A Perspective on Molecular Structure and Bond-Breaking in Radiation Damage in Serial Femtosecond Crystallography


1
Department of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden
2
Center for Free-Electron Laser Science, Deutsches Elektronen-Synchrotron, Notkestraße 85, DE-22607 Hamburg, Germany
3
School of Science, RMIT University, Melbourne, VIC 3000, Australia
*
Authors to whom correspondence should be addressed.
Crystals 2020, 10(7), 585; https://doi.org/10.3390/cryst10070585
Submission received: 15 June 2020
/
Revised: 29 June 2020
/
Accepted: 30 June 2020
/
Published: 6 July 2020
(This article belongs to the Special Issue Time Resolved Crystallography)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:X-ray free-electron lasers (XFELs) have a unique capability for time-resolved studies of protein dynamics and conformational changes on femto- and pico-second time scales. The extreme intensity of X-ray pulses can potentially cause significant modifications to the sample structure during exposure. Successful time-resolved XFEL crystallography depends on the unambiguous interpretation of the protein dynamics of interest from the effects of radiation damage. Proteins containing relatively heavy elements, such as sulfur or metals, have a higher risk for radiation damage. In metaloenzymes, for example, the dynamics of interest usually occur at the metal centers, which are also hotspots for damage due to the higher atomic number of the elements they contain. An ongoing challenge with such local damage is to understand the residual bonding in these locally ionized systems and bond-breaking dynamics. Here, we present a perspective on radiation damage in XFEL experiments with a particular focus on the impacts for time-resolved protein crystallography. We discuss recent experimental and modelling results of bond-breaking and ion motion at disulfide bonding sites in protein crystals.
1. Introduction
Time-resolved protein crystallography has developed into a powerful capability of X-ray free-electron laser sources (XFELs) [1,2]. Femtosecond XFEL pulses provide substantially higher time-resolution than synchrotron sources [3] and permit experiments on proteins at room temperature [4]. One of the most significant advances of XFEL crystallography is the capacity to outrun radiation damage [5]. Although the total dose in XFEL experiments is far in excess of the synchrotron dose limits [6,7], the timescale of the interaction is so short that it does not adversely affect the quality of the Bragg diffraction in high-resolution experiments [8]. Those early experiments were a striking demonstration of the success of the “diffraction-before-destruction” principle [5].
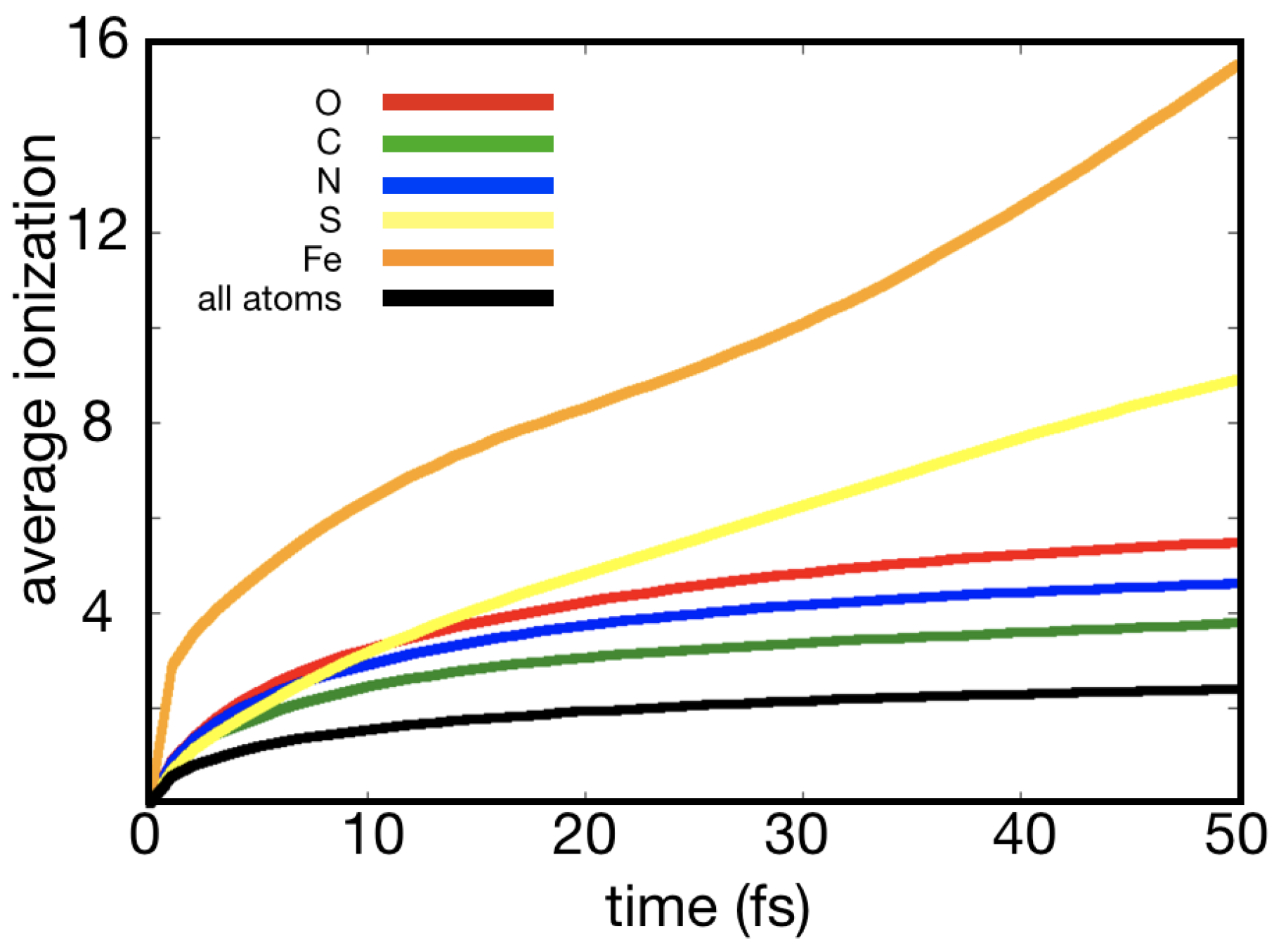
However, a closer examination of the physics of XFEL radiation damage suggests that radiation damage is an ongoing concern for the success of time-resolved XFEL crystallography experiments. The key to appreciating the specific issues for time-resolved crystallography lies in the distinction between “global” and “local” damage effects [9,10]. Global damage processes include the mean stochastic photoionization and thermal motion that affect all atoms equally in the sample, irrespective of their location. Because carbon, oxygen, and nitrogen have a similar atomic number and they make up a substantial proportion of biomolecules, the damage to these elements can be predominantly treated as global damage, as shown in Figure 1. Global damage is relatively well understood and it can be modelled with modified atomic form factors and a factor compensating for the non-equilibrium heating of the system [7,11]. These correction factors differ from the Debye-Waller (temperature) factors, since they describe systems that are far from thermal equilibrium, see Figure 2. They compare favourably to experiment and explain the self-gating effect of XFEL radiation damage [7]. Essentially, the Debye–Waller (temperature) factors from synchrotron-based protein crystallography model the thermal motion of atoms at a fixed temperature, whereas XFEL correction factors model the rapid heating of the sample by the XFEL pulse. The significance of X-ray induced heating effects depends on pulse duration, pulse energy, and the wavelength [11]. Early XFEL protein crystallography experiments identified beam conditions for which the heating effects were highly significant [7] and others for which they could be effectively ignored [8].
Local damage is not independent of the spatial arrangement of the atoms. It occurs when there is significant variation in the ionization rates of different elements or when certain ions exhibit reproducible, non-thermal motion [13]. Local damage is less well-understood, because it is element specific and depends on the local molecular environment around each atom [14]. However, it is local damage that remains an ongoing concern for time-resolved crystallography. This is because local damage has been predicted and observed in the vicinity of heavy ions that occur in, for example, metalloproteins. Local damage has been predicted in photosystem proteins [15] and in ferrodoxin [16]. These examples of site specific damage could lead to mistakes in structural interpretation. Time-resolved experiments are particularly sensitive, because changes as small as a few tenths of an Ångström can be interpreted as significant chemical changes. It is precisely at these very small length scales that we can not yet rule out the impact of local damage [17].
Local damage is also of importance for anomalous dispersion methods of structure determination [18]. Here, the selective absorption at a heavier element is exploited to determine the protein structure ab initio. It has been demonstrated with XFELs, with, for example, gadolinium [19], sulfur [20], and selenium [21].
The modelling of XFEL induced damage processes is complex and approximations are made to target the most relevant damage processes for an experiment. For example, damage simulations of global damage or of ionization rates can ignore the spatial location of the atoms. However, local damage is sensitive to the local molecular structure around damage hotspots. It is precisely this ionization of molecular structure that drives reproducible local ion motion. Therefore, molecular dynamics simulations that track ion motion have an increasing role to play in local damage simulation. Another approximation is that molecular bonds are broken by ionization. This is used in almost all modelling schemes, because the physics of bonding in ionized proteins is not sufficiently well known. However, local reproducible motion can be heavily affected by residual bonding and there are new research directions to incorporate these effects into XFEL damage simulations.
Bond-breaking dynamics have been studied with XFELs for small molecules in the gas phase [22,23,24]. In small molecule studies, molecular fragments can be measured with precision in order to produce a detailed understanding of the fragmentation process. Here, we are concerned with the role of molecular structure in XFEL damage to proteins and protein crystals. The key difference between a protein crystal and a gas phase protein molecule is that the crystals samples trap unbound electrons. The unbound electrons generate ionization cascades via electron-impact ionization, generating from a few to several hundred secondary electrons, depending on material and electron energy [25,26,27,28]. The unbound electrons also cause charge screening, which modifies the Coulomb interactions between ions [29]. Hence, bond-breaking and molecular effects in XFEL damage need to be investigated in protein samples independently of small molecule experiments.
Radiation damage becomes problematic for structural biology when it impacts the structural interpretation of the electron density. Most of the X-rays scattered to high angles interact with bound electrons and, hence, the recovered electron density is primarily determined by the location of the ions. The evidence so far for “damage free” experiments with XFELs [5,8] does not rule out local reproducible ion motion in the vicinity of a heavier element, so-called “damage hot spots”, at least on sub-Ångström scales. An ongoing concern is, thus, whether local damage could impact the interpretation of structure and dynamics of metal clusters in time-resolved studies of metaloproteins.
Here, we present a perspective on radiation damage in XFEL crystallography with a particular focus on the local damage that impacts time-resolved crystallography experiments. Previous reviews of XFEL damage have given significant attention to the progress in understanding global damage and how SFX has achieved its successes to date [5,10]. However, it is the subtler effects of local damage, which are not so well understood, which may have greater relevance to future time-resolved experiments, especially those that push the boundaries of high beam intensity and high time resolution. Our perspective is presented in two parts. In Section 2, we briefly summarize the key damage processes in XFEL-matter interactions. Section 3 deals how they are currently modeled and their particular role in local damage. Subsequently, in Section 4, we discuss the future directions in XFEL damage theory and modeling. These include the role of molecule effects and recent experimental evidence for their occurrence in XFEL experiments. We present new quantum simulations of a disulfide bond breaking to illustrate these future directions.
2. Overview of XFEL Damage Processes
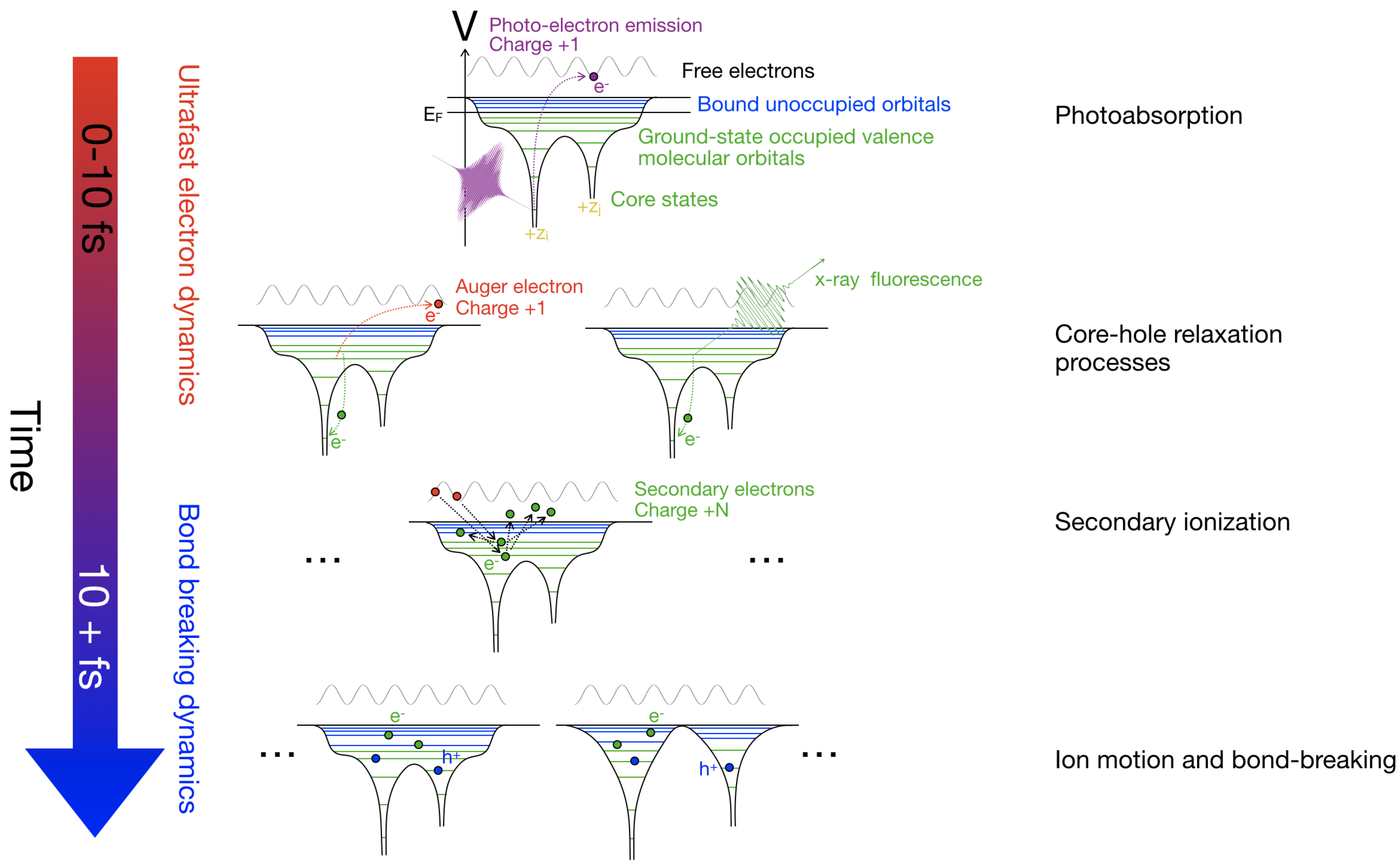
The most relevant damage processes in XFEL science are described in detail in References [10,30]. Here, we provide a brief overview of these processes highlighting the role each can play in experiments that are sensitive to local damage effects. Figure 3 illustrates the different processes described in the text.
2.1. Photoabsorption
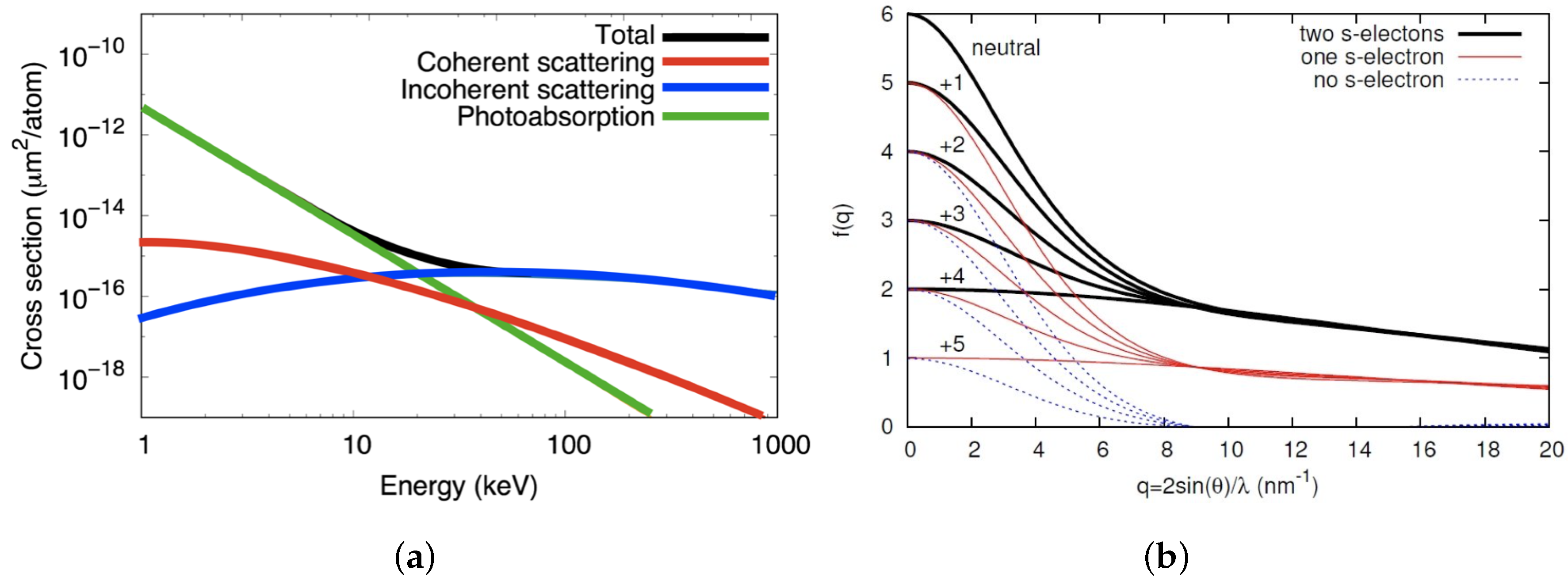
At the wavelengths that are typically used in XFEL experiments, the most likely interaction between X-rays and matter is photoabsorption [31]. The X-ray photon transfers all of its energy to an electron, which is ejected from the atom, leaving a positively charged ion behind. Figure 4 shows that, at 10 keV, the probability for photoabsorption of X-rays by a carbon atom is around ten times higher than that for coherent scattering.
Photoabsorption rates are highly dependent on atomic number. Light elements, such as carbon, nitrogen, and oxygen, have similar photoabsorption rates (this is reflected in Figure 1). When few heavier elements are present, the photoionization of these light elements can be treated globally with an average scaling the atomic scattering factor. More detail on this global modification is given in Section 3.1, below. Heavier elements that are common in proteins and nucleic acids, such as sulfur and phosphorous, or the metals that are contained by metalloproteins, have higher ionization rates. Hence, these heavier elements can function as a damage “hotspots”, where X-ray energy is deposited at much higher rates (see Figure 1). The key issue for crystallography is when this energy leads to reproducible structural modifications that impact accurate structure determination or the interpretation of that structure.
Damage significantly affects the ionization edges. In intense XFEL conditions, they are predicted to broaden and shift to smaller wavelengths [32].
2.2. Core Hole Relaxations Processes
Photoabsorption initiates a chain of subsequent ionization processes that ultimately leads to the destruction of the sample [33]. Photoabsorption is more likely from the core shells than the valence shells, so that the resulting ions are in excited states [11]. Light elements, such as carbon, decay most often from the core-hole state to the ground state via Auger decay [34,35], which results in a second electron (the Auger electron) being emitted. In biological molecules, Auger electrons have energies that are in the range 250 eV–2 keV [36], which is lower than the photoelectrons that account for the majority of the absorbed energy. The lifetimes for Auger decay of light elements with K-shell holes is up to 10 fs and the lifetimes of heavier elements are typically shorter. In heavier elements, radiative decay is more probable than Auger decay. The final state of the Auger process is an ion with core-shells populated, ready for further core-shell ionization. Auger decay along with photoionization has been included in damage modeling since the very first studies [33,37].
The different decay rates and mechanisms between heavy and light elements contribute to the elemental variation in the ionization rates and are, therefore, a contributing factor to local damage. A heavy element may go through a cycle of core-shell ionization, decay and reionization faster than a lighter element. Auger decay has a significantly larger probability than relaxation through X-ray fluorescence for most of the lighter elements relevant for biomolecules.
2.3. Secondary Ionization
Photoelectrons and Auger electrons further ionize the sample, initiating a cascade of low energy electrons (called secondary electrons) [38,39]. The number of secondary electrons is roughly proportional to the energy of the impact electron triggering the cascade [25]. All of the photoelectrons, Auger electrons, and secondary electrons interact with each other and rapidly thermalize. Although some of these electrons recombine with atoms, the average energy of these electrons is sufficiently high that the majority remain unbound for the duration of the pulse-sample interaction (<100 fs). The result is a plasma-like state of ions and a trapped electron gas. The thermalized electron gas is characterized by macroscopic variables, such as the density and temperature [40].
To understand the scale of secondary ionization, consider that, in a near neutral organic sample where the majority of atoms have bound outer shell electrons, an 8 keV photon electron causes up to 400 secondary ionization events [28]. The thermalization happens within tens of femtosecond, which means that 10–30 femtoseconds after a single photo-ionization event, more than 400 secondary ionization events have occurred in the sample [28].
2.4. Ion Motion and Bond-Breaking
As the sample is interacting with the X-rays, the motion of the ions can be affected in two different ways. Either by thermal motion, caused by ion-ion and ion-electron collision, or by electrostatic repulsion, where particles of the same charge repel each other.
Thermal motion of ions increases during the pulse-sample interaction as the trapped electron gas collides with the ions and heats them [41]. This motion is random and it can be effectively modeled as global damage with appropriate temperature factors. The effect of random motion is mitigated by the self-gating effect [7], so that measured Bragg diffraction has greater sensitivity to the initial undamaged crystal than the final disordered crystal.
Ion motion due to electrostatic repulsion occurs when the positively charged ions repel each other via Coulomb forces [33]. Atomic bonds are broken by ionization and remove bonding forces that keep atoms in chemically relevant locations. The motion driven by electrostatic forces can be either reproducible or random [16]. In the random case, the electrostatic forces are ultimately contributing to thermal motion. However, it is the reproducible motion that raises an issue for XFEL crystallography. At a damage hot spot, a heavier ion creates a repulsive potential that drives reproducible motion in the neighbouring ions. The Bragg diffraction is sensitive to the reproducible motion and it will impact the electron density that is recovered by crystallographic analysis.
3. Modelling
3.1. Global Damage Attenuation Factor with Ionization and Ion Motion
The effect of global damage on the Bragg diffraction can be modelled as an attenuation that only depends on the magnitude of the scattering vector (or scattering angle). The attenuation factor is due to the fact that the system is far from thermal equilibrium and is not the conventional Debye–Waller factor, commonly used in conventional X-ray crystallography. The q-dependence of the attenuation factor is not Gaussian in general. Instead, the precise form of the attenuation depends on the distribution of ion states in the sample and the time-dependence of the ion temperatures. The attenuation due to ionization and thermal motion appears in an expression for the diffracted intensity, , as follows:
where is the scattering vector, is classical electron radius, is solid angle of a pixel, is the molecular form factor, is the incident pulse intensity and the time-integral on the right hand side of Equation (1) represents the global correction factor for both ion motion, , and ionization, .
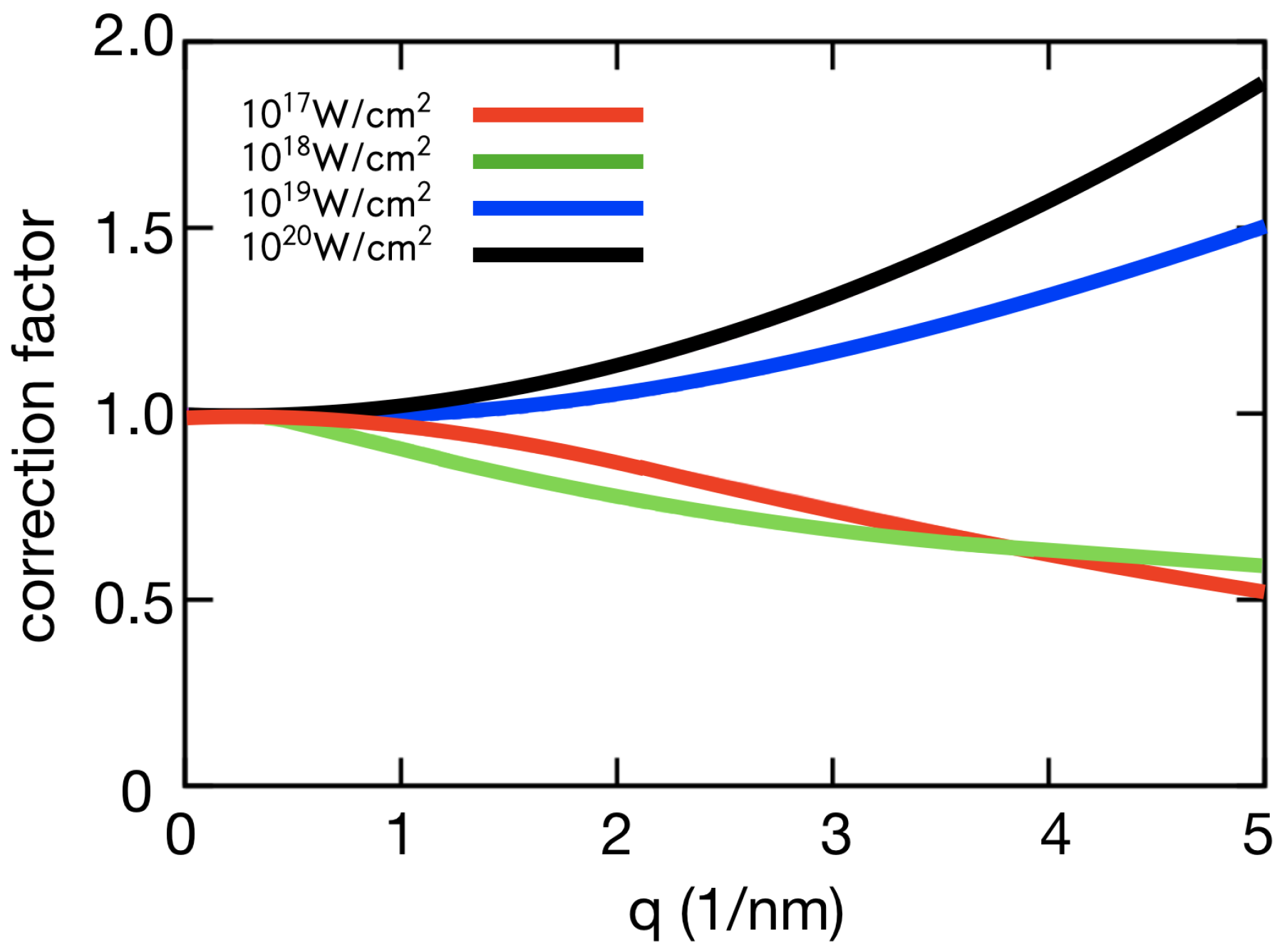
Core-shell electrons make up a greater fraction of the scattering factor at high resolution, as shown in Figure 4. Hence, core-shell ionization is expected to reduce high resolution peaks relative to low resolution peaks, adding to the initial effects of ion motion. Interestingly, valence shell ionization can have the opposite effect. Figure 4 shows that valence electrons contribute to the scattering factor at resolutions up to 3 Å and when valence ionization dominates low resolution peaks can be attenuated relative to high resolution peaks. This effect is predicted by the simulations [11]. At lower intensities ( W cm) ion motion and core-shell ionization dominate and high resolution peaks are most affected, but at higher intensities ( W cm) valence ionization dominates global damage and the low resolution peaks are more greatly affected.
3.2. Damage-Induced Coherence Loss of the Scattered X-rays
Another subtle effect of radiation damage is the dynamical changes in the electron density reduce the coherence of the scattered X-rays. This was predicted to have an impact on XFEL single particle imaging [43], but it also occurs to an extent in crystals [44]. Similar to local damage, damage-induced coherence loss is driven by the variations in the ionization rates of different elements. If the effect is strong, then the diffraction would not be modeled by a electron density, but by multiple real-space distributions known as “modes”. It has been predicted theoretically that a single mode case will be sufficient for most current experimental conditions at XFELs [43,45]. A subtle consequence of the coherence loss is that the physical interpretation of the dominant mode can deviate from the time-average of the electron density, particularly for the relatively heavier elements where the effect is greatest. Hence, the apparent number of bound electrons appearing in the experimentally recovered electron density may not simply be the time-average. This could affect how we relate experimental data back to the theoretical estimates.
3.3. Simulation Tools
The simulation of XFEL damage processes divide into two main approaches: (i) molecular dynamics (MD) [16,28,29,33,42,46,47] and (ii) rate equations [7,11,40,44,48,49]. The molecular dynamics approach simulates the motion of each ion and unbound electron in the sample, subject to Coulomb forces. The rates of ionization are required as input to the simulation to determine the time-dependent ionic charges, the number of unbound electrons, and the energy of the unbound electrons [40]. The earliest study of XFEL damage by Neutze et al. [33] used this approach with a simplistic ionization and bond-breaking model. The conclusions about the time-scales of ion motion are still valid. Since then, the MD approach has been developed in several advanced codes that model all of the above damage processes. The Cimarron Project [50], developed at Lawrence Livermore National Laboratory, simulates dense plasmas and it has been applied to biological samples [16]. The Xraypac [42] suite of programs was developed specifically for XFEL studies and it includes both a MD part, XMDYN, and an atomic ionization part, XATOM [51], which is based on nonrelativistic quantum electrodynamics and perturbation theory within the Hartree–Fock–Slater model. MD codes for XFEL simulations are computationally expensive because sub-attosecond time steps are required to model fast unbound electrons accurately, yet the total simulation time is typically around 100 fs. This places a practical limit on the size of the sample that can be simulated. Nevertheless, MD simulations are necessary when precise details of ion motion are required, as is often the case for local damage.
The rate equations approach to modelling XFEL damage primarily models radiation transfer, the distribution of ionization states for each elements and the statistical properties of the unbound electrons. By ignoring the spatial position of atoms and unbound electrons, rate-equations simulations are inexpensive to run and they are not limited in particle size. Hence, these simulations can facilitate calculations of ion scattering factors and global damage factors, which are insensitive to the ion locations. However, this is also their major disadvantage when compared to MD, as they cannot be used to model specific ion trajectories. As a type of continuum model, the rate-equations approach is highly similar to simulation models of plasmas and warm dense matter physics. The first application of the rate-equations approach to biological samples in FEL beams was conducted by Hau-Riege et al. in 2004 [37]. Non-local thermal equilibrium codes (NLTE), originally developed for warm dense matter physics applications, have been used to model damage in several SFX studies [5,7,11,52].
A recent development in XFEL damage simulation proposes to overcome the computational limitations of MD by combining it with the rate-equations approach. The details of this model are described in an another article in this issue [53] and in Reference [13]. The so-called hybrid plasma/MD model treats the ions as classical particles, but the electrons a continuum. The properties of the unbound electrons are incorporated into ion-ion potentials while using Debye screening. This approach enables faster simulations and larger sample volumes than the conventional MD approach.
Damage simulation based on quantum mechanical approaches, such as quantum molecular dynamics, is appealing, but, in many instances, too slow to sample a sufficient number of initial conditions and, hence, suffer from poor statistics. Approaches that are based on density-functional theory in conjunction with molecular dynamics in the adiabatic approximation, Born–Oppenheimer molecular dynamics, can account for quantum mechanical effects of bonding beyond the level of classical molecular dynamics. For a full account of damage processes, non-adiabatic processes have to be taken account, as the adiabatic approximation assumes that the electrons adjust instantaneously to the ionic positions. Recent implementations of time-dependent density-functional theory in conjunction with the delta-SCF method for describing excited states allow for treating e.g., peptides with account for non-adiabatic effects [54,55]. These simulations give a detailed description for the damage process, including the time-dependent electron density; hence, fine features of the diffraction patterns originating with excited bound electrons can be accounted for.
4. Outstanding Questions and Future Prospects Concerning Local Damage Processes
The theory and simulation tools reviewed in the previous section assumed that damage takes place at the atomic level. Bonds between atoms are generally assumed to be broken through ionization on femtosecond timescales before the ions have time to move significantly. Damage processes that are sensitive to the molecular environment are generally ignored. However, there is recent evidence from both theory and experiment that molecular effects play a non-negligible role in XFEL radiation damage. This includes damage at the sites of relatively heavy elements that is sensitive to the local molecular environment. Developing more sophisticated damage models to include these effects will be given increasing attention in the near future. In this section, we review the recent theoretical developments in molecular nature of XFEL damage, including some previously unpublished quantum-mechanical simulations in order to illustrate the underlying physics.
4.1. Experimental Evidence for Local Damage and Molecular Damage Effects
In early SFX experiments, the electron densities were compared to synchrotron results of known structures to identify “damage free” experimental conditions and it was found that electron densities could be recovered by molecular replacement with sufficient accuracy for atomic modelling [8]. In this context, a damage-free result can be considered to be one in which the XFEL damage does not cause errors in the interpretation or the atomic structure. In studies with lower photon energies and higher photoionization cross-sections, a study of photo-system protein crystals found evidence of global damage due to ion heating [7] and a study of lysozyme crystals contained quantitative discrepancies with synchrotron data attributed to local damage in the SFX data [9].
The development of absorption-based methods of crystallography for XFELs allowed for new insights to be gained in local damage processes. Single-wavelength anomalous dispersion (SAD) experiments have been successful demonstrated with gadolinium [19], sulfur [20] and selenium [21]. The preferential absorption of X-rays by a relatively heavy element is sensitive to the relatively higher ionization of that element to the most common atoms in proteins (H, C, N, and O). Comparing electron densities at sulfur sites, for example, permitted useful comparisons between theoretical models and experiment, strongly confirming the preferential ionization [14]. Interestingly, early theoretical proposals to develop femtosecond multi-wavelength anomalous dispersion (MAD) methods [32] have not yet been realized experimentally, perhaps because of the complex changes to wavelength-dependence of the scattering factor due to the high levels of ionization.
To further pursue the question of local damage, targeted damage experiments have been conducted on high-pulse intensities on systems that are likely to produce these effects. These experiments are typically performed with a nanofocus beam (<300 nm focal spot size) which is around a factor of ten smaller than the typical SFX experiments with a micron-sized focus. A notable example is an experiment on ferrodoxin, which contains iron-sulfur clusters [56]. Reproducible non-random motion of the sulfur atoms was observed. This motion is observed as a deviation of the sulfur ions from their known equilibrium position. Another interesting result for this experiment was that the two iron sites were not equivalent, which indicated the importance of the molecular environment. The iron ions also showed a reduced sensitivity to change of X-ray wavelength above and below the iron edge, which may indicate the changing nature of the absorption edge with ionization state.
Another notable experiment studied protein crystals with a femtosecond X-ray pump and X-ray probe experiment to observe clear local motion of disulfide bonds, carbon-oxygen bonded atoms, and in carbonyl groups [13] (see Figure 5). Remarkably for temporal separations between the two pulses up to 100 fs, the crystals remained sufficiently well-ordered to recover electron densities at a high resolution. This meant that global damage due to the pump pulse was not large and did not “melt” the crystals before the probe pulse arrived. The low levels of global damage were confirmed by the stable B-factors that corresponded to random motion of less than an Ångström. Despite the low levels of global damage, the local motion of ions was clear and reproducible in the recovered electron densities. The disulfide bonds dissociate and rapidly accelerate, before decelerating due to interaction with the surrounding ions, as shown in Figure 5c,d. The displacement increases 2 Å in around 100 fs. The C-O bonds in the backbone increase slightly and then decrease again toward their original value. Elsewhere in the structure, the aromatic side chains show a loss of electron density in the phenyl ring and an asymmetric increase of density near the exterior of the ring. The theoretical simulations shown in Figure 5c,d reproduced the sulfur dissociation and emphasized the importance of Debye-screening and ion-ion interactions in determining the sulfur dynamics. The motion of C-O bonds and carbonyl groups are beyond current theoretical models and likely requires further understanding of molecular effects in highly ionized systems.
4.2. Molecular Effects in Ionization
In conventional crystallography, photoionization from the X-rays is relatively sparse and the damage that it generates to the structure occurs on picosecond timescales. However, in SFX, the situation is very different due to the high photon flux [7]. Atoms can be ionized several times during the short X-ray exposure, as seen in Figure 1. When a molecule is suddenly ionized several times, it is pushed far from its electronic ground state. The electrons remaining in the molecule will redistribute and this, in turn, affects the ability for the atoms to bond with each other. The electronic redistribution caused by the absence of electrons is fast, on the order of femtoseconds [57]. Even if the electron rearrangement is almost instantaneous, the heavier nuclei are relatively slow. The movement of the nuclei is ultimately what is referred to as radiation damage, since it is their positions that are interpreted as the molecular structure.
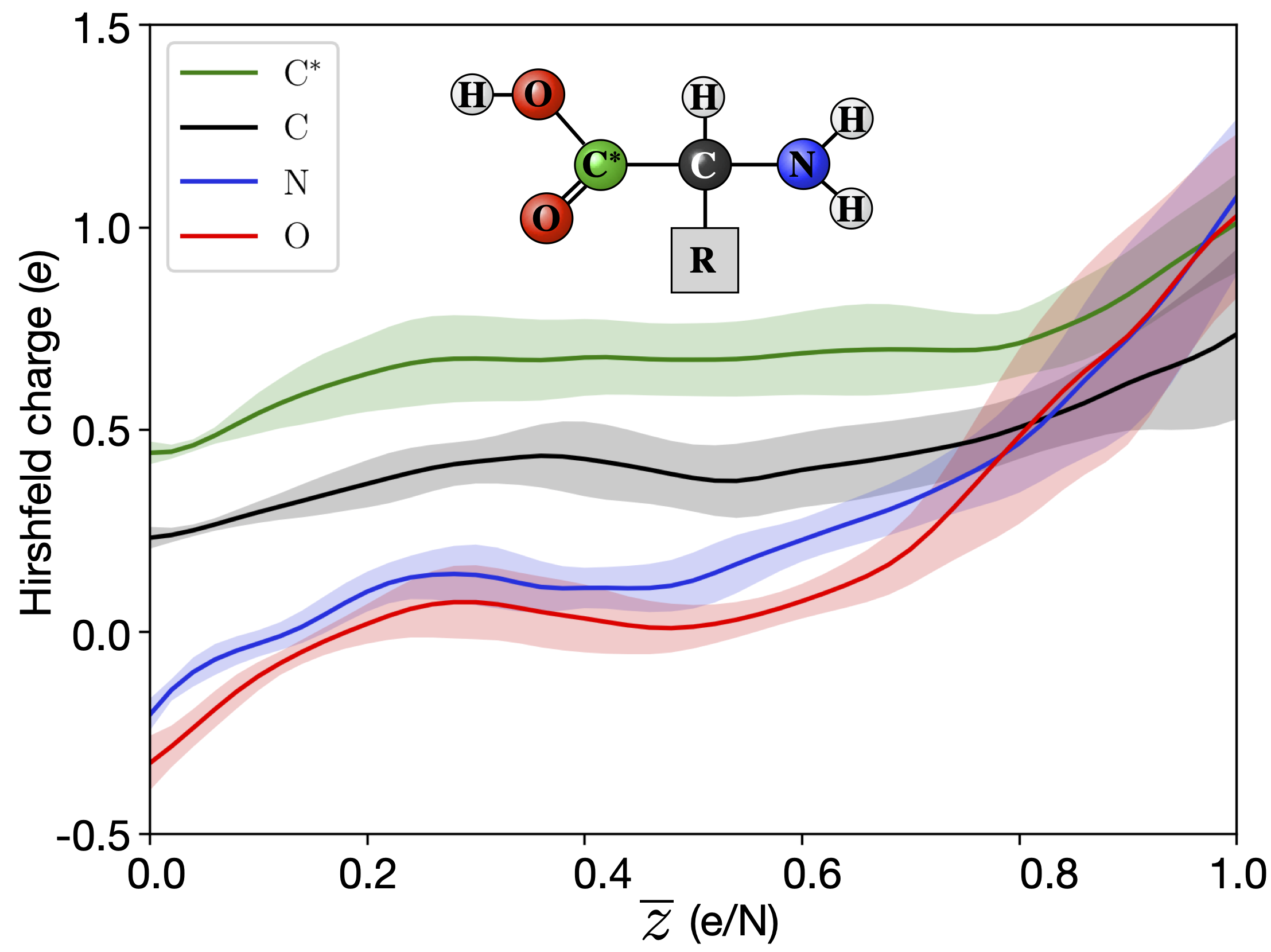
The electronic rearrangement only alters the position of the nuclei as a secondary effect due to the aforementioned influence of electronic states on bonding [30]. However, immediately upon ionization, it affects the scattering of the X-rays, and the intensity recorded at the detector. In an XFEL experiment, an average ionization of above one per atom is commonly reached. The differences in charge between oxygen, nitrogen and carbon can be different at these highly charged states, than in a neutral situation. For example, the oxygens in a carboxyl group normally has a lower charge than carbon. However, when we reach charge states of around one per atom, the oxygens instead get a higher charge than the carbon. QM/MM simulations of the ionization of amino acids have described theses effects [17], as illustrated in Figure 6. As can been seen in the figure this could result in an false interpretation of the structure from the diffracted image.
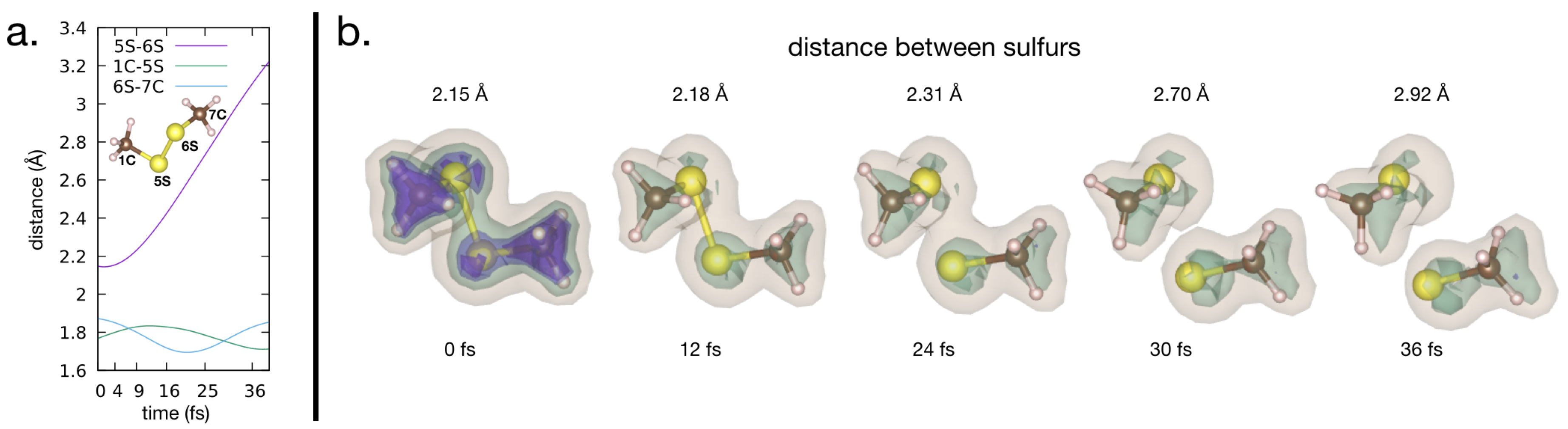
Carbon, oxygen and nitrogen all have similar photon ionization cross sections and, along with hydrogen, are the most common elements in proteins [37]. However, heavier atoms, like sulfur or metals, have a higher ionization cross section. Since these atoms with high cross sections are sparse in a protein, they create hot spots for ionization. The dynamics of atoms in these hot spots cannot be described by global damage, where the sample is described as a homogeneous soup of atoms. Instead, the dynamics on a molecular level are important. In a recent study, the dynamics of the sulfurs in lysozyme exposed to an XFEL pulse was studied [13]. From the experiment it clear that the sulfur-sulfur distance is elongated such that it is obvious that a bond between the sulfurs does not persist, and the sulfur bridge is broken, see Figure 5. This happens on a timescale of 100 fs.
To illustrate both how the electron distribution and nuclei are affected by the heavy ionization from an XFEL, we have conducted QM/MM simulations on a dimethyldisulfide molecule, see Figure 7. Dimethyldisulfide has a sulfur bridge, similar to what can be found in proteins, but it only has a methyl group attached to the sulfur, which makes it computationally favourable. The simulations were done in the same manner as in Grånäs et al. [17], with some minor modifications. It is assumed that the initial ultra-fast electron dynamics happen before significant bond breaking occurs. The ultra-fast dynamics include the photo-emitted core electron, florescence and Auger processes filling the core-hole, and initial cascade dynamics. See Figure 3 for a schematic overview. To focus solely on the bond-breaking dynamics, our QM/MM simulation does not include further photo-ionization processes after the initial ultra-fast dynamics.
This argument is used to start the quantum molecular dynamics simulations with only excited valence configurations considered, i.e., the approximation that all processes involving core-electrons are fast in relation to bond-breaking. Naturally, a multitude of initial conditions occur in an experiment. We sample the starting configurations from Born–Oppenheimer molecular dynamics based on pseudo-potential density functional theory (DFT), as implemented in the Siesta-package [54].
After the initial cascade effects, the molecule resides in an ionized state with a high likelihood of being excited (electrons not occupying the molecular orbitals of the ionized molecule according to the Aufbau principle). The number of possible excited states is very large. However, in case the molecule contains heavier ions, the probability of photoemission is higher in the vicinity of the heavy atom. Subsequently, we use the cross-section of molecular orbitals to the core hole to reduce the number of excited states we treat. We model these excited states according to the delta-SCF procedure. We use statistical trajectory sampling, where trajectories for ionized excited states as well as ionized ground-state (occupied according to the Aufbau principle) can be sampled using TD-DFT and Ehrenfest dynamics to allow for a multitude of initial conditions to be simulated [55].
In the example of the Dimethyldisulfide, we mimic a molecule that has been photo ionized at a sulfur core level, and than relaxed to a excited state via Auger decay to leave a hole in the low-lying valence molecular orbitals with high sulfur character. The molecule in this example is in the +1 state. Although the level of ionization is low, the excited valence configuration breaks the sulfur-sulfur bond. With an increasing level of ionization, we expect that the Coulomb-forces will dominate over the effects on bonding of the excited potential energy surface.
From our simulation it is obvious that the both the electronic structure and the structure of the molecule are affected on the timescales relevant in time resolved SFX experiments. In our simulation, we only have a singly ionized, excited sulfur, but still the sulfur bridge is broken within tenths of femtoseconds. The timescale of the bond breaking agrees well with those reported by Nass et al. [13], as well as in Beyerlein et al. [40], and very clearly indicates that local damage tied to atoms with high ionization cross sections, such as atoms or sulfurs, needs to be taken into account when analyzing data from time resolved SFX experiments.
5. Conclusions
We have reviewed XFEL damage processes with a particular focus on recent theoretical and experimental developments concerning local damage “hot spots” and molecular damage effects. The majority of XFEL damage theory has focused on assuming the bonds are broken early during the pulse-sample interaction, so that ions can be treated independently subject to electrostatic forces and thermal effects. However, recent experiments have provided new insights into how relatively heavy ions drive complex local dynamics. There is a growing understanding that damage modeling will need to develop as well to provide a complete picture of the molecular nature of damage. These molecular effects are most relevant to time-resolved SFX on, for example, metallo-proteins where the metal sites of most relevance of biological function are also those that are most susceptible to local damage. Fundamental studies of X-ray matter interactions will continue to support the progress of SFX for the foreseeable future.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/10/7/585/s1, Input/output for CRETIN calculations associated with Figure 1. Input/output for TDAP/Siesta calculations associated with Figure 7.
Author Contributions
Conceptualization, C.C. and A.V.M.; formal analysis, F.J.J. and O.G.; investigation, C.C., A.V.M., F.J.J. and O.G.; writing–original draft preparation, C.C., A.V.M. and O.G.; writing–review and editing, all authors; supervision, C.C. and O.G.; funding acquisition, C.C., O.G. and A.V.M. All authors have read and agreed to the published version of the manuscript.
Funding
The Swedish Research Council (2018-00740), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT), the Helmholtz Association through the Center for Free-Electron Laser Science at DESY, and the Australian Research Council’s Centre of Excellence Programme. A.V.M. and C.C. acknowledges the funding support from the Australian Research Council Discovery Project grant (DP190103027).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| XFEL | X-ray Free-Electron Laser |
| MD | Molecular Dynamics |
| MM | Molecular Mechanics |
| QM | Quantum Mechanics |
| SFX | Serial Femtosecond Crystallography |
| PSI | Photosystem I |
| NLTE | Non-Local Thermal Equilibrium |
| MAD | Multi-wavelength Anomalous Dispersion |
| DFT | Density Functional Theory |
| TD-DFT | Time Dependent-Density Functional Theory |
| average ionization |
References
- Kern, J.; Yachandra, V.K.; Yano, J. Metalloprotein structures at ambient conditions and in real-time: Biological crystallography and spectroscopy using X-ray free electron lasers. Curr. Opin. Struct. Biol. 2015, 34, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Levantino, M.; Yorke, B.A.; Monteiro, D.C.; Cammarata, M.; Pearson, A.R. Using synchrotrons and XFELs for time-resolved X-ray crystallography and solution scattering experiments on biomolecules. Curr. Opin. Struct. Biol. 2015, 35, 41–48. [Google Scholar] [CrossRef]
- Tenboer, J.; Basu, S.; Zatsepin, N.; Pande, K.; Milathianaki, D.; Frank, M.; Hunter, M.; Boutet, S.; Williams, G.J.; Koglin, J.E.; et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 2014, 346, 1242–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.; Weierstall, U.; Chapman, H. X-ray lasers for structural and dynamic biology. Rep. Prog. Phys. 2012, 75, 102601. [Google Scholar] [CrossRef] [Green Version]
- Chapman, H.N.; Caleman, C.; Timneanu, N. Diffraction before destruction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef]
- Barty, A.; Caleman, C.; Aquila, A.; Timneanu, N.; Lomb, L.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.M.; et al. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat. Photonics 2012, 6, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.M.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [Green Version]
- Lomb, L.; Barends, T.R.M.; Kassemeyer, S.; Aquila, A.; Epp, S.W.; Erk, B.; Foucar, L.; Hartmann, R.; Rudek, B.; Rolles, D.; et al. Radiation damage in protein serial femtosecond crystallography using an x-ray free-electron laser. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 214111. [Google Scholar] [CrossRef] [Green Version]
- Caleman, C.; Martin, A. Where Diffraction Stops and Destruction Begins. In X-ray Free-Electron Lasers—A Revolution in Structural Biology; Springer: Berlin, Germany, 2018; pp. 185–207. [Google Scholar]
- Caleman, C.; Tîmneanu, N.; Martin, A.V.; Jönsson, H.O.; Aquila, A.; Barty, A.; Scott, H.A.; White, T.A.; Chapman, H.N. Ultrafast self-gating Bragg diffraction of exploding nanocrystals in an X-ray laser. Opt. Express 2015, 23, 1213–1231. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, H.O.; Östlin, C.; Scott, H.A.; Chapmana, H.N.; Aplin, S.J.; Timneanu, N.; Caleman, C. FreeDam—A webtool for free-electron laser-induced damage in femtosecond X-ray crystallography. High Energy Density Phys. 2018, 26, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Nass, K.; Gorel, A.; Abdullah, M.M.; Martin, A.V.; Kloos, M.; Marinelli, A.; Aquila, A.; Barends, T.R.M.; Decker, F.J.; Bruce Doak, R.; et al. Structural dynamics in proteins induced by and probed with X-ray free-electron laser pulses. Nat. Commun. 2020, 11, 1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, L.; Son, S.K.; Klinge, M.; Bajt, S.; Barty, A.; Bean, R.; Betzel, C.; Beyerlein, K.R.; Caleman, C.; Doak, R.B.; et al. Electronic damage in S atoms in a native protein crystal induced by an intense X-ray free-electron laser pulse. Struct. Dyn. 2015, 2, 041703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.; Badawi, A.; Obayya, S.S. Radiation Damage in XFEL: Case study from the oxygen-evolving complex of Photosystem II. Sci. Rep. 2016, 6, 36492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hau-Riege, S.P.; Bennion, B.J. Reproducible radiation-damage processes in proteins irradiated by intense x-ray pulses. Phys. Rev. E 2015, 91, 022705. [Google Scholar] [CrossRef] [Green Version]
- Grånäs, O.; Timneanu, N.; Eliah Dawod, I.; Ragazzon, D.; Trygg, S.; Souvatzis, P.; Edvinsson, T.; Caleman, C. Femtosecond bond breaking and charge dynamics in ultracharged amino acids. J. Chem. Phys. 2019, 151, 144307. [Google Scholar] [CrossRef] [Green Version]
- Galli, L.; Barends, T.R.M.; Son, S.K.; White, T.A.; Barty, A.; Botha, S.; Boutet, S.; Caleman, C.; Doak, R.B.; Nanao, M.H.; et al. Crystal Structure of Gadolinium derivative of HEWL solved using Free-Electron Laser radiation. IUCrJ 2015, 2, 627–634. [Google Scholar] [CrossRef]
- Barends, T.R.M.; Foucar, L.; Botha, S.; Doak, R.B.; Shoeman, R.L.; Nass, K.; Koglin, J.E.; Williams, G.J.; Boutet, S.; Messerschmidt, M.; et al. De novo protein crystal structure determination from X-ray free-electron laser data. Nature 2014, 505, 244–247. [Google Scholar] [CrossRef]
- Nakane, T.; Song, C.; Suzuki, M.; Nango, E.; Kobayashi, J.; Masuda, T.; Inoue, S.; Mizohata, E.; Nakatsu, T.; Tanaka, T.; et al. Native sulfur/chlorine SAD phasing for serial femtosecond crystallography. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2519–2525. [Google Scholar] [CrossRef]
- Hunter, M.S.; Yoon, C.H.; DeMirci, H.; Sierra, R.G.; Dao, E.H.; Ahmadi, R.; Aksit, F.; Aquila, A.L.; Ciftci, H.; Guillet, S.; et al. Selenium single-wavelength anomalous diffraction de novo phasing using an X-ray-free electron laser. Nat. Commun. 2016, 7, 13388. [Google Scholar] [CrossRef] [Green Version]
- Erk, B.; Rolles, D.; Foucar, L.; Rudek, B.; Epp, S.W.; Cryle, M.; Bostedt, C.; Schorb, S.; Bozek, J.; Rouzee, A.; et al. Ultrafast Charge Rearrangement and Nuclear Dynamics upon Inner-Shell Multiple Ionization of Small Polyatomic Molecules. Phys. Rev. Lett. 2013, 110, 053003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudenko, A.; Inhester, L.; Hanasaki, K.; Li, X.; Robatjazi, S.J.; Erk, B.; Boll, R.; Toyota, K.; Hao, Y.; Vendrell, O.; et al. Femtosecond response of polyatomic molecules to ultra-intense hard X-rays. Nature 2017, 546, 129–132. [Google Scholar] [CrossRef]
- Fukuzawa, H.; Takanashi, T.; Kukk, E.; Motomura, K.; Wada, S.i.; Nagaya, K.; Ito, Y.; Nishiyama, T.; Nicolas, C.; Kumagai, Y.; et al. Real-time observation of X-ray-induced intramolecular and interatomic electronic decay in CH2I2. Nat. Commun. 2019, 10, 2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaja, B.; London, R.A.; Hajdu, J. Unified model of secondary electron cascades in diamond. J. Appl. Phys. 2005, 97, 064905. [Google Scholar] [CrossRef] [Green Version]
- Tîmneanu, N.; Caleman, C.; Hajdu, J.; van der Spoel, D. Auger Electron Cascades in Water and Ice. Chem. Phys. 2004, 299, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Caleman, C.; Ortiz, C.; Marklund, E.; Bultmark, F.; Gabrysch, M.; Parak, F.G.; Hajdu, J.; Klintenberg, M.; Tîmneanu, N. Radiation damage in biological material: Electronic properties and electron impact ionization in urea. EPL 2009, 85, 18005. [Google Scholar] [CrossRef] [Green Version]
- Caleman, C.; Huldt, G.; Maia, F.R.N.C.; Ortiz, C.; Parak, F.G.; Hajdu, J.; van der Spoel, D.; Chapman, H.N.; Timneanu, N. On the feasibility of nanocrystal imaging using intense and ultrashort X-ray pulses. ACS Nano 2011, 5, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Bergh, M.; Tîmneanu, N.; van der Spoel, D. Model for the dynamics of a water cluster in an x-ray free electron laser beam. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004, 70, 051904. [Google Scholar] [CrossRef]
- Hau-Riege, S. High Intensity X-rays—Interaction with Matter; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- Caleman, C.; Timneanu, N.; Martin, A.V.; White, T.A.; Scott, H.A.; Barty, A.; Aquila, A.; Chapman, H.N. Modeling of XFEL induced ionization and atomic displacement in protein nanocrystals. In Proceedings of the X-ray Free-Electron Lasers: Beam Diagnostics, Beamline Instrumentation, and Applications, San Diego, CA, USA, 13–16 August 2012; Volume 8504. [Google Scholar]
- Son, S.K.; Chapman, H.N.; Santra, R. Multiwavelength anomalous diffraction at high X-ray intensity. Phys. Rev. Lett. 2011, 107, 218102. [Google Scholar] [CrossRef] [PubMed]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Auger, P.; Ehrenfest, P.; Maze, R.; Daudin, J.; Fréon, R.A. Extensive Cosmic-Ray Showers. Rev. Mod. Phys. 1939, 11, 288–291. [Google Scholar] [CrossRef]
- Hubbell, J.H.; Veigele, W.J.; Briggs, E.A.; Brown, R.T.; Cromer, D.T.; Howerton, R.J. Atomic form factors, incoherent scattering functions, and photon scattering cross sections. J. Phys. Chem. Ref. Data 1975, 4, 471–538. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.C.; Vaughan, D. (Eds.) X-ray Data Booklet, 2nd ed.; Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2001. [Google Scholar]
- Hau-Riege, S.P.; London, R.A.; Szoke, A. Dynamics of biological molecules irradiated by short x-ray pulses. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004, 69, 051906. [Google Scholar] [CrossRef] [PubMed]
- Ziaja, B.; van der Spoel, D.; Szoke, A.; Hajdu, J. Radiation induced electron cascade in diamond and amorphous carbon. Int. Soc. Opt. Photonics 2001, 64, 214104. [Google Scholar]
- Ziaja, B.; Szoke, A.; van der Spoel, D.; Hajdu, J. Space-time evolution of electron cascades in diamond. Phys. Rev. B 2002, 66, 024116. [Google Scholar] [CrossRef] [Green Version]
- Beyerlein, K.R.; Jönsson, H.O.; Alonso-Mori, R.; Aquila, A.; Bajt, S.; Barty, A.; Bean, R.; Koglin, J.E.; Messerschmidt, M.; Ragazzon, D.; et al. Ultrafast nonthermal heating of water initiated by an X-ray Free-Electron Laser. Proc. Natl. Acad. Sci. USA 2018, 115, 5652–5657. [Google Scholar] [CrossRef] [Green Version]
- Book, D.L. NRL (Naval Research Laboratory) Plasma Formulary; Naval Research Lab.: Washington, DC, USA, 2007. [Google Scholar]
- Jurek, Z.; Son, S.K.; Ziaja, B.; Santra, R. XMDYN and XATOM: Versatile simulation tools for quantitative modeling of X-ray free-electron laser induced dynamics of matter. J. Appl. Crystallogr. 2016, 49, 1048–1056. [Google Scholar] [CrossRef]
- Quiney, H.M.; Nugent, K.A. Biomolecular imaging and electronic damage using X-ray free-electron lasers. Nat. Phys. 2011, 7, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.V.; Quiney, H.M. Coherence loss by sample dynamics and heterogeneity in x-ray laser diffraction. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 244001. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, U.; Kabachnik, N.M.; Weckert, E.; Vartanyants, I.A. Impact of ultrafast electronic damage in single-particle x-ray imaging experiments. Phys. Rev. B 2012, 86, 051911. [Google Scholar] [CrossRef] [Green Version]
- Jurek, Z.; Oszlányi, G.; Faigel, G. Imaging atom clusters by hard X-ray free-electron lasers. Europhys. Lett. 2004, 65, 491–497. [Google Scholar] [CrossRef]
- Östlin, C.; Timneanu, N.; Caleman, C.; Martin, A. Is Radiation Damage the Limiting Factor in Single Particle Imaging with X-ray Free-Electron Lasers? Struct. Dyn. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Bergh, M.; Huldt, G.; Tîmneanu, N.; Maia, F.R.N.C.; Hajdu, J. Feasibility of imaging living cells at subnanometer resolutions by ultrafast X-ray diffraction. Q. Rev. Biophys. 2008, 41, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Caleman, C.; Bergh, M.; Scott, H.A.; Spence, J.C.H.; Chapman, H.N.; Tîmneanu, N. Simulations of radiation damage in biomolecular nanocrystals induced by femtosecond X-ray pulses. J. Mod. Opt. 2011, 58, 1486–1497. [Google Scholar] [CrossRef]
- Graziani, F.R.; Batista, V.S.; Benedict, L.X.; Castor, J.I.; Chen, H.; Chen, S.N.; Fichtl, C.A.; Glosli, J.N.; Grabowski, P.E.; Graf, A.T.; et al. Large-scale molecular dynamics simulations of dense plasmas: The Cimarron Project. High Energy Density Phys. 2012, 8, 105–131. [Google Scholar] [CrossRef]
- Son, S.K.; Young, L.; Santra, R. Impact of hollow-atom formation on coherent X-ray scattering at high intensity. Phys. Rev. A 2011, 83, 033402. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, H.O.; Tîmneanu, N.; Östlin, C.; Scott, H.A.; Caleman, C. Simulations of radiation damage as a function of the temporal pulse profile in femtosecond X-ray protein crystallography. J. Synchrotron Radiat. 2015, 22, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.; Martin, A.; Quiney, H. Hybrid plasma/molecular-dynamics approach for efficient XFEL radiation damage simulations. Crystals 2012, 10, 478. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Kolesov, G.; Grånäs, O.; Hoyt, R.; Vinichenko, D.; Kaxiras, E. Real-Time TD-DFT with Classical Ion Dynamics: Methodology and Applications. J. Chem. Theory Comput. 2016, 12, 466–476. [Google Scholar] [CrossRef]
- Nass, K.; Foucar, L.; Barends, T.R.M.; Hartmann, E.; Botha, S.; Shoeman, R.L.; Doak, R.B.; Alonso-Mori, R.; Aquila, A.; Bajt, S.; et al. Indications of radiation damage in ferredoxin microcrystals using high-intensity X-FEL beams. J. Synchrotron Radiat. 2015, 22, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björneholm, O.; Bässler, M.; Ausmees, A.; Hjelte, I.; Wang, H.; Feifel, R.; Miron, C.; Piancastelli, M.N.; Svensson, S.; Sorensen, S.; et al. Doppler Splitting of In-Flight Auger Decay of Dissociating Oxygen Molecules: The Localization of Delocalized Core Holes. Phys. Rev. Lett. 2000, 84, 2826–2929. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
Figure 1.
Average number of electrons lost in a Photosystem I (PSI) crystal vs. time. Data shown are simulated using the plasma code CRETIN, as described in [12]; input and output files are available in the Supplementary Materials. The PSI crystal has the relative atomic content of , corresponding to a crystal containing 78% solvent water and a density of 1.077 g/cm. The simulations were done with parameters corresponding to what can be expected at an XFEL source—pulse duration: 50 fs, photon energy: 10 keV, radiant fluece: J/cm. The figure clearly shows how iron and sulfur reach much higher ionization states, which lead to so-called hot spots in the protein crystal.
Figure 1.
Average number of electrons lost in a Photosystem I (PSI) crystal vs. time. Data shown are simulated using the plasma code CRETIN, as described in [12]; input and output files are available in the Supplementary Materials. The PSI crystal has the relative atomic content of , corresponding to a crystal containing 78% solvent water and a density of 1.077 g/cm. The simulations were done with parameters corresponding to what can be expected at an XFEL source—pulse duration: 50 fs, photon energy: 10 keV, radiant fluece: J/cm. The figure clearly shows how iron and sulfur reach much higher ionization states, which lead to so-called hot spots in the protein crystal.

Figure 2.
Correction factor. For systems far from thermal equalibrium, the Debye–Waller factor does not model the global damage well. The correction factor, as presented in Barty et al. [7], includes both the non-thermal heating of the system and atomic form factors modified due to the ionization. Data shown are based on plasma simulations similar to those in Caleman et al. [11], with the following beam parameters: pulse intensities W/cm, pulse length 500 fs, photon energy 6 keV.
Figure 2.
Correction factor. For systems far from thermal equalibrium, the Debye–Waller factor does not model the global damage well. The correction factor, as presented in Barty et al. [7], includes both the non-thermal heating of the system and atomic form factors modified due to the ionization. Data shown are based on plasma simulations similar to those in Caleman et al. [11], with the following beam parameters: pulse intensities W/cm, pulse length 500 fs, photon energy 6 keV.

Figure 3.
Schematic figure of relaxation processes and their approximate time-scales. The initial absorption of an X-ray photon results in the emission of a photoelectron, increasing the molecular charge by one. Heavier elements have a larger number of core-states than light elements, and the absorption cross-section is substantially higher, hence it is more likely for the primary photoabsorption event to occur on a heavier element (Section 2.1). Subsequently, the core-hole is relaxed. For elements lighter than arsenic, the Auger process is more likely (Section 2.2), the emission of the Auger electron further ionizes the molecule. The emitted electron scatters with bound electrons, producing cascades of low energy electrons. The higher electron density of heavier elements makes the cascade more common around a heavy nucleus (Section 2.3. Molecules consisting only of light elements have limited cascades. At this stage, the molecule can reach a highly ionized state. The photo-induced processes leads to ion dynamics, due to the loss of electrons, and the excited state populations, the molecular bonds might break (Section 2.4). The relaxation processes branch out to a large number of possible outcomes, contributing to the challenge of accurately simulating radiation damage.
Figure 3.
Schematic figure of relaxation processes and their approximate time-scales. The initial absorption of an X-ray photon results in the emission of a photoelectron, increasing the molecular charge by one. Heavier elements have a larger number of core-states than light elements, and the absorption cross-section is substantially higher, hence it is more likely for the primary photoabsorption event to occur on a heavier element (Section 2.1). Subsequently, the core-hole is relaxed. For elements lighter than arsenic, the Auger process is more likely (Section 2.2), the emission of the Auger electron further ionizes the molecule. The emitted electron scatters with bound electrons, producing cascades of low energy electrons. The higher electron density of heavier elements makes the cascade more common around a heavy nucleus (Section 2.3. Molecules consisting only of light elements have limited cascades. At this stage, the molecule can reach a highly ionized state. The photo-induced processes leads to ion dynamics, due to the loss of electrons, and the excited state populations, the molecular bonds might break (Section 2.4). The relaxation processes branch out to a large number of possible outcomes, contributing to the challenge of accurately simulating radiation damage.

Figure 4.
(a) Cross sections vs. energy. Atomic cross sections of neutral carbon for photoabsorption, coherent scattering, and incoherent (Compton) scattering. At 10 keV photoabsorption is the dominating interaction. (b) Ionic scattering factors. The ionic scattering factors for carbon in different ionization states. Reproduced with permission from Caleman et al. [11]. The term “s-electrons” in the legend refers to the number of electrons occupying the 1 s shell of the ion.
Figure 4.
(a) Cross sections vs. energy. Atomic cross sections of neutral carbon for photoabsorption, coherent scattering, and incoherent (Compton) scattering. At 10 keV photoabsorption is the dominating interaction. (b) Ionic scattering factors. The ionic scattering factors for carbon in different ionization states. Reproduced with permission from Caleman et al. [11]. The term “s-electrons” in the legend refers to the number of electrons occupying the 1 s shell of the ion.

Figure 5.
Pump-probe serial crystallography. (a) The dissociation of sulfur bridge in a thaumatin crystal observed with pump-probe crystallography at LCLS. (b) Isomorphic different density maps between the two-pulse experiment minus a single pulse measurement with positive density in green and isomorphic different density map of a -sheet region in thaumatin, showing peaks near the carbonyl oxygen atoms. (c) Simulation of S-S dissociation in thaumatin with a molecular dynamics code (XMDYN) and (d) a simulation of lysozyme with a hybrid plasma-MD approach. Both simulations agree on the importance of Debye screening and ion-ion interactions in determining the sulfur dynamics. Reproduced from Nass et al. [13] under a CC4.0 international licence: http://creativecommons.org/licenses/by/4.0/.
Figure 5.
Pump-probe serial crystallography. (a) The dissociation of sulfur bridge in a thaumatin crystal observed with pump-probe crystallography at LCLS. (b) Isomorphic different density maps between the two-pulse experiment minus a single pulse measurement with positive density in green and isomorphic different density map of a -sheet region in thaumatin, showing peaks near the carbonyl oxygen atoms. (c) Simulation of S-S dissociation in thaumatin with a molecular dynamics code (XMDYN) and (d) a simulation of lysozyme with a hybrid plasma-MD approach. Both simulations agree on the importance of Debye screening and ion-ion interactions in determining the sulfur dynamics. Reproduced from Nass et al. [13] under a CC4.0 international licence: http://creativecommons.org/licenses/by/4.0/.

Figure 6.
Local charge redistribution. The deviation from the neutral charge state, described by the Hirshfeld charge [58], per ionization stage () for amino acids. In some parts of the molecule, the charge density is increased due to the reduced electronic screening caused by the ionization. This provides stability to certain bonds to high levels of ionization. Illustration is from Grånäs et al. [17].
Figure 6.
Local charge redistribution. The deviation from the neutral charge state, described by the Hirshfeld charge [58], per ionization stage () for amino acids. In some parts of the molecule, the charge density is increased due to the reduced electronic screening caused by the ionization. This provides stability to certain bonds to high levels of ionization. Illustration is from Grånäs et al. [17].

Figure 7.
Bond breaking. QM/MM simulation of a doubly charged dimethyldisulfide ion, resulting in a breaking of the Sulfur-bridge. (a) Distances between the atoms vs. time. (b) The electron density is illustrated as an iso-surface, and clearly shows how the electron density shared between the two sulfur atoms disappears after the ionization. The simulations were done in a similar manner as in Grånäs et al. [17]; input and output files are available in the Supplementary Materials.
Figure 7.
Bond breaking. QM/MM simulation of a doubly charged dimethyldisulfide ion, resulting in a breaking of the Sulfur-bridge. (a) Distances between the atoms vs. time. (b) The electron density is illustrated as an iso-surface, and clearly shows how the electron density shared between the two sulfur atoms disappears after the ionization. The simulations were done in a similar manner as in Grånäs et al. [17]; input and output files are available in the Supplementary Materials.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Caleman, C.; Jares Junior, F.; Grånäs, O.; Martin, A.V. A Perspective on Molecular Structure and Bond-Breaking in Radiation Damage in Serial Femtosecond Crystallography. Crystals 2020, 10, 585. https://doi.org/10.3390/cryst10070585
AMA Style
Caleman C, Jares Junior F, Grånäs O, Martin AV. A Perspective on Molecular Structure and Bond-Breaking in Radiation Damage in Serial Femtosecond Crystallography. Crystals. 2020; 10(7):585. https://doi.org/10.3390/cryst10070585
Chicago/Turabian StyleCaleman, Carl, Francisco Jares Junior, Oscar Grånäs, and Andrew V. Martin. 2020. "A Perspective on Molecular Structure and Bond-Breaking in Radiation Damage in Serial Femtosecond Crystallography" Crystals 10, no. 7: 585. https://doi.org/10.3390/cryst10070585
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.