groups. Following a nitrogen purge to clear excess reactants and reaction byproducts, the NH3 pulse initiates another transamination exchange reaction. During the ammonia pulse, titanium is formally reduced from +4 to +3. This mechanism is supported by results from in situ FTIR (high vacuum, <5 × 10−7 torr) and quartz crystal microbalance (QCM) vacuum (1 torr) studies of the half-cycle reactions as well as analysis of the resulting films by XRD, XPS, and AFM.
The results presented by Elam and colleagues also demonstrated that Ti(NMe2)4 deposition was not a self-limiting reaction [157], a critical requirement for ALD processes [158]. It was found that NH3 did not replace all of the methylamino groups, resulting in a build-up of carbon within the coating. Besides being detrimental to certain applications, the carbon contamination was also expected to lower film density and facilitate the oxidation of TiN upon exposure to air [157]. Using the QCM mass data during a full pulse cycle of [Ti(NMe2)4—purge—NH3—purge], researchers predicted that Ti(NMe2)x* surface species could retain one to three of the original dimethylamino ligands (x = 1, 2, or 3), resulting in the incorporation of up to two dimethylamino ligands into the TiN film per Ti unit.
Zr(NMe2)4, and the other alkylamino complexes, have a formal oxidation state of +4 on zirconium versus a +3 state in ZrN. Oxidation is thought to occur in the NH3 step (Equation (10b)); however, as noted above, an incomplete reaction with NH3 can leave residual ligands in the coating [157]. Ammonia is a strong transamination reagent but a relatively weak reducing agent. Adding stronger reducing compounds to the ammonia stream, such as <2 mol% hydrazine in ammonia, has been shown to decrease carbon contamination, improve crystallinity, and reduce deposition temperatures to <300 °C in CVD TiN [159]. Hydrazine decomposes into NH2• radical, forming NH* and NH2* surface sites more readily than NH3 [159,160]. It is important to note that bonding in ZrN is largely metallic [161], and traditional ionic valences are not present [161]. However, ammonia is still required to be reduced to atomic nitrogen and the remaining alkylamino complexes removed from the surface; thus, some redox chemistry can be expected in the reaction mechanism.
Understanding of the deposition mechanisms for ZrN from ZrCl4-NH3 and Zr(NMe2)4-NH3 has been assisted greatly by surface chemistry studies on TiN depositions in complementary precursor systems. From ALD TiN studies, it was identified that TiN (and ZrN) likely forms from MCl4-NH3 by thermal decomposition of metal chloride-ammonia adducts; alternatively, metal nitrides deposited from dialkylamido complexes and ammonia form via transamination exchange reactions at the substrate surface. ZrN deposition from dialkylamido complexes might be further assisted by adopting some of the advancements in TiN deposition, such as adding dilute amounts of hydrazine (or similar compounds) to improve the reducing strength of the nitrogen source(s).
5. Zirconium Dioxide
Zirconia (ZrO2) coatings are used in microelectronics and similar applications as an oxidation-stable resistor. CVD preparation of ZrO2 can be accomplished by oxidation of ZrCl4 [162], hydrolysis of ZrCl4 by water vapor, or by forming H2O in situ from CO2 and H2 [163], among others [164], with deposition temperatures between 800 and 1550 °C [163,164,165]. It was noted that both the ZrCl4-O2 and ZrCl4-H2O systems are very reactive and prone to homogeneous nucleation and condensation at 800–950 °C [163]. The ZrCl4-CO2-H2 system is more controllable at intermediate temperatures (e.g., 900–1200 °C) and thus is the preferred precursor system in this temperature regime.
5.1. ZrCl4-CO2-H2
Deposition of ZrO2 from ZrCl4-CO2-H2 proceeds overall by [164] as follows:
Here, Equation (12a) is regarded as the rate-limiting step [163,167] in the deposition of ZrO2 due to slow reaction rates up to 1100 °C [164], as determined by experimental analysis of the reaction from 950 to 1650 °C.
Equation (12a) is also the reverse water-gas shift (rWGS) reaction [168,169,170,171]. The rWGS reaction can be modeled as either a homogenous gas phase or a heterogenous surface-catalyzed reaction. The following homogeneous reaction is known as the Bradford mechanism (Equation (13)) [169]:
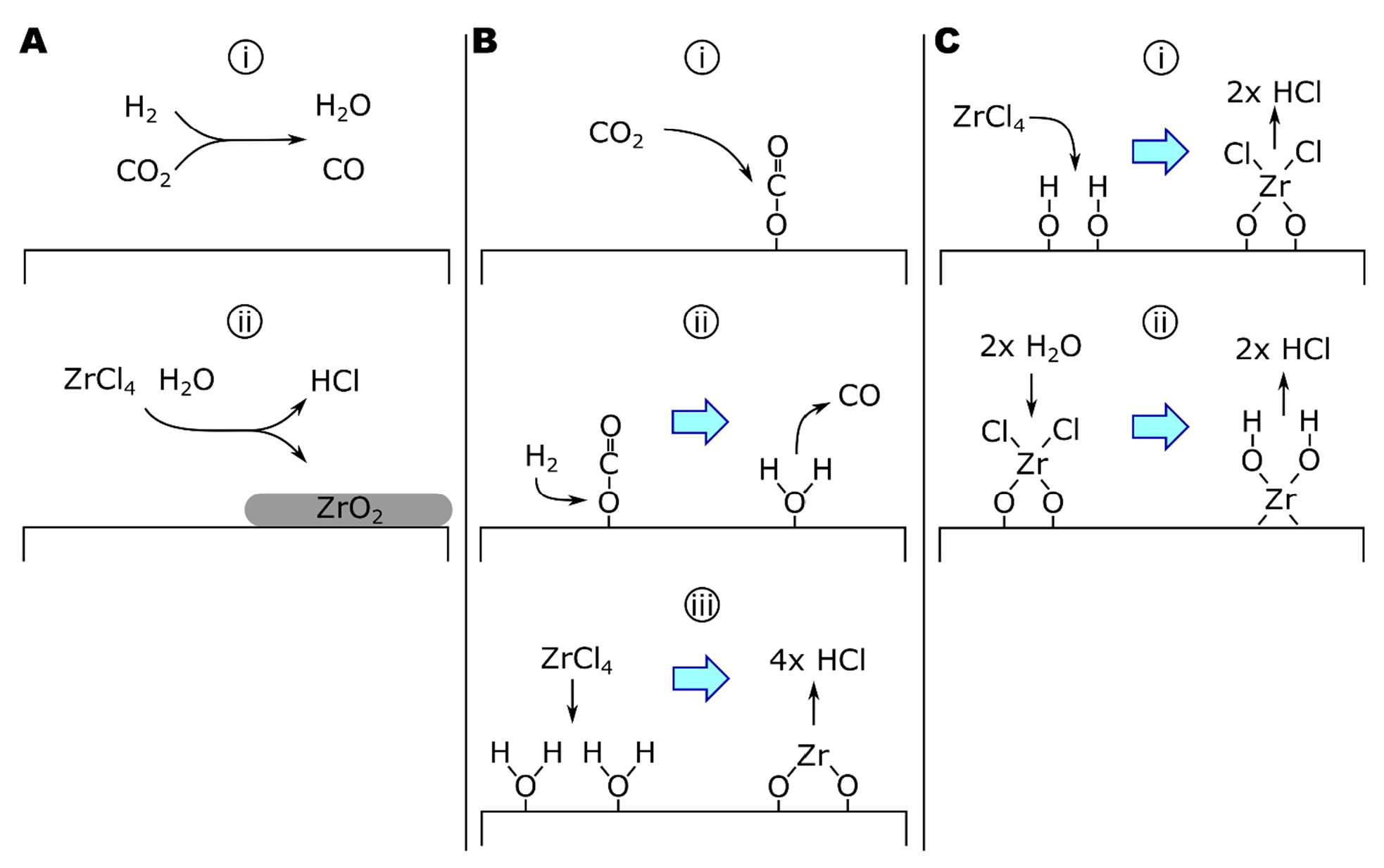
where M is any molecule in the gas phase. Notice that this mechanism is catalyzed by atomic hydrogen. Bradford’s study used low-pressure explosions, seeing temperature and pressure ranges of 500–4000 °C and ~0–475 torr, respectively. Importantly, general features of this mechanism are supported by studies at conditions more consistent with CVD. For example, Graven and Long, studying the CO2 + H2 reaction at 900 °C and 760 torr in either empty or packed quartz vessels, found evidence for a homogeneous reaction, which includes either atomic or radical species [171]. Additionally, Holgate and Tester suggested that hydroxyl radicals are also critical to the reaction mechanism based on modeling studies of 28 elementary reactions [172]. A reaction scheme for homogeneous ZrO2 deposition is given in Figure 3A.
The second mechanism specifies a surface-catalyzed mechanism based on analysis of apparent reaction kinetics (Equation (14) and Figure 3B) as follows [164]:
where the heterogeneous formation of H2O on ZrO2 (Equation (14b)) was identified as the rate-determining step in this mechanism. Notice that the combination of Equations (14a) and (14b) is equivalent to Equation (12a) and that Equation (14c) varies from Equation (12b) only by the substitution of gaseous water for surface-adsorbed water. Equation (14) is consistent with mechanisms for catalyzed rWGS reactions [173,174]. Copper-catalyzed rWGS has been proposed to proceed by forming a cupric or cuprous oxide surface species from CO2, evolving CO. Cu(2)O reacts with H2 to form H2O [173,174]. A surface-catalyzed mechanism is supported by the relatively low apparent activation energy (Ea) of the process. Sipp, and colleagues reported a reaction Ea of 40 kJ/mol for reaction temperatures of 1050–1650 °C and total reaction pressures >37 torr [164]. In comparison, homogeneous rWGS reaction kinetics have reported Ea of 326 kJ/mol [168], while the activation energy of alumina-catalyzed rWGS reactions can be as low as 67 kJ/mol.
5.2. ZrCl4-H2O
The growth of ZrO2 coatings by ALD has also been studied, typically using ZrCl4 or ZrI4 as the zirconium source and H2O or H2O2 as the oxygen source [175,176,177,178,179,180]. Rahtu, and colleagues studied the ZrCl4 + D2O system by QCM and quadrupole mass spectroscopy (QMS) from 250 to 375 °C, observing both the mass change of the surface and the pressure of deuterated reaction products (i.e., DCl) during each step of the ZrCl4-purge-D2O-purge cycle. Analysis of the results suggested that the mechanism proceeds as follows (Equation (15) and Figure 3C) [175]:
where ZrCl2 forms a bridging complex between two O* surface sites in Equation (15a). From 400 to 500 °C (the upper-temperature limit of the study), Rahtu and colleagues reported singly bound zirconium chloride complexes based on the reduced partial pressures of DCl in the effluent gas following ZrCl4 exposures compared to lower-temperature exposures. The change in reaction mechanism was attributed to dehydroxylation of the surface at higher temperatures [175].
The vapor deposition of ZrO2 is perhaps the best-studied zirconium system in terms of kinetics and surface reaction mechanics. Both ZrCl4-H2O and ZrCl4-CO2-H2 systems have accepted mechanisms based on kinetics and surface chemistry studies. Studying the surface chemistry occurring during ZrCl4-CO2-H2 deposition or the chemistry of the rWGS reaction over zirconium could offer insights to unify the mechanisms between these two ZrO2 deposition systems. Existent studies on the interaction between H2O and D2O vapor on zirconium surfaces, reviewed elsewhere [45], has observed autocatalytic oxidation of the zirconium surface to as low as −50 °C [181].
6. Zirconium Diboride
Zirconium and hafnium diborides (ZrB2, HfB2) are of interest for use in high-temperature applications [41,182] as well as microelectronics [33]. The deposition of group IV diborides can be accomplished by either independent reactants (e.g., ZrCl4-BCl3-H2 or TiCl4-B2H6-H2) [183,184,185,186] or single-source precursors (e.g., Zr(BH4)4) [187,188,189].
The earliest reports of ZrB2 deposition utilized the ZrCl4-BCl3-H2 system [190,191] or ZrCl4-BBr3-H2 [136]. The all-chloride system continues to be one of the most common processes employed [183,192,193]. A complementary system has been demonstrated for the deposition of TiB2 (i.e., TiCl4-BCl3-H2) [184,194,195], as well as TiCl4-B2H6-H2 [185]. Interest in deposition temperatures below 1000 °C has led to the development of a halogen-free single-source deposition system, Zr(BH4)4 [187,188,189], which also remains in use [196,197].
6.1. ZrB2 from Independent Precursors
In the all-halide deposition of ZrB2, multiple researchers have suggested that BCl3 reduction is independent of zirconium [183,186,193,198], implying that zirconium and boron deposit independently and not cooperatively. The reduction of ZrCl4 in the presence of H2 has been discussed previously, vide supra, with undefined mechanics. The second half of the deposition, the reduction of BCl3 or BBr3 by H2, has been investigated more thoroughly.
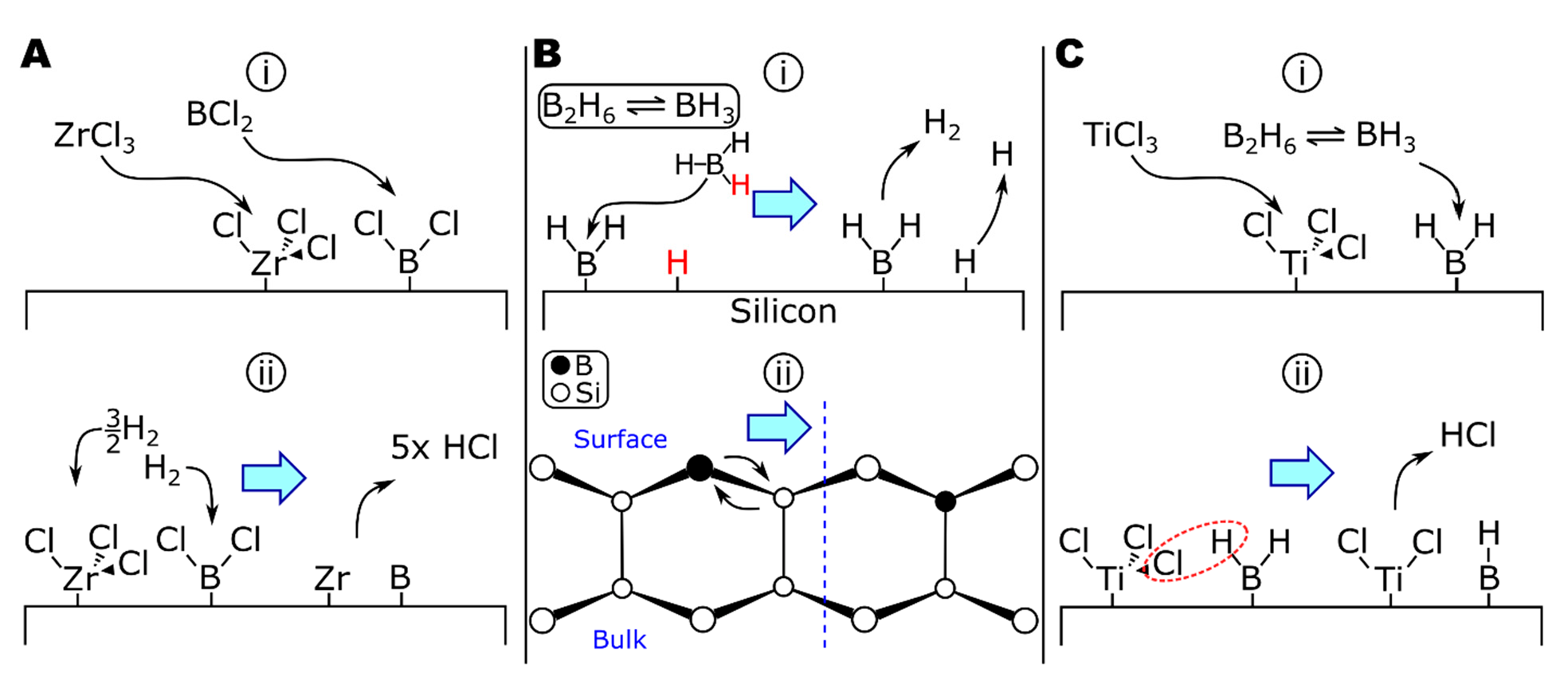
A theoretical examination of the deposition of boron by BCl3-H2 and BBr3-H2 by Naslain and colleagues calculated the equilibrium conditions of the systems in the presence of nonreactive (tungsten or boron), reactive (titanium), and partially reactive (tantalum) substrates [199]. At equilibrium on nonreactive substrates, the partial pressure of boron subchlorides (BCl2, BCl) is significant (0.049 and 0.0019 torr, respectively, at 1030 °C). The subchlorides are likely to adsorb to the substrate surface (Figure 4A, step (i)), similar to the deposition of BH3 via -BH2 surface groups [200,201]. Thermodynamics calculations of the ZrCl4-BCl3-H2 system suggest BHCl2 to be another important boron-containing intermediate [198] and would likely adsorb via a similar mechanism to other boron chlorides or hydrides. Boron (di)chloride surface groups could then be reduced by molecular or atomic hydrogen (PH = 6.5 × 10−4 torr at 1030 °C) [199] to form HCl (Figure 4A, step (ii)). This is in agreement with the ZrCl4-BCl3-H2 study, which saw increased reaction efficiencies as the amount of hydrogen increased [198]. When comparing nonreactive and reactive substrates in the deposition of borohalides [199], the most significant difference in the mechanism when depositing onto reactive substrates was substrate corrosion, for example, generating TiCl4 when depositing onto titanium. Substrate corrosion could be mitigated by very hydrogen-rich gas compositions (e.g., [BCl3]/[H2] < 1.7 × 10−5 at 1030 °C, 760 torr) [199]. Etching of zirconium by BCl3 has been reported, for instance, in the remote plasma-enhanced CVD of Ar-BCl3 onto zircaloy-4 to grow ZrB2 thin films [183,192]. In a codeposition system (e.g., ZrCl4-BCl3-H2), the presence of zirconium chlorides in the vapor phase should limit zirconium corrosion via chlorination from BCl3, as seen in the codeposition of TiCl4 and SiCl4 for TiSi2 [202], which is discussed in more detail in the following section.
Boron can also be deposited from boranes. Diborane (B2H6) is frequently used to grow pure boron coatings [201,203,204] and codeposit to form diborides [185] or as a dopant [200,205,206,207,208,209,210,211]. The deposition of boron from diborane proceeds overall as follows (Equation (16)) [203]:
The growth of pure boron from boranes can be quite complicated, with homogeneous gas-phase reactions forming progressively higher boranes [212]. The allotropes of boron all include icosahedral B12 clusters, whereas boron in diborides (i.e., TiB2, ZrB2, and HfB2) forms a two-dimensional network [213], suggesting a different growth mechanism could be occurring in the diborides. The deposition of diborane onto silicon has been extensively studied due to diborane’s use as a CVD precursor in the fabrication of p-type doped silicon electronics. From the results of various studies on the Si-B2H6 system, including thermally programmed desorption (TPD) [214], experimental reaction kinetics probing radical chemistries [205], and scanning tunneling microscopy [206,207,208,209], the surface reactions of diborane on silicon have been developed (Figure 4B). The first step in this mechanism is the gas-phase decomposition of diborane to borane (Equation (17a)), which can adsorb directly to the surface (Equation (17b)), shown in Figure 4B step (i). There is evidence that, at room temperature, diborane can bind to the surface intact, forming *B2H5 and *H surface species [209]. Surface-bound *B2H5 decomposes in ~30 min to *BH2 groups and additional H (as surface groups or free species). Hydrogen evolves from *BH2 units, resulting in surface *B (Equation (17c)); atomic H desorbs at T > 427 °C [209]. The final step involves the diffusion of boron into the bulk of the substrate (Figure 4B step ii) [206,207,211]. At moderate temperatures (T > 500 °C), boron can diffuse into silicon over several hundred angstroms and over a 1000 Å layer at 1000 °C [210]. In comparison to silicon, zirconium and boron have a much greater size difference and less covalent bond character, increasing the diffusivity of boron in zirconium relative to silicon.
There appear to be no published studies of ZrB2 deposition from zirconium halides and boranes; however, TiB2 has been deposited from TiCl4-B2H6-H2 [185,215] and can provide insights into ZrB2 deposition. While surface chemistry studies have not been reported, the authors noted that no chlorine was detected in the deposits. In contrast, all-chloride systems (e.g., TiCl4-BCl3-H2) are reported to trap chlorine within the deposit to the detriment of material properties [184,194]. While the reduction in chlorine contamination could be from the increased deposition temperature when using diborane (≥1150 °C) [185], compared to ~950 °C for the all-chloride system [184,194], the colocation of *Ti-Cl and *B-H surface species could enable surface reactions to form Ti-B surface bonds by elimination of HCl (Figure 4C).
6.2. ZrB2 from Single-Source Precursor
Interest in using transition metal diboride coatings (i.e., TiB2, HfB2, and ZrB2) for electronics applications necessitated a deposition technique at low temperatures (<600 °C) and with no halogen contamination. Three independent research groups nearly simultaneously published on the deposition of the metal diborides from metal tetrahydroborates—Zr(BH4)4 [187,188,189], Hf(BH4)4 [187,189], and Ti(BH4)3(dme), dme = 1,2-dimethoxyethane [187]. Early work was accomplished in greaseless equipment [187,188,189], similar to that developed for researching borane chemistry [213]. More recent reports indicate commercial CVD reactors can be used as well [196,197].
Deposits are amorphous below 500 °C [187,188,189,196,197] but can be crystalline above this temperature [196]. Most reports indicate that the stoichiometry of deposits is related to the deposition temperature, with lower temperatures producing boron-poor deposits and higher temperatures producing boron-rich deposits. Rice and Woodin reported ZrB1.6 at 265 °C and ZrB3.1 at 398 °C. However, the results can vary significantly depending on the deposition parameters and reactor characteristics. For example, Jensen and colleagues stated that all deposits >250 °C had a B/Zr > 2, [187] Jayaraman and colleagues found B/Hf of 2 from 250 to 900 °C, [196] and Sulyaeva and colleagues found B/Zr of 4-15 from 100 to 300 °C [197]. In addition to the deposition temperature, residence time and composition of dilution gases (e.g., Ar or H2) could also have a significant effect on deposit stoichiometry [188], but studies of such effects have not been reported to date.
Currently, the only deposition mechanism proposed is that ZrB2 forms via a metal hydride intermediate, followed by reductive elimination of H2 [187]. The intermediate structure was proposed to be similar to compounds found in solution chemistry, such as (C5H5)2Zr(H)(BH4) or (BH4)LHf(µ-H)3HfL(BH4)2 (L = N [Si(CH3)2CH2P(CH3)2]2) [216,217]. Formation of the hydride intermediate would include the elimination of BH3 [217]. A temperature programmed reaction (TPR) study [196] involving in situ mass spectrometry of the vapor phase over a silicon wafer as it was heated from room temperature to 800 °C in the presence of 0.1–1 mtorr of Hf(BH4)4, identified both BHx and B2Hx fragments; however, only data down to m/z 10 were published. The onset of deposition was observed at 190 °C with an activation energy of 41 kJ/mol. As noted by the researchers, both BHx and B2Hx fragments are related to the deposition and decomposition byproducts of Hf(BH4)4 [196]. Both Zr(BH4)4 and Hf(BH4)4 are 12-coordinate, tetrahedral molecules where borane anions are tridentate [218,219,220]. In contrast, Ti(BH4)3(dme) is 8-coordinate with bidentate boranes, although it is also tetrahedral [187]. As reviewed elsewhere [220], transition metal tetrahydroborates are known to undergo a variety of intramolecular reactions, primarily due to the dynamic motion in –(BH4) ligands. Hydrogen exchange reactions between ligands directly or involving a zirconium- or hafnium-hydride intermediate have been observed [221]. Additional TPR studies with a wider mass spectrometry detection window, in situ XPS reaction analysis [222,223], or low-temperature neutron scattering [220] might give more information on the nature of reaction intermediates and the deposition mechanism in general.
7. Zirconium Silicides
Zirconium disilicide (ZrSi2) has been studied for use as a neutron-tolerant cladding material for nuclear power generation [17,18], and the silicides of titanium, hafnium, and zirconium are also of interest in electronics applications [30,31,202,224,225]. Reports on the vapor deposition of zirconium silicides are very limited. The only instances found are in a historic review of CVD precursor systems [1] and a recent report on the Zr-Si-C codeposition system [104]. ZrSi and ZrSi2 were found to codeposit with ZrC and SiC from ZrCl4-CH3SiCl3-CH4-H2 at 1200 °C and 50 torr, depending primarily on precursor gas ratio [104]. Computational analysis of the Zr-Si-C system by minimization of Gibbs free energy predicted ZrSix deposition from 1150 to 1350 °C (maximum temperature limit studied) and from 0 to 160 torr within the precursor composition window reported [104]. Zirconium silicides can also grow from the reaction of SiCl4-H2 on zirconium (1100–1500 °C, 760 torr). While not verified experimentally, ZrCl4-SiCl4-H2 is also expected to produce silicides [1].
Using the CVD of TiSi2 as an analog for ZrSi2 and HfSi2, silicide coatings may also be deposited by TiCl4-H2, TiCl4-SiH4, or TiCl4 alone onto Si [202]. The general reaction proceeds according to (Equation (18)) as follows [202]:
Note that the silicon substrate reduces TiCl4, becoming etched in the process; etching rates of up to 2 µm/min have been reported [202]. TiSi2 forms by diffusion of silicon into titanium; silicon is the dominant diffusion species in metal silicides at deposition temperatures (e.g., 900 °C) [31,225,226]. Sputter-coating hafnium onto silicon substrates at lower temperatures has been found to form Hf-Si phases via the diffusion of Si into Hf as well [225]. From 550–750 °C, HfSi was the only silicide formed and was related to the diffusion of silicon from the Hf-Si interface. At higher temperatures (750–900 °C), HfSi2 was found to form within the HfSi layer rather than from additional Si diffusion (i.e., forming at the Hf-Si interface) [225]. The etching of Si in the CVD of TiCl4 can be inhibited or eliminated by introducing another reducing agent and/or silicon source, for example, H2 or SiH4 to generate HCl instead of SiCl4 [202]. In the absence of a gaseous silicon source, silicon substrates must be carefully treated prior to the deposition to remove the native oxide present [202], as SiO2 is more stable than TiSi2 (ΔHf,SiO2 = −910.7 kJ/mol vs. ΔHf,TiSi2 = −160.5 kJ/mol at 25 °C) [118,227].
8. Conclusions
Zirconium compounds are utilized in a variety of fields, with applications in microelectronics, nuclear technologies, and aerospace. With contemporary interest in zirconium-based coatings [17,104,153,197], further understanding of the chemistry and mechanisms involved in material growth is critical to material adoption and scaling to industrial production. A summary of the key findings follows:
The vapor deposition of zirconium by ZrCl4-H2 is an important factor in several of the compounds covered in this review. Introducing an additional reactant can produce ZrC (CH4-H2), ZrN (N2-H2 or NH3-H2), ZrO2 (CO2-H2), ZrB2 (BCl3-H2), or ZrSix (SiCl4-H2). Research on the ZrCl4-H2 deposition mechanism could benefit the optimization of CVD for these materials.
The deposition of metallic zirconium via disproportionation of zirconium halides (van Arkel-de Boer process) is well understood and is supported by a series of studies on the three main halides of zirconium. The deposition of ZrC by ZrCl4-CH4-H2 is the most common, but other halides or hydrocarbons can be used successfully. However, the two most prominent mechanisms for ZrC growth are insufficient. In contrast, both ZrN and ZrO2 have well-supported mechanisms. ZrN by ZrCl4-NH3-H2 likely grows by the deposition of Zr(m) with concomitant nitriding; ZrO2 similarly grows Zr(m) and is oxidized by O2 or H2O directly (at low temperature) or by H2O formed in situ from CO2-H2 reactions. The growth of ZrB2 by ZrCl4-BCl3-H2 is most common, though other zirconium or boron halides have been used. Thermodynamic calculations suggest boron subhalides and hydroborohalides are important to the surface reaction. Analogous studies on TiB2 also suggest that the codeposition of ZrCl4 with B2H6 might improve the quality of the final deposit. The CVD of ZrSix coatings is the least studied of the compounds reviewed, with no accepted mechanisms in the literature. Diffusion studies on TiSi2 and HfSix suggest bulk or surface diffusion of silicon into Zr to be important to ZrSix growth.
The deposition of several zirconium compounds have benefitted from organometallic (ZrN from Zr(NMe2)4-(NH3)) or reactive single-source (ZrB2 from Zr(BH4)4) precursors. These precursors are more reactive than ZrX4, significantly reducing the substrate temperatures required for coating growth. Decreased substrate temperatures (e.g., <500 °C) expand the range of substrates that can be utilized, and correspondingly expand the potential applications that these coatings can be incorporated. Eliminating a halide source can also improve the electronic properties of the resulting coat.
Insight into the CVD deposition mechanisms of several precursor systems was also assisted by comparison to complementary titanium-based materials and/or by analysis of ALD mechanisms. The similarities between the Group IV compounds are useful for establishing reaction steps, though a comparison of actual kinetics via experimentation is important to clarify potentially significant differences. Additionally, the combination of in situ metrics and reduced kinetic rates in ALD enables access to vapor deposition mechanics not elucidated via CVD, providing a platform for detailed studies of deposition mechanisms.
* Notice of Copyright: This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (www.energy.gov/sites/prod/files/2014/08/f18/DOE_Public_Access%20Plan_FINAL.pdf (accessed on 18 January 2023)).
Author Contributions
Conceptualization, B.W.L. and D.J.M.; methodology, B.W.L.; software, B.W.L.; investigation, B.W.L.; resources, D.J.M.; writing—original draft preparation, B.W.L.; writing—review and editing, B.W.L. and D.J.M.; visualization, B.W.L.; funding acquisition, D.J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was sponsored by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC for the US Department of Energy under contract DE-AC05-00OR22725.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Jake McMurray (MSTD, ORNL; now at Kairos Power) for developing the database used in calculating thermodynamic equilibria diagrams as well as Hanns Gietl and Chanaka Gamaralalage (MSTD, ORNL) for their support in writing this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Campbell, I.E.; Powell, C.F.; Nowicki, D.H.; Gonser, B.W. The Vapor-Phase Deposition of Refractory Materials. J. Electrochem. Soc. 1949, 96, 318. [Google Scholar] [CrossRef]
- Lemaignan, C. Zirconium Alloys: Properties and Characteristics. In Material Properties/Oxide Fuels for Light Water Reactors and Fast Neutron Reactors, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 217–232. [Google Scholar]
- Ogata, T. Metal Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 1–40. [Google Scholar]
- Wooding, S.J.; Bacon, D.J. A molecular dynamics study of displacement cascades in α-zirconium. Philos. Mag. A 1997, 76, 1033–1051. [Google Scholar] [CrossRef]
- Bacon, D.J.; Gao, F.; Osetsky, Y.N. The primary damage state in fcc, bcc and hcp metals as seen in molecular dynamics simulations. J. Nucl. Mater. 2000, 276, 1–12. [Google Scholar] [CrossRef]
- Voskoboinikov, R.E.; Osetsky, Y.N.; Bacon, D.J. Statistics of primary damage creation in high-energy displacement cascades in copper and zirconium. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2006, 242, 68–70. [Google Scholar] [CrossRef]
- Stoller, R.E. Primary Radiation Damage Formation. In Basic Aspects of Radiation Effects in Solids/Basic Aspects of Multi-Scale Modeling, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 293–332. [Google Scholar]
- Yamanaka, S.; Yamada, K.; Kurosaki, K.; Uno, M.; Takeda, K.; Anada, H.; Matsuda, T.; Kobayashi, S. Characteristics of zirconium hydride and deuteride. J. Alloy. Compd. 2002, 330–332, 99–104. [Google Scholar] [CrossRef]
- Yamanaka, S.; Yamada, K.; Kurosaki, K.; Uno, M.; Takeda, K.; Anada, H.; Matsuda, T.; Kobayashi, S. Thermal properties of zirconium hydride. J. Nucl. Mater. 2001, 294, 94–98. [Google Scholar] [CrossRef]
- Yamanaka, S.; Yoshioka, K.; Uno, M.; Katsura, M.; Anada, H.; Matsuda, T.; Kobayashi, S. Isotope effects on the physicochemical properties of zirconium hydride. J. Alloy. Compd. 1999, 293–295, 908–914. [Google Scholar] [CrossRef]
- Olander, D.R.; Konashi, K.; Yamawaki, M. Uranium-Zirconium Hydride Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd: Amsterdam, The Netherlands, 2012; pp. 313–357. [Google Scholar]
- Minato, K.; Ogawa, T. Advanced Concepts in TRISO Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 216–236. [Google Scholar]
- Wagner, P. High-Temperature Fuel Technology for Nuclear Process Heat: ZrC-Containing Coated Particle Fuels and High-Density Graphite Fuel Matrices; LA-6984; Los Alamos Scientific Laboratory: Los Alamos, NM, USA, 1977.
- Arai, Y. Nitride Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 41–54. [Google Scholar]
- Arai, Y.; Minato, K. Fabrication and electrochemical behavior of nitride fuel for future applications. J. Nucl. Mater. 2005, 344, 180–185. [Google Scholar] [CrossRef]
- Canel, J.; Zaman, J.; Bettembourg, J.; Flem, M.; Poissonnet, S. Composite Zirconium Silicides Through an In Situ Process. Int. J. Appl. Ceram. Technol. 2006, 3, 23–31. [Google Scholar] [CrossRef]
- Cheol Lee, G.; Noh, H.; Yeom, H.; Jo, H.; Kyun Kim, T.; Kim, M.; Sridharan, K.; Sun Park, H. Zirconium-silicide coating on zircaloy-4 substrate for accident tolerance: Effects on oxidation resistance and boiling. Ann. Nucl. Energy 2019, 126, 350–358. [Google Scholar] [CrossRef]
- Yeom, H.; Lockhart, C.; Mariani, R.; Xu, P.; Corradini, M.; Sridharan, K. Evaluation of steam corrosion and water quenching behavior of zirconium-silicide coated LWR fuel claddings. J. Nucl. Mater. 2018, 499, 256–267. [Google Scholar] [CrossRef]
- Raison, P.E.; Haire, R.G. Structural investigation of the pseudo-ternary system AmO2–Cm2O3–ZrO2 as potential materials for transmutation. J. Nucl. Mater. 2003, 320, 31–35. [Google Scholar] [CrossRef]
- Poeml, P.; Konings, R.J.M.; Somers, J.; Wiss, T.; de Haas, G.J.L.M.; Klaassen, F.C. Inter Matrix Fuel. In Advanced Fuels/Fuel #ladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 237–256. [Google Scholar]
- Eggers, G.H. Method of Making ZrH Fuel Element. US407 1587A, 1978.
- Murata, Y. Cutting Tool Tips and Ceramics Containing HfN and ZrB2. US348 7594A, 1970.
- Hintermann, H.E. Tribological and protective coatings by chemical vapour deposition. Thin Solid Film. 1981, 84, 215–243. [Google Scholar] [CrossRef]
- Hirose, M.; Yasui, T.; Ochi, Y.; Nakagawa, M. Method of Forming a Decorative Metallic Nitride Coating. US442 0498A, 1982.
- Johnson, P.C.; Randhawa, H. Zirconium nitride films prepared by cathodic arc plasma deposition process. Surf. Coat. Technol. 1987, 33, 53–62. [Google Scholar] [CrossRef]
- Johansson, B.O.; Sundgren, J.E.; Helmersson, U.; Hibbs, M.K. Structure of reactively magnetron sputtered Hf-N films. Appl. Phys. Lett. 1984, 44, 670–672. [Google Scholar] [CrossRef]
- Karlsson, B.; Shimshock, R.P.; Seraphin, B.O.; Haygarth, J.C. Optical properties of CVD-coated TiN, ZrN and HfN. Sol. Energy Mater. 1983, 7, 401–411. [Google Scholar] [CrossRef]
- Schlegel, A.; Wachter, P.; Nickl, J.J.; Lingg, H. Optical properties of TiN and ZrN. J. Phys. C Solid State Phys. 1977, 10, 4889–4896. [Google Scholar] [CrossRef]
- Namavar, F.; Wang, G.; Cheung, C.L.; Sabirianov, R.F.; Zeng, X.C.; Mei, W.N.; Bai, J.; Brewer, J.R.; Haider, H.; Garvin, K.L. Thermal stability of nanostructurally stabilized zirconium oxide. Nanotechnology 2007, 18, 415702. [Google Scholar] [CrossRef]
- Lepselter, M.P.; Andrews, J.M. Ohmic Contacts to Silicon. In Ohmic Contacts to Semiconductors, Schwartz, B., Ed.; The Electrochemical Society: New York, NY, USA, 1969; pp. 159–186. [Google Scholar]
- Roy, S.; Paul, A. Growth of hafnium and zirconium silicides by reactive diffusion. Mater. Chem. Phys. 2014, 143, 1309–1314. [Google Scholar] [CrossRef]
- Sung, J.; Goedde, D.M.; Girolami, G.S.; Abelson, J.R. Remote-plasma chemical vapor deposition of conformal ZrB2 films at low temperature: A promising diffusion barrier for ultralarge scale integrated electronics. J. Appl. Phys. 2002, 91, 3904–3911. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.; Goedde, D.M.; Girolami, G.S.; Abelson, J.R. Diffusion Barrier Characteristics of Zirconium Diboride Films Grown by Remote Plasma CVD. MRS Proc. 1999, 563, 39–44. [Google Scholar] [CrossRef]
- Suni, I.; Mäenpää, M.; Nicolet, M.A.; Luomajärvi, M. Thermal Stability of Hafnium and Titanium Nitride Diffusion Barriers in Multilayer Contacts to Silicon. J. Electrochem. Soc. 2019, 130, 1215–1218. [Google Scholar] [CrossRef]
- Wittmer, M. Properties and microelectronic applications of thin films of refractory metal nitrides. J. Vac. Sci. Technol. A Vac. Surf. Film. 1985, 3, 1797–1803. [Google Scholar] [CrossRef]
- Mäenpää, M.; Suni, I.; Sigurd, D.; Finetti, M.; Nicolet, M.A. Stable Metallization Systems for Solar Cells. Phys. Status Solidi A 1982, 72, 763–769. [Google Scholar] [CrossRef]
- Tauber, R.N.; Dumbri, A.C.; Caffrey, R.E. Preparation and Properties of Pyrolytic Zirconium Dioxide Films. J. Electrochem. Soc. 1971, 118, 747. [Google Scholar] [CrossRef]
- Wilk, G.D.; Wallace, R.M.; Anthony, J.M. High-κ gate dielectrics: Current status and materials properties considerations. J. Appl. Phys. 2001, 89, 5243–5275. [Google Scholar] [CrossRef]
- Espinoza-Pérez, L.J.; López-Honorato, E.; González, L.A. Development of ZrO2 and YSZ coatings deposited by PE-CVD below 800 °C for the protection of Ni alloys. Ceram. Int. 2020, 46, 15621–15630. [Google Scholar] [CrossRef]
- Ohlhorst, C.W.; Glass, D.E.; Bruce, W.E., III; Lindell, M.C.; Vaugn, W.L.; Dirling, R.B., Jr.; Hogenson, P.A.; Nichols, J.M.; Risner, N.W.; Thompson, D.R. Development of X-43A Mach 10 Leading Edges. In Proceedings of the 56th International Astronautical Congress, Fukuoka, Japan, 17–21 October 2005. [Google Scholar]
- Fahrenholtz, W.G.; Hilmas, G.E. Oxidation of ultra-high temperature transition metal diboride ceramics. Int. Mater. Rev. 2012, 57, 61–72. [Google Scholar] [CrossRef]
- Fahrenholtz, W.G.; Hilmas, G.E. Ultra-high temperature ceramics: Materials for extreme environments. Scr. Mater. 2017, 129, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Fahrenholtz, W.G.; Hilmas, G.E.; Talmy, I.G.; Zaykoski, J.A. Refractory Diborides of Zirconium and Hafnium. J. Am. Ceram. Soc. 2007, 90, 1347–1364. [Google Scholar] [CrossRef]
- Blumenthal, W.B. The Element, Zirconium. In The Chemical Behavior of Zirconium; D. van Nostrand Co. Inc.: Princeton, NJ, USA, 1958; pp. 1–45. [Google Scholar]
- Stojilovic, N.; Bender, E.T.; Ramsier, R.D. Surface chemistry of zirconium. Prog. Surf. Sci. 2005, 78, 101–184. [Google Scholar] [CrossRef]
- Xu, L.; Xiao, Y.; van Sandwijk, A.; Xu, Q.; Yang, Y. Production of nuclear grade zirconium: A review. J. Nucl. Mater. 2015, 466, 21–28. [Google Scholar] [CrossRef]
- Lustman, B.; Kerze, F.J. (Eds.) The Metallurgy of Zirconium, 1st ed.; McGraw-Hill: New York, NY, USA, 1955; Volume 4. [Google Scholar]
- Blumenthal, W.B. The Chemical Behavior of Zirconium; D. Van Nostrand Co. Inc.: Princeton, NJ, USA, 1958. [Google Scholar]
- Pierson, H.O. Introduction and General Considerations. In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Saddle River, NJ, USA, 1999; pp. 25–35. [Google Scholar]
- Jairath, R.; Jain, A.; Tolles, R.D.; Hampden-Smith, M.J.; Kodas, T.T. Introduction. In The Chemistry of Metal CVD; Kodas, T.T., Hampden-Smith, M.J., Eds.; VCH Publishers Inc.: Weinheim, Germany, 1994; pp. 1–44. [Google Scholar]
- Pierson, H.O. Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Noyes Publications: Norwich, NY, USA, 1999; p. 506. [Google Scholar]
- Xu, Y.; Yan, X.-T. Chemical Vapour Deposition; Springer: London, UK, 2010; p. 352. [Google Scholar]
- Kodas, T.T.; Hampden-Smith, M.J. (Eds.) The Chemistry of Metal CVD.; Wiley: Weinheim, Germany, 1994. [Google Scholar]
- Pierson, H.O. CVD Processes and Equipment. In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Saddle River, NJ, USA, 1999; pp. 108–146. [Google Scholar]
- Xu, Y.; Yan, X.-T. Chemical Vapor Deposition Systems Design. In Chemical Vapour Deposition; Engineering Materials and Processes; Springer: London, UK, 2010; pp. 73–128. [Google Scholar]
- Pierson, H.O. Fundamentals of Chemical Vapor Deposition. In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Saddle River, NJ, USA, 1999; pp. 36–67. [Google Scholar]
- Thornton, J.A. High Rate Thick Film Growth. Annu. Rev. Mater. Sci. 1977, 7, 239–260. [Google Scholar] [CrossRef]
- Thornton, J.A. Influence of apparatus geometry and deposition conditions on the structure and topography of thick sputtered coatings. J. Vac. Sci. Technol. 1974, 11, 666–670. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, X.-T. Microstructure Evolution and Process Control. In Chemical Vapour Deposition; Engineering Materials and Processes; Springer: London, UK, 2010; pp. 215–270. [Google Scholar]
- Martinu, L.; Zabeida, O.; Klemberg-Sapieha, J.E. Plasma-Enhanced Chemical Vapor Deposition of Functional Coatings. In Handbook of Deposition Technologies for Films and Coatings; Martin, P.M., Ed.; Science, Applications and Technology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 392–465. [Google Scholar]
- Walton, S.G.; Greene, J.E. Plasmas in Deposition Processes. In Handbook of Deposition Technologies for Films and Coatings; Martin, P.M., Ed.; Science, Applications, and Technology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 32–92. [Google Scholar]
- Pierson, H.O. Metallo-Organic CVD (MOCVD). In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Norwich, NY, USA, 1999; pp. 84–107. [Google Scholar]
- Blumenthal, W.B. Interstitial Solutions and Intermetallic Compounds. In The Chemical Behavior of Zirconium; D. Van Nostrand Co, Inc: Princeton, NJ, USA, 1958; pp. 46–101. [Google Scholar]
- Fix, R.M.; Gordon, R.G.; Hoffman, D.M. Solution-phase reactivity as a guide to the low-temperature chemical vapor deposition of early-transition-metal nitride thin films. J. Am. Chem. Soc. 1990, 112, 7833–7835. [Google Scholar] [CrossRef]
- Wagner, P. Research, Development, and Production of Substoichiometric Zirconium Carbide for High-Temperature Insulation; LA-5224; Los Alamos National Laboratory: Los Alamos, NM, USA, 1973. [Google Scholar]
- Gasparrini, C.; Rana, D.S.; Le Brun, N.; Horlait, D.; Markides, C.N.; Farnan, I.; Lee, W.E. On the stoichiometry of zirconium carbide. Sci. Rep. 2020, 10, 6347. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Heitmann, T.W.; Fahrenholtz, W.G.; Hilmas, G.E. Synthesis of ZrCx with controlled carbon stoichiometry by low temperature solid state reaction. J. Eur. Ceram. Soc. 2019, 39, 2594–2600. [Google Scholar] [CrossRef]
- Miller, J.H.; Hunn, J.D.; Jolly, B.C.; Menchhofer, P.A. Development of ZrC Coating in a Fluidized Bed Chemical Vapor Deposition Furnace; ORNL/TM-2009/214; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 2009. [Google Scholar]
- Aylsworth, J.W. Art of Manufacturing Electrical Incandescing Conductors. U.S. Patent US553296A, 21 January 1896. [Google Scholar]
- Weintraub, E. Reduction of Chemical Compounds. U.S. Patent US1019394A, 5 March 1912. [Google Scholar]
- Langmuir, I. Chemical Reactions at Low Pressures. J. Am. Chem. Soc. 1915, 37, 1139–1167. [Google Scholar] [CrossRef]
- Van Arkel, A.E.; de Boer, J.H. Darstellung von reinem Titanium-, Zirkonium-, Hafnium- und Thoriummetall. Z. Fuer Anorg. Und Allg. Chem. 1925, 148, 345–350. [Google Scholar] [CrossRef]
- Van Arkel, A.E.; de Boer, J.H. Process of Precipitating Metals on an Incandescing Body. U.S. Patent US1671213A, 29 May 1928. [Google Scholar]
- Shapiro, Z.M. Iodide-decomposition process for production of zirconium. In The Metallurgy of Zirconium; Lustman, B., Kerze, F.J., Eds.; National Nuclear Energy Series; McGraw-Hill: New York, NY, USA, 1955; Volume 4, pp. 135–215. [Google Scholar]
- Campbell, I.E.; Sherwood, E.M.; Powell, C.F.; Jones, R.P. Protection of Uranium: Vapor-Deposited Coatings; Battelle Memorial Institute: Columbus, OH, USA, 1953. [Google Scholar]
- Kroll, W.J. How commercial titanium and zirconium were born. J. Frankl. Inst. 1955, 260, 169–192. [Google Scholar] [CrossRef]
- Sale, F.R. The transport and deposition reactions involved in the production of zirconium coatings from mixed iodide vapours. J. Less Common Met. 1969, 19, 53–62. [Google Scholar] [CrossRef]
- Bhatti, M.A.; Copley, D.B.; Shelton, R.A.J. A reinvestigation of the disproportionation of zirconium triiodide. J. Less Common Met. 1977, 55, 293–296. [Google Scholar] [CrossRef]
- Copley, D.B.; Shelton, R.A.J. The disproportionation and non-stoichiometry of zirconium trichloride. J. Less Common Met. 1970, 20, 359–366. [Google Scholar] [CrossRef]
- Normanton, A.S.; Shelton, R.A.J. The disproportionation of zirconium tribromide. J. Less Common Met. 1973, 32, 111–116. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—IV pyrocarbon deposition from methane in the low temperature regime. Carbon 1998, 36, 213–224. [Google Scholar] [CrossRef]
- Miller, J.A.; Melius, C.F. Kinetic and thermodynamic issues in the formation of aromatic compounds in flames of aliphatic fuels. Combust. Flame 1992, 91, 21–39. [Google Scholar] [CrossRef]
- Hu, C.; Shen, H.; Zhang, S.; Li, H. Methane pyrolysis in preparation of pyrolytic carbon: Thermodynamic and kinetic analysis by density functional theory. Chin. J. Aeronaut. 2020, 33, 1064–1073. [Google Scholar] [CrossRef]
- Pierson, J.F.; Czerwiec, T.; Belmonte, T.; Michel, H. Diagnostic of Ar–BCl3 microwave discharges by optical emission spectroscopy. Surf. Coat. Technol. 1997, 97, 749–754. [Google Scholar] [CrossRef]
- Pierson, J.F.; Belmonte, T.; Michel, H. Low temperature growth mechanism of zirconium diboride films synthesised in flowing microwave Ar–BCl3 post-discharges. Surf. Coat. Technol. 1999, 116–119, 1049–1054. [Google Scholar] [CrossRef]
- Ohshita, Y.; Ishitani, A.; Takada, T. Surface reaction mechanism of SiCl2 with carrier gas H2 in silicon vapor phase epitaxial growth. J. Cryst. Growth 1991, 108, 499–507. [Google Scholar] [CrossRef]
- Ohshita, Y.; Ishitani, A.; Takada, T. Theoretical studies of Si vapor-phase epitaxial growth by ab initio molecular-orbital calculations. Phys. Rev. B Condens. Matter 1990, 41, 12720–12727. [Google Scholar] [CrossRef]
- Wagner, P.; Wahman, L.A.; White, R.W.; Hollabaugh, C.M.; Reiswig, R.D. Factors influencing the chemical vapor deposition of ZrC. J. Nucl. Mater. 1976, 62, 221–228. [Google Scholar] [CrossRef]
- Hollabaugh, C.M.; Wahman, L.A.; Reiswig, R.D.; White, R.W.; Wagner, P. Chemical Vapor Deposition of ZrC Made by Reactions of ZrCl4 with CH4 and with C3H6. Nucl. Technol. 1977, 35, 527–535. [Google Scholar] [CrossRef]
- Ikawa, K. Vapor deposition of zirconium carbide-carbon composites by the chloride process. J. Less Common Met. 1972, 29, 233–239. [Google Scholar] [CrossRef]
- Ogawa, T.; Ikawa, K.; Iwamoto, K. Chemical vapor deposition of ZrC within a spouted bed by bromide process. J. Nucl. Mater. 1981, 97, 104–112. [Google Scholar] [CrossRef]
- Charollais, F.; Fonquernie, S.; Perrais, C.; Perez, M.; Dugne, O.; Cellier, F.; Harbonnier, G.; Vitali, M.-P. CEA and AREVA R&D on HTR fuel fabrication and presentation of the CAPRI experimental manufacturing line. Nucl. Eng. Des. 2006, 236, 534–542. [Google Scholar] [CrossRef]
- Reynolds, G.H. Chemical vapor deposition of ZrC on pyrocarbon-coated fuel particles. J. Nucl. Mater. 1974, 50, 215–216. [Google Scholar] [CrossRef]
- Wallace, T.C. Chemical Vapor Deposition of ZrC in Small Bore Carbon-Composite Tubes; LA-UR-73-692; LANL: Los Alamos, NM, USA, 1973.
- Caputo, A.J. Vapor Deposition of Metal Carbides; Y-1852 (pt 4); Oak Ridge Y-12 Plant: Oak Ridge, TN, USA, 1973. [Google Scholar]
- Don, J.; Wright, M.A. Investigations of Oxidation Protection Systems for Carbon-Carbon Composites Formed by Chemical Vapor Deposition and Plasma-Assisted Chemical Vapor Deposition Techniques; ADA232800; Bolling AFB: Washington, DC, USA, 1991. [Google Scholar]
- Samoilenko, V.G.; Pereselentseva, L.N. Deposition of zirconium carbide coatings acting as diffusion barriers in composites consisting of a metallic matrix and refractory metal fibers. Sov. Powder Metall. Met. Ceram. 1975, 14, 725–728. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, C.H.; Kim, D.J.; Park, J.Y. Effect of H2 dilution gas on the growth of ZrC during low pressure chemical vapor deposition in the ZrCl4–CH4–Ar system. Surf. Coat. Technol. 2008, 203, 87–90. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, C.H.; Kim, D.J.; Park, J.Y. Temperature dependency of the LPCVD growth of ZrC with the ZrCl4–CH4–H2 system. Surf. Coat. Technol. 2008, 203, 324–328. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Cheng, L.; Wang, Y. Morphologies and growth mechanisms of zirconium carbide films by chemical vapor deposition. J. Coat. Technol. Res. 2009, 6, 269–273. [Google Scholar] [CrossRef]
- Kim, J.G.; Park, S.J.; Park, J.Y.; Choi, D.J. The effect of temperature on the growth and properties of chemical vapor deposited ZrC films on SiC-coated graphite substrates. Ceram. Int. 2015, 41, 211–216. [Google Scholar] [CrossRef]
- Hollabaugh, C.M.; Reiswig, R.D.; Wagner, P.; Wahman, L.A.; White, R.W. A new method for coating microspheres with zirconium carbide and zirconium carbide-carbon graded coats. J. Nucl. Mater. 1975, 57, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Q.; Liu, J.; Zhang, L.; Cheng, L. Deposition Mechanism for Chemical Vapor Deposition of Zirconium Carbide Coatings. J. Am. Ceram. Soc. 2008, 91, 1249–1252. [Google Scholar] [CrossRef]
- Lamm, B.W.; McMurray, J.W.; Cakmak, E.; Lance, M.J.; Mitchell, D.J. Leveraging computational thermodynamics to guide SiC-ZrC chemical vapor deposition process development. Surf. Coat. Tech. 2022, 444, 128672. [Google Scholar] [CrossRef]
- Ikawa, K. Vapor deposition of zirconium carbide-carbon composites by the iodide process. J. Less Common Met. 1972, 27, 325–332. [Google Scholar] [CrossRef]
- Ikawa, K.; Iwamoto, K. Coating Microspheres with Zirconium Carbide-Carbon Alloy by Iodide Process. J. Nucl. Sci. Technol. 1974, 11, 263–267. [Google Scholar] [CrossRef]
- Ogawa, T.; Ikawa, K.; Iwamoto, K. Effect of gas composition on the deposition of ZrC-C mixtures: The bromide process. J. Mater. Sci. 1979, 14, 125–132. [Google Scholar] [CrossRef]
- Ikawa, K. Co-deposition of zirconium with carbon by the bromide process. J. Less Common Met. 1976, 44, 207–213. [Google Scholar] [CrossRef]
- Benzinger, W.; Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapour deposition of pyrocarbon: I. Fundamentals of kinetics and chemical reaction engineering. Carbon 1996, 34, 957–966. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—III pyrocarbon deposition from propylene and benzene in the low temperature regime. Carbon 1998, 36, 201–211. [Google Scholar] [CrossRef]
- Jackson, H.F.; Lee, W.E. Properties and Characteristics of ZrC. In Material Properties/Oxide Fuels of Light Water Reactors and Fast Neutron Reactors, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier: Amsterdam, The Netherlands, 2012; pp. 339–372. [Google Scholar]
- Liu, Q.M.; Zhang, L.T.; Meng, Z.X.; Cheng, L.F. Chemical Vapor Deposition (CVD) of ZrC Coatings from ZrCl4-C3H6-H2. Adv. Mater. Res. 2011, 189–193, 648–652. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, J.; Luan, X. Preparation of ZrC-SiC composite coatings by chemical vapor deposition and study of co-deposition mechanism. J. Mater. Sci. Technol. 2019, 35, 2942–2949. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Cheng, L.; Wang, Y. Chemical vapour deposition of zirconium carbide and silicon carbide hybrid whiskers. Mater. Lett. 2010, 64, 552–554. [Google Scholar] [CrossRef]
- Liu, Q.M.; Zhang, L.T.; Liu, J.; Wang, Y.G. Thermodynamic study on codeposition of ZrC-SiC from MTS-ZrCl4-CH4-H2. Inorg. Mater. 2010, 46, 1090–1095. [Google Scholar] [CrossRef]
- Grisdale, R.O.; Pfister, A.C.; Van Roosbroeck, W. Pyrolytic Film Resistors: Carbon and Borocarbon. Bell Syst. Tech. J. 1951, 30, 271–314. [Google Scholar] [CrossRef]
- Grisdale, R.O. The Formation of Black Carbon. J. Appl. Phys. 1953, 24, 1082–1091. [Google Scholar] [CrossRef]
- Haynes, W.M.; Lide, D.R.; Bruno, T.J. (Eds.) Thermochemistry, Electrochemistry, and Solution Chemistry. In CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Baker, F.B.; Storms, E.K.; Holley, C.E. Enthalpy of formation of zirconium carbide. J. Chem. Eng. Data 1969, 14, 244–246. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—II pyrocarbon deposition from ethylene, acetylene and 1,3-butadiene in the low temperature regime. Carbon 1998, 36, 177–199. [Google Scholar] [CrossRef]
- Tamari, N.; Kato, A. Catalytic effect of nickel on the growth of zirconium carbide whiskers by chemical vapor deposition. J. Less Common Met. 1978, 58, 147–160. [Google Scholar] [CrossRef]
- Loumagne, F.; Langlais, F.; Naslain, R. Reactional mechanisms of the chemical vapour deposition of SiC-based ceramics from CH3SiCl3-H2 gas precursor. J. Cryst. Growth 1995, 155, 205–213. [Google Scholar] [CrossRef]
- Fix, R.; Gordon, R.G.; Hoffman, D.M. Chemical vapor deposition of titanium, zirconium, and hafnium nitride thin films. Chem. Mater. 1991, 3, 1138–1148. [Google Scholar] [CrossRef]
- Motojima, S.; Kani, E.; Takahashi, Y.; Sugiyama, K. Impurity activated whisker growth of zirconium nitride by chemical vapour deposition. J. Mater. Sci. 1979, 14, 1495–1499. [Google Scholar] [CrossRef]
- Yajima, A.; Segawa, Y.; Matsuzaki, R.; Saeki, Y. Reaction Process of Zirconium Tetrachloride with Ammonia in the Vapor Phase and Properties of the Zirconium Nitride Formed. Bull. Chem. Soc. Jpn. 1983, 56, 2638–2642. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, K.; Pac, S.; Takahashi, Y.; Motojima, S. Low Temperature Deposition of Metal Nitrides by Thermal Decomposition of Organometallic Compounds. J. Electrochem. Soc. 1975, 122, 1545–1549. [Google Scholar] [CrossRef]
- Chiu, H.-T.; Huang, C.-C. Low-pressure chemical vapor deposition of titanium and zirconium carbonitride thin films from M(NEt2)4 (M = Ti and Zr). Mater. Lett. 1993, 16, 194–199. [Google Scholar] [CrossRef]
- Berndt, H.; Zeng, A.Q.; Stock, H.R.; Mayr, P. Zirconium carbonitride films produced by plasma-assisted metal organic chemical vapour deposition. Surf. Coat. Technol. 1995, 74–75, 369–374. [Google Scholar] [CrossRef]
- Arrieta, M. Low Temperature Chemical Vapor Deposition of Zirconium Nitride in a Fluidized Bed. Doctoral dissertation, Texas A&M University, College Station, TX, USA, 2012. [Google Scholar]
- Arrieta, M.Y.; Keiser, D.D.; Perez-Nunez, D.; McDeavitt, S.M. Fluidized Bed Chemical Vapor Deposition of Zirconium Nitride Films. Nucl. Technol. 2017, 199, 219–226. [Google Scholar] [CrossRef]
- Sudderth, L.; Perez-Nunez, D.; Keiser, D.; McDeavitt, S. Fabrication of ZrN Barrier Coatings for U-Mo Microspheres Via Fluidized Bed Chemical Vapor Deposition Using a Metalorganic Precursor. Nucl. Technol. 2018, 202, 81–93. [Google Scholar] [CrossRef]
- Mittemeijer, E.J. Fundamentals of Nitriding and Nitrocarburizing. In Steel Heat Treating Fundamentals and Processes; Dossett, J.L., Totten, G.E., Eds.; ASM International: Almere, The Netherlands, 2013; Volume 4A. [Google Scholar]
- Mittemeijer, E.J.; Slycke, J.T. Chemical Potentials and Activities of Nitrogen and Carbon Imposed by Gaseous Nitriding and Carburising Atmospheres. Surf. Eng. 1996, 12, 152–162. [Google Scholar] [CrossRef]
- Mittemeijer, E.J.; Somers, M.A.J. Thermodynamics, kinetics, and process control of nitriding. Surf. Eng. 1997, 13, 483–497. [Google Scholar] [CrossRef]
- Grabke, H.J. Reaktionen von Ammoniak, Stickstoff und Wasserstoff an der Oberfläche von Eisen I. Zur Kinetik der Nitrierung von Eisen mit NH3-H2-Gemischen und der Denitrierung. Ber. Bunsenges. Phys. Chem. 1968, 74, 533–541. [Google Scholar] [CrossRef]
- Agte, C.; Moers, K. Methoden zur Reindarstellung hochschmelzender Carbide, Nitride und Boride und Beschreibung einiger ihrer Eigenschaften. Z. Fuer Anorg. Allg. Chem. 1931, 198, 233–275. [Google Scholar] [CrossRef]
- Haynes, W.E.; Lide, D.R.; Bruno, T.J. Properties of the Elements and Inorganic Compounds. In CRC Handbook of Chemistry and Physics, 97th ed.; Haynes, W.M., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 4–71. [Google Scholar]
- Futamoto, M.; Yuito, I.; Kawabe, U. Hafnium carbide and nitride whisker growth by chemical vapor deposition. J. Cryst. Growth 1983, 61, 69–74. [Google Scholar] [CrossRef]
- Wagner, R.S.; Ellis, W.C. Vapor-Liquid-Solid Mechanism of Single Crystal Growth. Appl. Phys. Lett. 1964, 4, 89–90. [Google Scholar] [CrossRef]
- Laidler, K.J. Elementary Reactions in Solution. In Chemical Kinetics, 3rd ed.; Harper & Row: New York, NY, USA, 1987; pp. 183–228. [Google Scholar]
- Rabinowitch, E.; Wood, W.C. The collison mechanism and the primary photochemical process in solutions. Trans. Faraday Soc. 1936, 32, 1381–1387. [Google Scholar] [CrossRef]
- Franck, J.; Rabinowitsch, E. Some remarks about free radicals and the photochemistry of solutions. Trans. Faraday Soc. 1934, 30, 120–130. [Google Scholar] [CrossRef]
- Wedekind, E. Studien über das elementare Zirkonium II. Justus Liebigs Ann. Der Chem. 1913, 395, 149–194. [Google Scholar] [CrossRef] [Green Version]
- Alexander, P.P. Production of Zirconium Nitride. U.S. Patent US2461019A, 8 February 1949. [Google Scholar]
- Tibbetts, G.G. Role of nitrogen atoms in "ion-nitriding". J. Appl. Phys. 1974, 45, 5072–5073. [Google Scholar] [CrossRef]
- Roliński, E.; Konieczny, A.; Sharp, G. Influence of nitriding mechanisms on surface roughness of plasma and gas nitrided/nitrocarburized gray cast iron. Heat Treat. Prog. 2007, 7, 39–46. [Google Scholar]
- Hudis, M. Study of ion-nitriding. J. Appl. Phys. 1973, 44, 1489–1496. [Google Scholar] [CrossRef]
- Michel, H.; Czerwiec, T.; Gantois, M.; Ablitzer, D.; Ricard, A. Progress in the analysis of the mechanisms of ion nitriding. Surf. Coat. Technol. 1995, 72, 103–111. [Google Scholar] [CrossRef]
- Dietrich, H.; Geng, P.; Jacobi, K.; Ertl, G. Sticking coefficient for dissociative adsorption of N2 on Ru single-crystal surfaces. J. Chem. Phys. 1996, 104, 375–381. [Google Scholar] [CrossRef]
- Smith, T. Effect of Surface Coverage and Temperature on the Sticking Coefficient. J. Chem. Phys. 1964, 40, 1805–1812. [Google Scholar] [CrossRef]
- Rie, K.T.; Wöhle, J.; Gebauer, A. Synthesis of thin coatings by plasma-assisted chemical vapour deposition using metallo-organic compounds as precursors. Surf. Coat. Technol. 1993, 59, 202–206. [Google Scholar] [CrossRef]
- Becker, J.S.; Kim, E.; Gordon, R.G. Atomic Layer Deposition of Insulating Hafnium and Zirconium Nitrides. Chem. Mater. 2004, 16, 3497–3501. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Jamison, L.; Seidman, D.N.; Mohamed, W.; Bei, Y.; Pellin, M.J.; Yacout, A.M. Nanocrystalline ZrN thin film development via atomic layer deposition for U-Mo powder. J. Nucl. Mater. 2019, 526, 151770. [Google Scholar] [CrossRef]
- Bradley, D.C.; Thomas, I.M. Metallo-organic compounds containing metal–nitrogen bonds. Part I. Some dialkylamino-derivatives of titanium and zirconium. J. Chem. Soc. 1960, 0, 3857–3861. [Google Scholar] [CrossRef]
- Bradley, D.C.; Torrible, E.G. Metallo-Organic Compounds Containing Metal–Nitrogen Bonds: Part IV. Some Bis-(Primary Amino)-Titanium Compounds. Can. J. Chem. 1963, 41, 134–138. [Google Scholar] [CrossRef]
- Bartlett, R.K. The reaction of primary amines with tetrakis(diethylamino)zirconium. J. Inorg. Nucl. Chem. 1966, 28, 2448–2449. [Google Scholar] [CrossRef]
- Elam, J.W.; Schuisky, M.; Ferguson, J.D.; George, S.M. Surface chemistry and film growth during TiN atomic layer deposition using TDMAT and NH3. Thin Solid Film. 2003, 436, 145–156. [Google Scholar] [CrossRef]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef]
- Amato-Wierda, C.; Wierda, D.A. Chemical vapor deposition of titanium nitride thin films from tetrakis(dimethylamido)titanium and hydrazine as a coreactant. J. Mater. Res. 2000, 15, 2414–2424. [Google Scholar] [CrossRef]
- Truong, C.M.; Chen, P.J.; Corneille, J.S.; Oh, W.S.; Goodman, D.W. Low-Pressure Deposition of TiN Thin Films from a Tetrakis(dimethylamido)titanium Precursor. J. Phys. Chem. 1995, 99, 8831–8842. [Google Scholar] [CrossRef]
- Becker, K.; Ebert, F. Die Kristallstruktur einiger binärer Carbide und Nitride. Z. Phys. 1925, 31, 268–272. [Google Scholar] [CrossRef]
- Wahl, G.; Schlosser, S.; Schmaderer, F. Kinetics of the chlorination of Y and Zr and the deposition of Y- and Zr-oxides by reaction of the chlorides with oxygen. In Proceedings of the The Seventh International Conference on Chemical Vapor Deposition, Los Angeles, CA, USA, 14–19 October 1979; pp. 536–543. [Google Scholar]
- Brennfleck, K.; Fitzer, E.; Mack, G. Basic study of CVD of ZrO2-layers on metal substrates. In Proceedings of the 8th International Conference on Chemical Vapor Deposition, Gouvieux, France, 15–18 September 1981; pp. 672–684. [Google Scholar]
- Sipp, E.; Langlais, F.; Naslain, R. Kinetics of deposition of zirconia-based ceramics from ZrCl4-H2-CO2-Ar gas mixtures. J. Alloy. Compd. 1992, 186, 65–76. [Google Scholar] [CrossRef]
- Minet, J.; Langlais, F.; Naslain, R. Chemical vapor infiltration of zirconia within the pore network of fibrous ceramic materials from ZrCl4H2CO2 gas mixtures. Compos. Sci. Technol. 1990, 37, 79–107. [Google Scholar] [CrossRef]
- Minet, J.; Langlais, F.; Naslain, R.; Bernard, C. On the chemical vapour deposition of zirconia from ZrCl4-H2-CO2-Ar gas mixtures: I. A thermodynamic approach. J. Less Common Met. 1986, 119, 219–235. [Google Scholar] [CrossRef]
- Minet, J.; Langlais, F.; Naslain, R. On the chemical vapour deposition of zirconia from ZrCl4-H2-CO2-Ar gas mixture: II. An experimental approach. J. Less Common Met. 1987, 132, 273–287. [Google Scholar] [CrossRef]
- Tingey, G.L. Kinetics of the Water—Gas Equilibrium Reaction. I. The Reaction of Carbon Dioxide with Hydrogen. J. Phys. Chem. 1966, 70, 1406–1412. [Google Scholar] [CrossRef]
- Bradford, B.W. The water-gas reaction in low-pressure explosions. J. Chem. Soc. (Resumed) 1933, 1557–1563. [Google Scholar] [CrossRef]
- Bustamante, F.; Enick, R.M.; Cugini, A.V.; Killmeyer, R.P.; Howard, B.H.; Rothenberger, K.S.; Ciocco, M.V.; Morreale, B.D.; Chattopadhyay, S.; Shi, S. High-temperature kinetics of the homogeneous reverse water-gas shift reaction. AIChE J. 2004, 50, 1028–1041. [Google Scholar] [CrossRef]
- Graven, W.M.; Long, F.J. Kinetics and Mechanisms of the Two Opposing Reactions of the Equilibrium CO + H2O = CO2 + H2. J. Am. Chem. Soc. 1954, 76, 2602–2607. [Google Scholar] [CrossRef]
- Holgate, H.R.; Tester, J.W. Oxidation of hydrogen and carbon monoxide in sub- and supercritical water: Reaction kinetics, pathways, and water-density effects. 2. Elementary reaction modeling. J. Phys. Chem. 1994, 98, 810–822. [Google Scholar] [CrossRef]
- Fujita, S. Mechanism of the reverse water gas shift reaction over Cu/ZnO catalyst. J. Catal. 1992, 134, 220–225. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Mechanism of CO formation in reverse water-gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Rahtu, A.; Ritala, M. Reaction mechanism studies on the zirconium chloride–water atomic layer deposition process. J. Mater. Chem. 2002, 12, 1484–1489. [Google Scholar] [CrossRef]
- Kukli, K.; Forsgren, K.; Aarik, J.; Uustare, T.; Aidla, A.; Niskanen, A.; Ritala, M.; Leskelä, M.; Hårsta, A. Atomic layer deposition of zirconium oxide from zirconium tetraiodide, water and hydrogen peroxide. J. Cryst. Growth 2001, 231, 262–272. [Google Scholar] [CrossRef]
- Kukli, K.; Forsgren, K.; Ritala, M.; Leskela, M.; Aarik, J.; Harsta, A. Dielectric Properties of Zirconium Oxide Grown by Atomic Layer Deposition from Iodide Precursor. J. Electrochem. Soc. 2001, 148, F227. [Google Scholar] [CrossRef]
- Aarik, J.; Aidla, A.; Mändar, H.; Uustare, T.; Sammelselg, V. Growth kinetics and structure formation of ZrO2 thin films in chloride-based atomic layer deposition process. Thin Solid Film. 2002, 408, 97–103. [Google Scholar] [CrossRef]
- Ritala, M.; Leskelä, M. Zirconium dioxide thin films deposited by ALE using zirconium tetrachloride as precursor. Appl. Surf. Sci. 1994, 75, 333–340. [Google Scholar] [CrossRef]
- Houssa, M.; Tuominen, M.; Naili, M.; Afanas’ev, V.; Stesmans, A.; Haukka, S.; Heyns, M.M. Trap-assisted tunneling in high permittivity gate dielectric stacks. J. Appl. Phys. 2000, 87, 8615–8620. [Google Scholar] [CrossRef]
- Li, B.; Griffiths, K.; Zhang, C.S.; Norton, P.R. The autocatalytic decomposition of water on Zr(0001). Surf. Sci. 1997, 384, 70–80. [Google Scholar] [CrossRef]
- Wuchina, E.; Opila, E.; Opeka, M.; Fahrenholtz, W.G.; Talmy, I.G. UHTCs: Ultra-High Temperature Ceramic Materials for Extreme Environment Applications. Interface 2007, 16, 30–36. [Google Scholar] [CrossRef]
- Pierson, J.F.; Belmonte, T.; Michel, H. Thermodynamic and experimental study of low temperature ZrB2 chemical vapor deposition. Le J. De Phys. IV 2001, 11, Pr3-85–Pr83-91. [Google Scholar] [CrossRef]
- Pierson, H.O.; Mullendore, A.W. Thick boride coatings by chemical vapor deposition. Thin Solid Film. 1982, 95, 99–104. [Google Scholar] [CrossRef]
- Mukaida, M.; Goto, T.; Hirai, T. Preferred orientation of TiB2 plates prepared by CVD of the TiCl4 + B2H6 system. J. Mater. Sci. 1991, 26, 6613–6617. [Google Scholar] [CrossRef]
- Wang, A.; Malé, G. Experimental investigation of the ZrBClH CVD system. J. Eur. Ceram. Soc. 1993, 11, 241–251. [Google Scholar] [CrossRef]
- Jensen, J.A.; Gozum, J.E.; Pollina, D.M.; Girolami, G.S. Titanium, zirconium, and hafnium tetrahydroborates as "tailored" CVD precursors for metal diboride thin films. J. Am. Chem. Soc. 1988, 110, 1643–1644. [Google Scholar] [CrossRef]
- Rice, G.W.; Woodin, R.L. Zirconium Borohydride as a Zirconium Boride Precursor. J. Am. Ceram. Soc. 1988, 71, C-181–C-183. [Google Scholar] [CrossRef]
- Wayda, A.L.; Schneemeyer, L.F.; Opila, R.L. Low-temperature deposition of zirconium and hafnium boride films by thermal decomposition of the metal borohydrides (M[BH4]4). Appl. Phys. Lett. 1988, 53, 361–363. [Google Scholar] [CrossRef]
- Kaufman, L.; Clougherty, E.V. Investigation of Boride Compounds for Very High Temperature Applications Part 2; RTD-TDR-63-4096; ManLabs Inc.: Cambridge, MA, USA, 1965. [Google Scholar]
- Clougherty, E.V.; Hill, R.J.; Rhodes, W.H.; Peters, E.T. Processing and Characterization; ManLabs Inc.: Cambridge, MA, USA; Avco Corp.: Greenwich, CT, USA, 1970. [Google Scholar]
- Pierson, J.F.; Belmonte, T.; Czerwiec, T.; Hertz, D.; Michel, H. Low temperature ZrB2 remote plasma enhanced chemical vapor deposition. Thin Solid Film. 2000, 359, 68–76. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, P.; Wei, C.; Han, W.; Zhang, X.; Xu, B. ZrB 2 grains synthesized on graphite by chemical vapor deposition. J. Alloy. Compd. 2017, 698, 27–32. [Google Scholar] [CrossRef]
- Caputo, A.J.; Lackey, W.J.; Wright, I.G.; Angelini, P. Chemical Vapor Deposition of Erosion-Resistant TiB2 Coatings. J. Electrochem. Soc. 1985, 132, 2274–2280. [Google Scholar] [CrossRef]
- Choy, K.L.; Derby, B. The CVD of TiB2 Protective Coating on SiC Monofilament Fibres. Le J. De Phys. IV 1991, 2, C2-697–C2-703. [Google Scholar] [CrossRef]
- Jayaraman, S.; Yang, Y.; Kim, D.Y.; Girolami, G.S.; Abelson, J.R. Hafnium diboride thin films by chemical vapor deposition from a single source precursor. J. Vac. Sci. Technol. A: Vac. Surf. Film. 2005, 23, 1619–1625. [Google Scholar] [CrossRef] [Green Version]
- Sulyaeva, V.S.; Shestakov, V.A.; Rumyantsev, Y.M.; Kosinova, M.L. Synthesis of Zirconium Diboride Films and ZrB2/BCxNy Heterostructures. Inorg. Mater. 2018, 54, 133–139. [Google Scholar] [CrossRef]
- Wang, A.; Male, G. Thermodynamics of the heterogeneous system ZrCl4—BCl3—H2. Calphad 1992, 16, 243–254. [Google Scholar] [CrossRef]
- Naslain, R.; Thebault, J.; Hagenmuller, P.; Bernard, C. The thermodynamic approach to boron chemical vapour deposition based on a computer minimization of the total gibbs free energy. J. Less Common Met. 1979, 67, 85–100. [Google Scholar] [CrossRef]
- Lengyel, I.; Jensen, K.F. A chemical mechanism for in situ boron doping during silicon chemical vapor deposition. Thin Solid Film. 2000, 365, 231–241. [Google Scholar] [CrossRef]
- Lamborn, D.R.; Snyder, D.W.; Xi, X.X.; Redwing, J.M. Modeling studies of the chemical vapor deposition of boron films from B2H6. J. Cryst. Growth 2007, 299, 358–364. [Google Scholar] [CrossRef]
- Reynolds, G.J.; Cooper III, C.B.; Gaczi, P.J. Selective titanium disilicide by low-pressure chemical vapor deposition. J. Appl. Phys. 1989, 65, 3212–3218. [Google Scholar] [CrossRef]
- Mohammadi, V.; de Boer, W.B.; Nanver, L.K. Temperature dependence of chemical-vapor deposition of pure boron layers from diborane. Appl. Phys. Lett. 2012, 101, 111906. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, V.; de Boer, W.B.; Nanver, L.K. An analytical kinetic model for chemical-vapor deposition of pureB layers from diborane. J. Appl. Phys. 2012, 112, 113501. [Google Scholar] [CrossRef]
- Komatsu, S.; Moriyoshi, Y. Influence of atomic hydrogen on the growth reactions of amorphous boron films in a low-pressure B2H6+He+H2plasma. J. Appl. Phys. 1988, 64, 1878–1884. [Google Scholar] [CrossRef]
- Wang, Y.; Hamers, R.J. Boron-induced reconstructions of Si(001) investigated by scanning tunneling microscopy. J. Vac. Sci. Technol. A Vac. Surf. Film. 1995, 13, 1431–1437. [Google Scholar] [CrossRef]
- Wang, Y.; Hamers, R.J.; Kaxiras, E. Atomic structure and bonding of boron-induced reconstructions on Si(001). Phys Rev Lett 1995, 74, 403–406. [Google Scholar] [CrossRef]
- Hamers, R.J.; Wang, Y. Atomically-Resolved Studies of the Chemistry and Bonding at Silicon Surfaces. Chem. Rev. 1996, 96, 1261–1290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, J.; Hamers, R.J. Combined scanning tunneling microscopy and infrared spectroscopy study of the interaction of diborane with Si(001). J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. 1996, 14, 1038–1042. [Google Scholar] [CrossRef]
- Headrick, R.L.; Weir, B.E.; Bevk, J.; Freer, B.S.; Eaglesham, D.J.; Feldman, L.C. Influence of surface reconstruction on the orientation of homoepitaxial silicon films. Phys. Rev. Lett. 1990, 65, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Headrick, R.L.; Levi, A.F.; Luftman, H.S.; Kovalchick, J.; Feldman, L.C. Electrical conduction in the Si(111):B-(sqrt 3 x sqrt 3)R30 degrees/a-Si interface reconstruction. Phys. Rev. B Condens. Matter 1991, 43, 14711–14714. [Google Scholar] [CrossRef] [PubMed]
- Fernández, H.; Grotewold, J.; Previtali, C.M. Thermal decomposition of diborane. Part I. The decomposition mechanism at low conversion and temperature and the inhibiting effect of accumulated hydrogen. J. Chem. Soc. Dalton Trans. 1973, 20, 2090–2095. [Google Scholar] [CrossRef]
- Grimes, R.N. Boron. In Advanced Inorganic Chemistry, 6th ed.; Cotton, F.A., Wilkinson, G., Murillo, C.A., Bochmann, M., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1999; pp. 131–174. [Google Scholar]
- Yu, M.L.; Vitkavage, D.J.; Meyerson, B.S. Doping reaction of PH3 and B2H6 with Si(100). J. Appl. Phys. 1986, 59, 4032–4037. [Google Scholar] [CrossRef]
- Pierson, H.O.; Mullendore, A.W. The chemical vapor deposition of TiB2 from diborane. Thin Solid Film. 1980, 72, 511–516. [Google Scholar] [CrossRef]
- James, B.D.; Nanda, R.K.; Walbridge, M.G.H. Reactions of Lewis bases with tetrahydroborate derivatives of the Group IVa elements. Preparation of new zirconium hydride species. Inorg. Chem. 1967, 6, 1979–1983. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Rettig, S.J.; Westerhaus, A.; Williams, H.D. Synthesis, stability, and fluxional behavior of binuclear mixed-hydride-tetrahydroborate complexes of hafnium(IV): X-ray crystal structure of [[(Me2PCH2SiMe2)2N]Hf(BH4)2](.mu.-H)3[Hf(BH4)[N(SiMe2CH2PMe2)2. Inorg. Chem. 1985, 24, 4316–4325. [Google Scholar] [CrossRef]
- Bird, P.H.; Churchill, M.R. The crystal structure of zirconium(IV) borohydride (at –160°). Chem. Commun. 1967, 403. [Google Scholar] [CrossRef]
- Hedberg, K.; Plato, V. Electron-diffraction investigation of zirconium tetraborohydride, Zr(BH4)4. Inorg. Chem. 1971, 10, 590–594. [Google Scholar] [CrossRef]
- Marks, T.J.; Kolb, J.R. Covalent transition metal, lanthanide, and actinide tetrahydroborate complexes. Chem. Rev. 1977, 77, 263–293. [Google Scholar] [CrossRef]
- Marks, T.J.; Kolb, J.R. Dynamic interligand hydrogen transfer in some η5-cyclopentadienylzirconium and -hafnium tetrahydroborates. J. Am. Chem. Soc. 1975, 97, 3397–3401. [Google Scholar] [CrossRef]
- Temperton, R.H.; Gibson, A.; O’Shea, J.N. In situ XPS analysis of the atomic layer deposition of aluminium oxide on titanium dioxide. Phys. Chem. Chem. Phys. 2019, 21, 1393–1398. [Google Scholar] [CrossRef]
- Kokkonen, E.; Kaipio, M.; Nieminen, H.E.; Rehman, F.; Miikkulainen, V.; Putkonen, M.; Ritala, M.; Huotari, S.; Schnadt, J.; Urpelainen, S. Ambient pressure x-ray photoelectron spectroscopy setup for synchrotron-based in situ and operando atomic layer deposition research. Rev. Sci. Instrum. 2022, 93, 013905. [Google Scholar] [CrossRef]
- Kircher, C.J.; Mayer, J.W.; Tu, K.N.; Ziegler, J.F. Analysis of formation of hafnium silicide on silicon. Appl. Phys. Lett. 1973, 22, 81–83. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Mayer, J.W.; Kircher, C.J.; Tu, K.N. Kinetics of the formation of hafnium silicides on silicon. J. Appl. Phys. 1973, 44, 3851–3857. [Google Scholar] [CrossRef]
- Murarka, S.P. Thermodynamic Considerations. In Silicides for VLSI Applications; Academic Press: New York, NY, USA, 1983; pp. 71–98. [Google Scholar]
- Kematick, R.J.; Myers, C.E. Thermodynamics of the Phase Formation of the Titanium Silicides. Chem. Mater. 1996, 8, 287–291. [Google Scholar] [CrossRef]
Figure 1.
Graph of the moles of zirconium chlorides in ZrCl4-H2 mixtures at equilibrium as P°H2 increases, at a total pressure of 50 torr. Line styles (shown in black) indicate P°H2. Calculated in FactSage™ 8.1 (Thermfact Ltd. and GTT-Technologies).
Figure 1.
Graph of the moles of zirconium chlorides in ZrCl4-H2 mixtures at equilibrium as P°H2 increases, at a total pressure of 50 torr. Line styles (shown in black) indicate P°H2. Calculated in FactSage™ 8.1 (Thermfact Ltd. and GTT-Technologies).

Scheme 1.
The deposition of ZrC by the diffusion mechanism. PyC deposition (i) is followed by zirconium deposition (ii). Deposited zirconium diffuses into PyC to form ZrC (iii).
Scheme 1.
The deposition of ZrC by the diffusion mechanism. PyC deposition (i) is followed by zirconium deposition (ii). Deposited zirconium diffuses into PyC to form ZrC (iii).

Scheme 2.
The deposition of ZrC by the droplet mechanism. (i) PyC droplets deposit on the substrate surface. (ii) Surface-bound or gaseous zirconium chlorides diffuse into droplets. (iii) Atomic chlorine evolves from the droplets, forming ZrC—atomic chlorine reacts with hydrogen to form HCl.
Scheme 2.
The deposition of ZrC by the droplet mechanism. (i) PyC droplets deposit on the substrate surface. (ii) Surface-bound or gaseous zirconium chlorides diffuse into droplets. (iii) Atomic chlorine evolves from the droplets, forming ZrC—atomic chlorine reacts with hydrogen to form HCl.

Figure 2.
(A) The moles of hydrocarbons in CH4-H2 mixtures are at equilibrium as P°H2 increases, at a total pressure of 50 torr. Line styles (shown in black) indicate P°H2. (B) Moles of PyC deposited from 50 mol CH4 in CH4-H2 mixtures as P°H2 increases. Calculated in FactSage™ 8.1 (Thermfact Ltd. (Mont-Royal, QC, Canada) and GTT-Technologies (Herzogenrath, Germany). (C) Steady-state deposition rate of PyC from several hydrocarbons as the initial partial pressure of hydrogen increases; data taken from Refs [81,110,120], with lines added to highlight trends.
Figure 2.
(A) The moles of hydrocarbons in CH4-H2 mixtures are at equilibrium as P°H2 increases, at a total pressure of 50 torr. Line styles (shown in black) indicate P°H2. (B) Moles of PyC deposited from 50 mol CH4 in CH4-H2 mixtures as P°H2 increases. Calculated in FactSage™ 8.1 (Thermfact Ltd. (Mont-Royal, QC, Canada) and GTT-Technologies (Herzogenrath, Germany). (C) Steady-state deposition rate of PyC from several hydrocarbons as the initial partial pressure of hydrogen increases; data taken from Refs [81,110,120], with lines added to highlight trends.

Scheme 3.
ALD of TiN. (i) The TDMAT pulse reacts with surface amides and imides. Following the inert gas purge, the NH3 pulse (ii) reduces titanium and regenerates the amide/imide surface.
Scheme 3.
ALD of TiN. (i) The TDMAT pulse reacts with surface amides and imides. Following the inert gas purge, the NH3 pulse (ii) reduces titanium and regenerates the amide/imide surface.

Figure 3.
The growth of ZrO2 by ZrCl4-CO2-H2 via (A) homogeneous or (B) heterogeneous rWGS mechanics. (C) The growth of ZrO2 by ZrCl4-H2O.
Figure 3.
The growth of ZrO2 by ZrCl4-CO2-H2 via (A) homogeneous or (B) heterogeneous rWGS mechanics. (C) The growth of ZrO2 by ZrCl4-H2O.

Figure 4.
The deposition of boron in (A) ZrCl4-BCl3-H2, (B) B2H6, and (C) TiCl4-B2H6-H2.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lamm, B.W.; Mitchell, D.J. Chemical Vapor Deposition of Zirconium Compounds: A Review. Coatings 2023, 13, 266. https://doi.org/10.3390/coatings13020266
AMA Style
Lamm BW, Mitchell DJ. Chemical Vapor Deposition of Zirconium Compounds: A Review. Coatings. 2023; 13(2):266. https://doi.org/10.3390/coatings13020266
Chicago/Turabian StyleLamm, Benjamin Weitkamp, and David Joseph Mitchell. 2023. "Chemical Vapor Deposition of Zirconium Compounds: A Review" Coatings 13, no. 2: 266. https://doi.org/10.3390/coatings13020266
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.