Roles of Elongator Dependent tRNA Modification Pathways in Neurodegeneration and Cancer

 , , , and
, , , and
Abstract
:1. Epitranscriptomic Transfer RNA Regulation
2. Disease Related Transfer RNA Modifications
3. Elongator Dependent Transfer RNA Modifications
4. U34 Modifications and Neurodegeneration
5. A Role of Protein Aggregation in Neurodegeneration
6. U34 Modification in Cancer
7. Phosphoregulation of Elongator Involving Kti12 and Kti14/Hrr25
8. Elongator Regulation via Kti11/Dph3 and Kti13
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- El Yacoubi, B.; Bailly, M.; de Crecy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Licht, K.; Jantsch, M.F. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J. Cell Biol. 2016, 213, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Laxman, S.; Sutter, B.M.; Wu, X.; Kumar, S.; Guo, X.; Trudgian, D.C.; Mirzaei, H.; Tu, B.P. Sulfur amino acids regulate translational capacity and metabolic homeostasis through modulation of tRNA thiolation. Cell 2013, 154, 416–429. [Google Scholar] [CrossRef]
- Chan, C.T.; Pang, Y.L.; Deng, W.; Babu, I.R.; Dyavaiah, M.; Begley, T.J.; Dedon, P.C. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012, 3, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.T.; Deng, W.; Li, F.; DeMott, M.S.; Babu, I.R.; Begley, T.J.; Dedon, P.C. Highly predictive reprogramming of tRNA modifications is linked to selective expression of codon-biased genes. Chem. Res. Toxicol. 2015, 28, 978–988. [Google Scholar] [CrossRef]
- Alings, F.; Sarin, L.P.; Fufezan, C.; Drexler, H.C.; Leidel, S.A. An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA 2015, 21, 202–212. [Google Scholar] [CrossRef]
- Han, L.; Kon, Y.; Phizicky, E.M. Functional importance of Psi38 and Psi39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA 2015, 21, 188–201. [Google Scholar] [CrossRef]
- Lin, H.; Miyauchi, K.; Harada, T.; Okita, R.; Takeshita, E.; Komaki, H.; Fujioka, K.; Yagasaki, H.; Goto, Y.I.; Yanaka, K.; et al. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018, 9, 1875. [Google Scholar] [CrossRef]
- Liu, F.; Clark, W.; Luo, G.; Wang, X.; Fu, Y.; Wei, J.; Wang, X.; Hao, Z.; Dai, Q.; Zheng, G.; et al. ALKBH1-mediated tRNA demethylation regulates translation. Cell 2016, 167, 816.e16–828.e16. [Google Scholar] [CrossRef]
- Asano, K.; Suzuki, T.; Saito, A.; Wei, F.Y.; Ikeuchi, Y.; Numata, T.; Tanaka, R.; Yamane, Y.; Yamamoto, T.; Goto, T.; et al. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res. 2018, 46, 1565–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Hartmann, M.; Schuster, I.; Bender, S.; Thuring, K.L.; Helm, M.; Katze, J.R.; Nellen, W.; Lyko, F.; Ehrenhofer-Murray, A.E. Dynamic modulation of Dnmt2-dependent tRNA methylation by the micronutrient queuine. Nucleic Acids Res. 2015, 43, 10952–10962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuorto, F.; Legrand, C.; Cirzi, C.; Federico, G.; Liebers, R.; Muller, M.; Ehrenhofer-Murray, A.E.; Dittmar, G.; Grone, H.J.; Lyko, F. Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lunse, C.E.; Morl, M. tRNA modifications: Impact on structure and thermal adaptation. Biomolecules 2017, 7. [Google Scholar] [CrossRef]
- Torres, A.G.; Batlle, E.; Ribas de Pouplana, L. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Bednarova, A.; Hanna, M.; Durham, I.; VanCleave, T.; England, A.; Chaudhuri, A.; Krishnan, N. Lost in translation: Defects in transfer RNA modifications and neurological disorders. Front. Mol. Neurosci. 2017, 10, 135. [Google Scholar] [CrossRef]
- Sokolowski, M.; Klassen, R.; Bruch, A.; Schaffrath, R.; Glatt, S. Cooperativity between different tRNA modifications and their modification pathways. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 409–418. [Google Scholar] [CrossRef]
- Kirino, Y.; Suzuki, T. Human mitochondrial diseases associated with tRNA wobble modification deficiency. RNA Biol. 2005, 2, 41–44. [Google Scholar] [CrossRef]
- Bohnsack, M.T.; Sloan, K.E. The mitochondrial epitranscriptome: The roles of RNA modifications in mitochondrial translation and human disease. Cell. Mol. Life Sci. CMLS 2018, 75, 241–260. [Google Scholar] [CrossRef]
- Karlsborn, T.; Tukenmez, H.; Chen, C.; Bystrom, A.S. Familial dysautonomia (FD) patients have reduced levels of the modified wobble nucleoside mcm5s2U in tRNA. Biochem. Biophys. Res. Commun. 2014, 454, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Slaugenhaupt, S.A.; Blumenfeld, A.; Gill, S.P.; Leyne, M.; Mull, J.; Cuajungco, M.P.; Liebert, C.B.; Chadwick, B.; Idelson, M.; Reznik, L.; et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet. 2001, 68, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.L.; Coli, R.; Daly, I.W.; Kichula, E.A.; Rork, M.J.; Volpi, S.A.; Ekstein, J.; Rubin, B.Y. Familial dysautonomia is caused by mutations of the IKAP gene. Am. J. Hum. Genet. 2001, 68, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.S.; Srivastava, S.; Farwell, K.D.; Lu, H.M.; Zeng, W.; Lu, H.; Chao, E.C.; Fatemi, A. ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am. J. Med Genet. Part A 2015, 167, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.L.; Lemmens, R.; Miskiewicz, K.; Broom, W.J.; Hansen, V.K.; van Vught, P.W.; Landers, J.E.; Sapp, P.; Van Den Bosch, L.; Knight, J.; et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 2009, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Bento-Abreu, A.; Jager, G.; Swinnen, B.; Rue, L.; Hendrickx, S.; Jones, A.; Staats, K.A.; Taes, I.; Eykens, C.; Nonneman, A.; et al. Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum. Mol. Genet. 2018, 27, 1276–1289. [Google Scholar] [CrossRef]
- Begley, U.; Sosa, M.S.; Avivar-Valderas, A.; Patil, A.; Endres, L.; Estrada, Y.; Chan, C.T.; Su, D.; Dedon, P.C.; Aguirre-Ghiso, J.A.; et al. A human tRNA methyltransferase 9-like protein prevents tumour growth by regulating LIN9 and HIF1-alpha. EMBO Mol. Med. 2013, 5, 366–383. [Google Scholar] [CrossRef]
- Shimada, K.; Nakamura, M.; Anai, S.; De Velasco, M.; Tanaka, M.; Tsujikawa, K.; Ouji, Y.; Konishi, N. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res. 2009, 69, 3157–3164. [Google Scholar] [CrossRef]
- Takeoka, S.; Unoki, M.; Onouchi, Y.; Doi, S.; Fujiwara, H.; Miyatake, A.; Fujita, K.; Inoue, I.; Nakamura, Y.; Tamari, M. Amino-acid substitutions in the IKAP gene product significantly increase risk for bronchial asthma in children. J. Hum. Genet. 2001, 46, 57–63. [Google Scholar] [CrossRef]
- Close, P.; Gillard, M.; Ladang, A.; Jiang, Z.; Papuga, J.; Hawkes, N.; Nguyen, L.; Chapelle, J.P.; Bouillenne, F.; Svejstrup, J.; et al. DERP6 (ELP5) and C3ORF75 (ELP6) regulate tumorigenicity and migration of melanoma cells as subunits of Elongator. J. Biol. Chem. 2012, 287, 32535–32545. [Google Scholar] [CrossRef] [PubMed]
- Rapino, F.; Delaunay, S.; Rambow, F.; Zhou, Z.; Tharun, L.; De Tullio, P.; Sin, O.; Shostak, K.; Schmitz, S.; Piepers, J.; et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 2018, 558, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, S.; Rapino, F.; Tharun, L.; Zhou, Z.; Heukamp, L.; Termathe, M.; Shostak, K.; Klevernic, I.; Florin, A.; Desmecht, H.; et al. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J. Exp. Med. 2016, 213, 2503–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, M.P.; Shaw, M.; Weiner, C.L.; Hobson, L.; Stark, Z.; Rose, K.; Kalscheuer, V.M.; Gecz, J.; Phizicky, E.M. Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum. Mutat. 2015, 36, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y.; Goto, Y.; Campos, Y.; Arenas, J.; Suzuki, T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 2005, 102, 7127–7132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasukawa, T.; Suzuki, T.; Ueda, T.; Ohta, S.; Watanabe, K. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem. 2000, 275, 4251–4257. [Google Scholar] [CrossRef]
- Kopajtich, R.; Nicholls, T.J.; Rorbach, J.; Metodiev, M.D.; Freisinger, P.; Mandel, H.; Vanlander, A.; Ghezzi, D.; Carrozzo, R.; Taylor, R.W.; et al. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 2014, 95, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Suzuki, T.; Ishii, N.; Ueda, T.; Ohta, S.; Watanabe, K. Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNA(Lys) with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett. 2000, 467, 175–178. [Google Scholar] [CrossRef]
- Guan, M.X.; Yan, Q.; Li, X.; Bykhovskaya, Y.; Gallo-Teran, J.; Hajek, P.; Umeda, N.; Zhao, H.; Garrido, G.; Mengesha, E.; et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006, 79, 291–302. [Google Scholar] [CrossRef]
- Zeharia, A.; Shaag, A.; Pappo, O.; Mager-Heckel, A.M.; Saada, A.; Beinat, M.; Karicheva, O.; Mandel, H.; Ofek, N.; Segel, R.; et al. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 2009, 85, 401–407. [Google Scholar] [CrossRef]
- Edvardson, S.; Prunetti, L.; Arraf, A.; Haas, D.; Bacusmo, J.M.; Hu, J.F.; Ta-Shma, A.; Dedon, P.C.; de Crecy-Lagard, V.; Elpeleg, O. tRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur. J. Hum. Genet. EJHG 2017, 25, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Rao, J.; Mollet, G.; Schapiro, D.; Daugeron, M.C.; Tan, W.; Gribouval, O.; Boyer, O.; Revy, P.; Jobst-Schwan, T.; et al. Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly. Nat. Genet. 2017, 49, 1529–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Potapov, V.A.; Smetanina, S.A.; Bel’chikova, L.N.; Suplotova, L.A.; Nosikov, V.V. The carriage of risk variants of CDKAL1 impairs beta-cell function in both diabetic and non-diabetic patients and reduces response to non-sulfonylurea and sulfonylurea agonists of the pancreatic KATP channel. Acta Diabetol. 2011, 48, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, V.; Chen, Y.; Elkahloun, A.; Dutra, A.; Pak, E.; Chandrasekharappa, S. Chromosome 8 BAC array comparative genomic hybridization and expression analysis identify amplification and overexpression of TRMT12 in breast cancer. Genes Chromosom. Cancer 2007, 46, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Han, L.; Faqeih, E.; Ewida, N.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 2016, 135, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, H.A.; Al-Shamsi, A.M.; Ali, B.R.; Al-Gazali, L. A null variant in PUS3 confirms its involvement in intellectual disability and further delineates the associated neurodevelopmental disease. Clin. Genet. 2018, 94, 586–587. [Google Scholar] [CrossRef]
- De Brouwer, A.P.M.; Abou Jamra, R.; Kortel, N.; Soyris, C.; Polla, D.L.; Safra, M.; Zisso, A.; Powell, C.A.; Rebelo-Guiomar, P.; Dinges, N.; et al. Variants in PUS7 Cause Intellectual Disability with Speech Delay, Microcephaly, Short Stature, and Aggressive Behavior. Am. J. Hum. Genet. 2018, 103, 1045–1052. [Google Scholar] [CrossRef]
- Abbasi-Moheb, L.; Mertel, S.; Gonsior, M.; Nouri-Vahid, L.; Kahrizi, K.; Cirak, S.; Wieczorek, D.; Motazacker, M.M.; Esmaeeli-Nieh, S.; Cremer, K.; et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 847–855. [Google Scholar] [CrossRef]
- Martinez, F.J.; Lee, J.H.; Lee, J.E.; Blanco, S.; Nickerson, E.; Gabriel, S.; Frye, M.; Al-Gazali, L.; Gleeson, J.G. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med Genet. 2012, 49, 380–385. [Google Scholar] [CrossRef]
- Fahiminiya, S.; Almuriekhi, M.; Nawaz, Z.; Staffa, A.; Lepage, P.; Ali, R.; Hashim, L.; Schwartzentruber, J.; Abu Khadija, K.; Zaineddin, S.; et al. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin. Genet. 2014, 86, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Watt, F.M. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. CB 2006, 16, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Vachon, C.M.; Sellers, T.A.; Carlson, E.E.; Cunningham, J.M.; Hilker, C.A.; Smalley, R.L.; Schaid, D.J.; Kelemen, L.E.; Couch, F.J.; Pankratz, V.S. Strong evidence of a genetic determinant for mammographic density, a major risk factor for breast cancer. Cancer Res. 2007, 67, 8412–8418. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Dragoni, I.; Chin, S.F.; Spiteri, I.; Kurowski, A.; Provenzano, E.; Green, A.; Ellis, I.O.; Grimmer, D.; Teschendorff, A.; et al. Genomic gain of 5p15 leads to over-expression of Misu (NSUN2) in breast cancer. Cancer Lett. 2010, 289, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Hijazi, H.; Al-Dosari, M.S.; Shaheen, R.; Hashem, A.; Aldahmesh, M.A.; Mohamed, J.Y.; Kentab, A.; Salih, M.A.; Awaji, A.; et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J. Med. Genet. 2013, 50, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Yarham, J.W.; Lamichhane, T.N.; Pyle, A.; Mattijssen, S.; Baruffini, E.; Bruni, F.; Donnini, C.; Vassilev, A.; He, L.; Blakely, E.L.; et al. Defective i6A37 modification of mitochondrial and cytosolic tRNAs results from pathogenic mutations in TRIT1 and its substrate tRNA. PLoS Genet. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed]
- Spinola, M.; Falvella, F.S.; Galvan, A.; Pignatiello, C.; Leoni, V.P.; Pastorino, U.; Paroni, R.; Chen, S.; Skaug, V.; Haugen, A.; et al. Ethnic differences in frequencies of gene polymorphisms in the MYCL1 region and modulation of lung cancer patients’ survival. Lung Cancer 2007, 55, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinola, M.; Galvan, A.; Pignatiello, C.; Conti, B.; Pastorino, U.; Nicander, B.; Paroni, R.; Dragani, T.A. Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene 2005, 24, 5502–5509. [Google Scholar] [CrossRef]
- Yue, Z.; Li, H.T.; Yang, Y.; Hussain, S.; Zheng, C.H.; Xia, J.; Chen, Y. Identification of breast cancer candidate genes using gene co-expression and protein-protein interaction information. Oncotarget 2016, 7, 36092–36100. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Abdel-Salam, G.M.; Guy, M.P.; Alomar, R.; Abdel-Hamid, M.S.; Afifi, H.H.; Ismail, S.I.; Emam, B.A.; Phizicky, E.M.; Alkuraya, F.S. Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 2015, 16, 210. [Google Scholar] [CrossRef]
- Leschziner, G.D.; Coffey, A.J.; Andrew, T.; Gregorio, S.P.; Dias-Neto, E.; Calafato, M.; Bentley, D.R.; Kinton, L.; Sander, J.W.; Johnson, M.R. Q8IYL2 is a candidate gene for the familial epilepsy syndrome of Partial Epilepsy with Pericentral Spikes (PEPS). Epilepsy Res. 2011, 96, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Genin, A.; Lambert, N.; Desir, J.; Pirson, I.; Abdulkarim, B.; Simonis, N.; Drielsma, A.; Marselli, L.; Marchetti, P.; et al. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet. 2013, 9, e1003888. [Google Scholar] [CrossRef] [PubMed]
- Gillis, D.; Krishnamohan, A.; Yaacov, B.; Shaag, A.; Jackman, J.E.; Elpeleg, O. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J. Med. Genet. 2014, 51, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Zung, A.; Kori, M.; Burundukov, E.; Ben-Yosef, T.; Tatoor, Y.; Granot, E. Homozygous deletion of TRMT10A as part of a contiguous gene deletion in a syndrome of failure to thrive, delayed puberty, intellectual disability and diabetes mellitus. Am. J. Med. Genet. Part A 2015, 167, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Yew, T.W.; McCreight, L.; Colclough, K.; Ellard, S.; Pearson, E.R. tRNA methyltransferase homologue gene TRMT10A mutation in young adult-onset diabetes with intellectual disability, microcephaly and epilepsy. Diabet. Med. 2016, 33, e21–e25. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.M.; Thomas, J.; Ross, D.T.; Seitz, R.S.; Ring, B.Z.; Beck, R.A.; Pedersen, H.C.; Munro, A.; Kunkler, I.H.; Campbell, F.M.; et al. Mammostrat as a tool to stratify breast cancer patients at risk of recurrence during endocrine therapy. Breast Cancer Res. BCR 2010, 12, R47. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Casas, K.; Mengesha, E.; Inbal, A.; Fischel-Ghodsian, N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet. 2004, 74, 1303–1308. [Google Scholar] [CrossRef]
- Torres, A.G.; Pineyro, D.; Filonava, L.; Stracker, T.H.; Batlle, E.; Ribas de Pouplana, L. A-to-I editing on tRNAs: Biochemical, biological and evolutionary implications. FEBS Lett. 2014, 588, 4279–4286. [Google Scholar] [CrossRef] [Green Version]
- Esberg, A.; Huang, B.; Johansson, M.J.; Bystrom, A.S. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 2006, 24, 139–148. [Google Scholar] [CrossRef]
- Nedialkova, D.D.; Leidel, S.A. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef]
- Tukenmez, H.; Xu, H.; Esberg, A.; Bystrom, A.S. The role of wobble uridine modifications in +1 translational frameshifting in eukaryotes. Nucleic Acids Res. 2015, 43, 9489–9499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klassen, R.; Bruch, A.; Schaffrath, R. Independent suppression of ribosomal +1 frameshifts by different tRNA anticodon loop modifications. RNA Biol. 2017, 14, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Bhatt, M.J.; Farabaugh, P.J. Codon-specific effects of tRNA anticodon loop modifications on translational misreading errors in the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 2018, 46, 10331–10339. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Schaffrath, R. Collaboration of tRNA modifications and elongation factor eEF1A in decoding and nonsense suppression. Sci. Rep. 2018, 8, 12749. [Google Scholar] [CrossRef] [PubMed]
- Schaffrath, R.; Leidel, S.A. Wobble uridine modifications—a reason to live, a reason to die?! RNA Biol. 2017, 14, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.J.O.; Xu, F.; Bystrom, A.S. Elongator—a tRNA modifying complex that promotes efficient translational decoding. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.J.; Esberg, A.; Huang, B.; Bjork, G.R.; Bystrom, A.S. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008, 28, 3301–3312. [Google Scholar] [CrossRef]
- Huang, B.; Johansson, M.J.; Bystrom, A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 2005, 11, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Otero, G.; Fellows, J.; Li, Y.; de Bizemont, T.; Dirac, A.M.; Gustafsson, C.M.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell 1999, 3, 109–118. [Google Scholar] [CrossRef]
- Winkler, G.S.; Petrakis, T.G.; Ethelberg, S.; Tokunaga, M.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. RNA polymerase II elongator holoenzyme is composed of two discrete subcomplexes. J. Biol. Chem. 2001, 276, 32743–32749. [Google Scholar] [CrossRef]
- Chen, C.; Huang, B.; Eliasson, M.; Ryden, P.; Bystrom, A.S. Elongator complex influences telomeric gene silencing and DNA damage response by its role in wobble uridine tRNA modification. PLoS Genet. 2011, 7, e1002258. [Google Scholar] [CrossRef] [PubMed]
- Karlsborn, T.; Tukenmez, H.; Mahmud, A.K.; Xu, F.; Xu, H.; Bystrom, A.S. Elongator, a conserved complex required for wobble uridine modifications in eukaryotes. RNA Biol. 2014, 11, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R.; Bandau, S.; Stark, M.J. A conserved and essential basic region mediates tRNA binding to the Elp1 subunit of the Saccharomyces cerevisiae Elongator complex. Mol. Microbiol. 2014, 92, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lin, Z.; Li, F.; Diao, W.; Dong, C.; Zhou, H.; Xie, X.; Wang, Z.; Shen, Y.; Long, J. Dimerization of elongator protein 1 is essential for Elongator complex assembly. Proc. Natl. Acad. Sci. USA 2015, 112, 10697–10702. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.; Letoquart, J.; Faux, C.; Taylor, N.M.; Seraphin, B.; Muller, C.W. The Elongator subcomplex Elp456 is a hexameric RecA-like ATPase. Nat. Struct. Mol. Biol. 2012, 19, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.; Zabel, R.; Kolaj-Robin, O.; Onuma, O.F.; Baudin, F.; Graziadei, A.; Taverniti, V.; Lin, T.Y.; Baymann, F.; Seraphin, B.; et al. Structural basis for tRNA modification by Elp3 from Dehalococcoides mccartyi. Nat. Struct. Mol. Biol. 2016, 23, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Setiaputra, D.T.; Cheng, D.T.; Lu, S.; Hansen, J.M.; Dalwadi, U.; Lam, C.H.; To, J.L.; Dong, M.Q.; Yip, C.K. Molecular architecture of the yeast Elongator complex reveals an unexpected asymmetric subunit arrangement. EMBO Rep. 2017, 18, 280–291. [Google Scholar] [CrossRef]
- Dauden, M.I.; Jaciuk, M.; Muller, C.W.; Glatt, S. Structural asymmetry in the eukaryotic Elongator complex. FEBS Lett. 2018, 592, 502–515. [Google Scholar] [CrossRef]
- Paraskevopoulou, C.; Fairhurst, S.A.; Lowe, D.J.; Brick, P.; Onesti, S. The Elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol. Microbiol. 2006, 59, 795–806. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Jablonowski, D.; Zhou, X.; Ren, X.; Hong, X.; Schaffrath, R.; Zhu, J.K.; Gong, Z. Mutations in ABO1/ELO2, a subunit of holo-Elongator, increase abscisic acid sensitivity and drought tolerance in Arabidopsis thaliana. Mol. Cell. Biol. 2006, 26, 6902–6912. [Google Scholar] [CrossRef]
- Lin, F.J.; Shen, L.; Jang, C.W.; Falnes, P.O.; Zhang, Y. Ikbkap/Elp1 deficiency causes male infertility by disrupting meiotic progression. PLoS Genet. 2013, 9, e1003516. [Google Scholar] [CrossRef] [PubMed]
- Mehlgarten, C.; Jablonowski, D.; Wrackmeyer, U.; Tschitschmann, S.; Sondermann, D.; Jager, G.; Gong, Z.; Bystrom, A.S.; Schaffrath, R.; Breunig, K.D. Elongator function in tRNA wobble uridine modification is conserved between yeast and plants. Mol. Microbiol. 2010, 76, 1082–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvadurai, K.; Wang, P.; Seimetz, J.; Huang, R.H. Archaeal Elp3 catalyzes tRNA wobble uridine modification at C5 via a radical mechanism. Nat. Chem. Biol. 2014, 10, 810–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittschieben, B.O.; Otero, G.; de Bizemont, T.; Fellows, J.; Erdjument-Bromage, H.; Ohba, R.; Li, Y.; Allis, C.D.; Tempst, P.; Svejstrup, J.Q. A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol. Cell 1999, 4, 123–128. [Google Scholar] [CrossRef]
- Dalwadi, U.; Yip, C.K. Structural insights into the function of Elongator. Cell. Mol. Life Sci. CMLS 2018, 75, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Dauden, M.I.; Kosinski, J.; Kolaj-Robin, O.; Desfosses, A.; Ori, A.; Faux, C.; Hoffmann, N.A.; Onuma, O.F.; Breunig, K.D.; Beck, M.; et al. Architecture of the yeast Elongator complex. EMBO Rep. 2017, 18, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Glatt, S. tRNA Modification by Elongator Protein 3 (Elp3). In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 1–9. [Google Scholar]
- Fichtner, L.; Frohloff, F.; Burkner, K.; Larsen, M.; Breunig, K.D.; Schaffrath, R. Molecular analysis of KTI12/TOT4, a Saccharomyces cerevisiae gene required for Kluyveromyces lactis zymocin action. Mol. Microbiol. 2002, 43, 783–791. [Google Scholar] [CrossRef]
- Jablonowski, D.; Schaffrath, R. Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem. Soc. Trans. 2007, 35, 1533–1537. [Google Scholar] [CrossRef]
- Fichtner, L.; Frohloff, F.; Jablonowski, D.; Stark, M.J.; Schaffrath, R. Protein interactions within Saccharomyces cerevisiae Elongator, a complex essential for Kluyveromyces lactis zymocicity. Mol. Microbiol. 2002, 45, 817–826. [Google Scholar] [CrossRef]
- Huang, B.; Lu, J.; Bystrom, A.S. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 2008, 14, 2183–2194. [Google Scholar] [CrossRef]
- Frohloff, F.; Fichtner, L.; Jablonowski, D.; Breunig, K.D.; Schaffrath, R. Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 2001, 20, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Frohloff, F.; Jablonowski, D.; Fichtner, L.; Schaffrath, R. Subunit communications crucial for the functional integrity of the yeast RNA polymerase II elongator (gamma-toxin target (TOT)) complex. J. Biol. Chem. 2003, 278, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, D.; Frohloff, F.; Fichtner, L.; Stark, M.J.; Schaffrath, R. Kluyveromyces lactis zymocin mode of action is linked to RNA polymerase II function via Elongator. Mol. Microbiol. 2001, 42, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Kalhor, H.R.; Clarke, S. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol. Cell. Biol. 2003, 23, 9283–9292. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, D.; Zink, S.; Mehlgarten, C.; Daum, G.; Schaffrath, R. tRNAGlu wobble uridine methylation by Trm9 identifies Elongator’s key role for zymocin-induced cell death in yeast. Mol. Microbiol. 2006, 59, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Studte, P.; Zink, S.; Jablonowski, D.; Bar, C.; von der Haar, T.; Tuite, M.F.; Schaffrath, R. tRNA and protein methylase complexes mediate zymocin toxicity in yeast. Mol. Microbiol. 2008, 69, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichtner, L.; Jablonowski, D.; Schierhorn, A.; Kitamoto, H.K.; Stark, M.J.R.; Schaffrath, R. Elongator’s toxin-target (TOT) function is nuclear localization sequence dependent and suppressed by post-translational modification. Mol. Microbiol. 2003, 49, 1297–1307. [Google Scholar] [CrossRef]
- Nakai, Y.; Nakai, M.; Hayashi, H. Thio-modification of yeast cytosolic tRNA requires a ubiquitin-related system that resembles bacterial sulfur transfer systems. J. Biol. Chem. 2008, 283, 27469–27476. [Google Scholar] [CrossRef]
- Nakai, Y.; Nakai, M.; Lill, R.; Suzuki, T.; Hayashi, H. Thio modification of yeast cytosolic tRNA is an iron-sulfur protein-dependent pathway. Mol. Cell. Biol. 2007, 27, 2841–2847. [Google Scholar] [CrossRef]
- Nakai, Y.; Umeda, N.; Suzuki, T.; Nakai, M.; Hayashi, H.; Watanabe, K.; Kagamiyama, H. Yeast Nfs1p is involved in thio-modification of both mitochondrial and cytoplasmic tRNAs. J. Biol. Chem. 2004, 279, 12363–12368. [Google Scholar] [CrossRef]
- Schlieker, C.D.; Van der Veen, A.G.; Damon, J.R.; Spooner, E.; Ploegh, H.L. A functional proteomics approach links the ubiquitin-related modifier Urm1 to a tRNA modification pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 18255–18260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidel, S.; Pedrioli, P.G.; Bucher, T.; Brost, R.; Costanzo, M.; Schmidt, A.; Aebersold, R.; Boone, C.; Hofmann, K.; Peter, M. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 2009, 458, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Noma, A.; Sakaguchi, Y.; Suzuki, T. Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res. 2009, 37, 1335–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judes, A.; Bruch, A.; Klassen, R.; Helm, M.; Schaffrath, R. Sulfur transfer and activation by ubiquitin-like modifier system Uba4*Urm1 link protein urmylation and tRNA thiolation in yeast. Microb. Cell 2016, 3, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Judes, A.; Ebert, F.; Bar, C.; Thuring, K.L.; Harrer, A.; Klassen, R.; Helm, M.; Stark, M.J.; Schaffrath, R. Urmylation and tRNA thiolation functions of ubiquitin-like Uba4.Urm1 systems are conserved from yeast to man. FEBS Lett. 2015, 589, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Leihne, V.; Kirpekar, F.; Vagbo, C.B.; van den Born, E.; Krokan, H.E.; Grini, P.E.; Meza, T.J.; Falnes, P.O. Roles of Trm9- and ALKBH8-like proteins in the formation of modified wobble uridines in Arabidopsis tRNA. Nucleic Acids Res. 2011, 39, 7688–7701. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.P.; Phizicky, E.M. Two-subunit enzymes involved in eukaryotic post-transcriptional tRNA modification. RNA Biol. 2014, 11, 1608–1618. [Google Scholar] [CrossRef] [Green Version]
- Letoquart, J.; van Tran, N.; Caroline, V.; Aleksandrov, A.; Lazar, N.; van Tilbeurgh, H.; Liger, D.; Graille, M. Insights into molecular plasticity in protein complexes from Trm9-Trm112 tRNA modifying enzyme crystal structure. Nucleic Acids Res. 2015, 43, 10989–11002. [Google Scholar] [CrossRef] [Green Version]
- Lentini, J.M.; Ramos, J.; Fu, D. Monitoring the 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) modification in eukaryotic tRNAs via the gamma-toxin endonuclease. RNA 2018, 24, 749–758. [Google Scholar] [CrossRef]
- Van Tran, N.; Muller, L.; Ross, R.L.; Lestini, R.; Letoquart, J.; Ulryck, N.; Limbach, P.A.; de Crecy-Lagard, V.; Cianferani, S.; Graille, M. Evolutionary insights into Trm112-methyltransferase holoenzymes involved in translation between archaea and eukaryotes. Nucleic Acids Res. 2018, 46, 8483–8499. [Google Scholar] [CrossRef]
- Bourgeois, G.; Letoquart, J.; van Tran, N.; Graille, M. Trm112, a protein activator of methyltransferases modifying actors of the eukaryotic translational apparatus. Biomolecules 2017, 7. [Google Scholar] [CrossRef]
- Pedrioli, P.G.; Leidel, S.; Hofmann, K. Urm1 at the crossroad of modifications. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Mizushima, N.; Noda, T.; Ohsumi, Y. A protein conjugation system in yeast with homology to biosynthetic enzyme reaction of prokaryotes. J. Biol. Chem. 2000, 275, 7462–7465. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, A.G.; Schorpp, K.; Schlieker, C.; Buti, L.; Damon, J.R.; Spooner, E.; Ploegh, H.L.; Jentsch, S. Role of the ubiquitin-like protein Urm1 as a noncanonical lysine-directed protein modifier. Proc. Natl. Acad. Sci. USA 2011, 108, 1763–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, Y.; Harada, A.; Hashiguchi, Y.; Nakai, M.; Hayashi, H. Arabidopsis molybdopterin biosynthesis protein Cnx5 collaborates with the ubiquitin-like protein Urm11 in the thio-modification of tRNA. J. Biol. Chem. 2012, 287, 30874–30884. [Google Scholar] [CrossRef] [PubMed]
- Shigi, N. Posttranslational modification of cellular proteins by a ubiquitin-like protein in bacteria. J. Biol. Chem. 2012, 287, 17568–17577. [Google Scholar] [CrossRef]
- Maupin-Furlow, J.A. Prokaryotic ubiquitin-like protein modification. Annu. Rev. Microbiol. 2014, 68, 155–175. [Google Scholar] [CrossRef]
- Scheidt, V.; Judes, A.; Bar, C.; Klassen, R.; Schaffrath, R. Loss of wobble uridine modification in tRNA anticodons interferes with TOR pathway signaling. Microb. Cell 2014, 1, 416–424. [Google Scholar] [CrossRef] [Green Version]
- Zinshteyn, B.; Gilbert, W.V. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 2013, 9, e1003675. [Google Scholar] [CrossRef]
- Khoshnood, B.; Dacklin, I.; Grabbe, C. Urm1: An essential regulator of JNK signaling and oxidative stress in Drosophila melanogaster. Cell. Mol. Life Sci. CMLS 2016, 73, 1939–1954. [Google Scholar] [CrossRef]
- Schmitz, J.; Chowdhury, M.M.; Hanzelmann, P.; Nimtz, M.; Lee, E.Y.; Schindelin, H.; Leimkuhler, S. The sulfurtransferase activity of Uba4 presents a link between ubiquitin-like protein conjugation and activation of sulfur carrier proteins. Biochemistry 2008, 47, 6479–6489. [Google Scholar] [CrossRef] [PubMed]
- Termathe, M.; Leidel, S.A. The Uba4 domain interplay is mediated via a thioester that is critical for tRNA thiolation through Urm1 thiocarboxylation. Nucleic Acids Res. 2018, 46, 5171–5181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, M.W.; Wuebbens, M.M.; Rajagopalan, K.V.; Schindelin, H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature 2001, 414, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Iyer, L.M.; Aravind, L. Natural history of the E1-like superfamily: Implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins 2009, 75, 895–910. [Google Scholar] [CrossRef] [Green Version]
- Marelja, Z.; Stocklein, W.; Nimtz, M.; Leimkuhler, S. A novel role for human Nfs1 in the cytoplasm: Nfs1 acts as a sulfur donor for MOCS3, a protein involved in molybdenum cofactor biosynthesis. J. Biol. Chem. 2008, 283, 25178–25185. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.M.; Dosche, C.; Lohmannsroben, H.G.; Leimkuhler, S. Dual role of the molybdenum cofactor biosynthesis protein MOCS3 in tRNA thiolation and molybdenum cofactor biosynthesis in humans. J. Biol. Chem. 2012, 287, 17297–17307. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Salvesen, G.S.; Wolf, D.A. Urm1 couples sulfur transfer to ubiquitin-like protein function in oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1749–1750. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, R.; Moore, M.J.; Al-Chalabi, A.; Brown, R.H., Jr.; Robberecht, W. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 2010, 33, 249–258. [Google Scholar] [CrossRef]
- Goffena, J.; Lefcort, F.; Zhang, Y.; Lehrmann, E.; Chaverra, M.; Felig, J.; Walters, J.; Buksch, R.; Becker, K.G.; George, L. Elongator and codon bias regulate protein levels in mammalian peripheral neurons. Nat. Commun. 2018, 9, 889. [Google Scholar] [CrossRef]
- Kojic, M.; Gaik, M.; Kiska, B.; Salerno-Kochan, A.; Hunt, S.; Tedoldi, A.; Mureev, S.; Jones, A.; Whittle, B.; Genovesi, L.A.; et al. Elongator mutation in mice induces neurodegeneration and ataxia-like behavior. Nat. Commun. 2018, 9, 3195. [Google Scholar] [CrossRef]
- Koplin, A.; Preissler, S.; Ilina, Y.; Koch, M.; Scior, A.; Erhardt, M.; Deuerling, E. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J. Cell Biol. 2010, 189, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klassen, R.; Ciftci, A.; Funk, J.; Bruch, A.; Butter, F.; Schaffrath, R. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Res. 2016, 44, 10946–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laguesse, S.; Creppe, C.; Nedialkova, D.D.; Prevot, P.P.; Borgs, L.; Huysseune, S.; Franco, B.; Duysens, G.; Krusy, N.; Lee, G.; et al. A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev. Cell 2015, 35, 553–567. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Ruggero, D. A wobbly road to drug resistance in melanoma: tRNA-modifying enzymes in translation reprogramming. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Fakruddin, M.; Wei, F.Y.; Suzuki, T.; Asano, K.; Kaieda, T.; Omori, A.; Izumi, R.; Fujimura, A.; Kaitsuka, T.; Miyata, K.; et al. Defective mitochondrial tRNA taurine modification activates global proteostress and leads to mitochondrial disease. Cell Rep. 2018, 22, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Bruch, A.; Klassen, R.; Schaffrath, R. Unfolded protein response suppression in yeast by loss of tRNA modifications. Genes 2018, 9. [Google Scholar] [CrossRef]
- Thiaville, P.C.; Legendre, R.; Rojas-Benitez, D.; Baudin-Baillieu, A.; Hatin, I.; Chalancon, G.; Glavic, A.; Namy, O.; de Crecy-Lagard, V. Global translational impacts of the loss of the tRNA modification t6A in yeast. Microb. Cell 2016, 3, 29–45. [Google Scholar] [CrossRef]
- Ohshio, I.; Kawakami, R.; Tsukada, Y.; Nakajima, K.; Kitae, K.; Shimanoe, T.; Saigo, Y.; Hase, H.; Ueda, Y.; Jingushi, K.; et al. ALKBH8 promotes bladder cancer growth and progression through regulating the expression of survivin. Biochem. Biophys. Res. Commun. 2016, 477, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vazquez, J.; Vargas-Perez, I.; Sanso, M.; Buhne, K.; Carmona, M.; Paulo, E.; Hermand, D.; Rodriguez-Gabriel, M.; Ayte, J.; Leidel, S.; et al. Modification of tRNA(Lys) UUU by elongator is essential for efficient translation of stress mRNAs. PLoS Genet. 2013, 9, e1003647. [Google Scholar] [CrossRef]
- Dewez, M.; Bauer, F.; Dieu, M.; Raes, M.; Vandenhaute, J.; Hermand, D. The conserved Wobble uridine tRNA thiolase Ctu1-Ctu2 is required to maintain genome integrity. Proc. Natl. Acad. Sci. USA 2008, 105, 5459–5464. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhou, W.; Ji, Y.; Shen, J.; Zhu, X.; Yu, H.; Guo, J.; Pang, Z.; Wei, W. Elongator promotes the migration and invasion of hepatocellular carcinoma cell by the phosphorylation of AKT. Int. J. Biol. Sci. 2018, 14, 518–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladang, A.; Rapino, F.; Heukamp, L.C.; Tharun, L.; Shostak, K.; Hermand, D.; Delaunay, S.; Klevernic, I.; Jiang, Z.; Jacques, N.; et al. Elp3 drives Wnt-dependent tumor initiation and regeneration in the intestine. J. Exp. Med. 2015, 212, 2057–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikman, H.; Nymark, P.; Vayrynen, A.; Jarmalaite, S.; Kallioniemi, A.; Salmenkivi, K.; Vainio-Siukola, K.; Husgafvel-Pursiainen, K.; Knuutila, S.; Wolf, M.; et al. CDK4 is a probable target gene in a novel amplicon at 12q13.3-q14.1 in lung cancer. Genes Chromosom. Cancer 2005, 42, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hirata, S.; Sato, S.; Koga, S.; Fujii, M.; Qi, G.; Ogawa, I.; Takata, T.; Shimamoto, F.; Tatsuka, M. Frequent increased gene copy number and high protein expression of tRNA (cytosine-5-)-methyltransferase (NSUN2) in human cancers. DNA Cell Biol. 2012, 31, 660–671. [Google Scholar] [CrossRef]
- Okamoto, M.; Fujiwara, M.; Hori, M.; Okada, K.; Yazama, F.; Konishi, H.; Xiao, Y.; Qi, G.; Shimamoto, F.; Ota, T.; et al. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet. 2014, 10, e1004639. [Google Scholar] [CrossRef]
- Lu, L.; Zhu, G.; Zeng, H.; Xu, Q.; Holzmann, K. High tRNA transferase NSUN2 gene expression is associated with poor prognosis in head and neck squamous carcinoma. Cancer Investig. 2018, 36, 246–253. [Google Scholar] [CrossRef]
- Gu, C.; Begley, T.J.; Dedon, P.C. tRNA modifications regulate translation during cellular stress. FEBS Lett. 2014, 588, 4287–4296. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.T.; Dyavaiah, M.; DeMott, M.S.; Taghizadeh, K.; Dedon, P.C.; Begley, T.J. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010, 6, e1001247. [Google Scholar] [CrossRef]
- Patil, A.; Dyavaiah, M.; Joseph, F.; Rooney, J.P.; Chan, C.T.; Dedon, P.C.; Begley, T.J. Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle 2012, 11, 3656–3665. [Google Scholar] [CrossRef] [Green Version]
- Mehlgarten, C.; Schaffrath, R. Mutant casein kinase I (Hrr25p/Kti14p) abrogates the G1 cell cycle arrest induced by Kluyveromyces lactiszymocin in budding yeast. Mol. Genet. Genom. MGG 2003, 269, 188–196. [Google Scholar] [CrossRef]
- Jablonowski, D.; Butler, A.R.; Fichtner, L.; Gardiner, D.; Schaffrath, R.; Stark, M.J. Sit4p protein phosphatase is required for sensitivity of Saccharomyces cerevisiae to Kluyveromyces lactis zymocin. Genetics 2001, 159, 1479–1489. [Google Scholar] [PubMed]
- Jablonowski, D.; Taubert, J.E.; Bar, C.; Stark, M.J.; Schaffrath, R. Distinct subsets of Sit4 holophosphatases are required for inhibition of Saccharomyces cerevisiae growth by rapamycin and zymocin. Eukaryot. Cell 2009, 8, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fattah, W.; Jablonowski, D.; Di Santo, R.; Thuring, K.L.; Scheidt, V.; Hammermeister, A.; Ten Have, S.; Helm, M.; Schaffrath, R.; Stark, M.J. Phosphorylation of Elp1 by Hrr25 is required for elongator-dependent tRNA modification in yeast. PLoS Genet. 2015, 11, e1004931. [Google Scholar] [CrossRef] [PubMed]
- Mehlgarten, C.; Jablonowski, D.; Breunig, K.D.; Stark, M.J.; Schaffrath, R. Elongator function depends on antagonistic regulation by casein kinase Hrr25 and protein phosphatase Sit4. Mol. Microbiol. 2009, 73, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonowski, D.; Fichtner, L.; Stark, M.J.; Schaffrath, R. The yeast elongator histone acetylase requires Sit4-dependent dephosphorylation for toxin-target capacity. Mol. Biol. Cell 2004, 15, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Bauer, F.; Matsuyama, A.; Candiracci, J.; Dieu, M.; Scheliga, J.; Wolf, D.A.; Yoshida, M.; Hermand, D. Translational control of cell division by Elongator. Cell Rep. 2012, 1, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Dedon, P.C.; Begley, T.J. A system of RNA modifications and biased codon use controls cellular stress response at the level of translation. Chem. Res. Toxicol. 2014, 27, 330–337. [Google Scholar] [CrossRef]
- Cheong, J.K.; Virshup, D.M. Casein kinase 1: Complexity in the family. Int. J. Biochem. Cell Biol. 2011, 43, 465–469. [Google Scholar] [CrossRef]
- Schafer, T.; Maco, B.; Petfalski, E.; Tollervey, D.; Bottcher, B.; Aebi, U.; Hurt, E. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature 2006, 441, 651–655. [Google Scholar] [CrossRef]
- Ray, P.; Basu, U.; Ray, A.; Majumdar, R.; Deng, H.; Maitra, U. The Saccharomyces cerevisiae 60S ribosome biogenesis factor Tif6p is regulated by Hrr25p-mediated phosphorylation. J. Biol. Chem. 2008, 283, 9681–9691. [Google Scholar] [CrossRef]
- Ghalei, H.; Schaub, F.X.; Doherty, J.R.; Noguchi, Y.; Roush, W.R.; Cleveland, J.L.; Stroupe, M.E.; Karbstein, K. Hrr25/CK1delta-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J. Cell Biol. 2015, 208, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.R.; White, J.H.; Folawiyo, Y.; Edlin, A.; Gardiner, D.; Stark, M.J. Two Saccharomyces cerevisiae genes which control sensitivity to G1 arrest induced by Kluyveromyces lactis toxin. Mol. Cell. Biol. 1994, 14, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelissen, H.; Clarke, J.H.; De Block, M.; De Block, S.; Vanderhaeghen, R.; Zielinski, R.E.; Dyer, T.; Lust, S.; Inze, D.; Van Lijsebettens, M. DRL1, a homolog of the yeast TOT4/KTI12 protein, has a function in meristem activity and organ growth in plants. Plant Cell 2003, 15, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Mehlgarten, C.; Prochaska, H.; Hammermeister, A.; Abdel-Fattah, W.; Wagner, M.; Krutyholowa, R.; Jun, S.E.; Kim, G.T.; Glatt, S.; Breunig, K.D.; et al. Use of a yeast tRNase killer toxin to diagnose Kti12 motifs required for tRNA modification by elongator. Toxins 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.E.; Cho, K.H.; Hwang, J.Y.; Abdel-Fattah, W.; Hammermeister, A.; Schaffrath, R.; Bowman, J.L.; Kim, G.T. Comparative analysis of the conserved functions of Arabidopsis DRL1 and yeast KTI12. Mol. Cells 2014. [Google Scholar] [CrossRef]
- Petrakis, T.G.; Sogaard, T.M.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Physical and functional interaction between Elongator and the chromatin-associated Kti12 protein. J. Biol. Chem. 2005, 280, 19454–19460. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, L.; Schaffrath, R. KTI11 and KTI13, Saccharomyces cerevisiae genes controlling sensitivity to G1 arrest induced by Kluyveromyces lactis zymocin. Mol. Microbiol. 2002, 44, 865–875. [Google Scholar] [CrossRef]
- Bar, C.; Zabel, R.; Liu, S.; Stark, M.J.; Schaffrath, R. A versatile partner of eukaryotic protein complexes that is involved in multiple biological processes: Kti11/Dph3. Mol. Microbiol. 2008, 69, 1221–1233. [Google Scholar] [CrossRef]
- Zabel, R.; Bar, C.; Mehlgarten, C.; Schaffrath, R. Yeast alpha-tubulin suppressor Ats1/Kti13 relates to the Elongator complex and interacts with Elongator partner protein Kti11. Mol. Microbiol. 2008, 69, 175–187. [Google Scholar] [CrossRef]
- Glatt, S.; Zabel, R.; Vonkova, I.; Kumar, A.; Netz, D.J.; Pierik, A.J.; Rybin, V.; Lill, R.; Gavin, A.C.; Balbach, J.; et al. Structure of the Kti11/Kti13 heterodimer and its double role in modifications of tRNA and eukaryotic elongation factor 2. Structure 2015, 23, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Kolaj-Robin, O.; McEwen, A.G.; Cavarelli, J.; Seraphin, B. Structure of the Elongator cofactor complex Kti11/Kti13 provides insight into the role of Kti13 in Elongator-dependent tRNA modification. FEBS J. 2015, 282, 819–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Leppla, S.H. Retroviral insertional mutagenesis identifies a small protein required for synthesis of diphthamide, the target of bacterial ADP-ribosylating toxins. Mol. Cell 2003, 12, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Bodley, J.W.; Livingston, D.M. Diphtheria toxin-resistant mutants of Saccharomyces cerevisiae. Mol. Cell. Biol. 1985, 5, 3357–3360. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Milne, G.T.; Kuremsky, J.G.; Fink, G.R.; Leppla, S.H. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol. Cell. Biol. 2004, 24, 9487–9497. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, A.M., Jr. Diphtheria toxin. Annu. Rev. Biochem. 1977, 46, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Van Ness, B.G.; Barrowclough, B.; Bodley, J.W. Recognition of elongation factor 2 by diphtheria toxin is not solely defined by the presence of diphthamide. FEBS Lett. 1980, 120, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Uthman, S.; Liu, S.; Giorgini, F.; Stark, M.J.; Costanzo, M.; Schaffrath, R. Diphtheria disease and genes involved in formation of diphthamide, key effector of the diphtheria toxin. In Insight and Control of Infectious Disease in Global Scenario; InTech: London, UK, 2012; pp. 333–356. [Google Scholar]
- Uthman, S.; Bar, C.; Scheidt, V.; Liu, S.; ten Have, S.; Giorgini, F.; Stark, M.J.; Schaffrath, R. The amidation step of diphthamide biosynthesis in yeast requires DPH6, a gene identified through mining the DPH1-DPH5 interaction network. PLoS Genet. 2013, 9, e1003334. [Google Scholar] [CrossRef]
- Schaffrath, R.; Abdel-Fattah, W.; Klassen, R.; Stark, M.J. The diphthamide modification pathway from Saccharomyces cerevisiae—revisited. Mol. Microbiol. 2014, 94, 1213–1226. [Google Scholar] [CrossRef]
- Tsuda-Sakurai, K.; Miura, M. The hidden nature of protein translational control by diphthamide—the secrets under the leather. J. Biochem. 2018. [Google Scholar] [CrossRef]
- Ortiz, P.A.; Ulloque, R.; Kihara, G.K.; Zheng, H.; Kinzy, T.G. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J. Biol. Chem. 2006, 281, 32639–32648. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathne, P.D.; Koh, C.S.; Grant, T.; Grigorieff, N.; Korostelev, A.A. Ensemble cryo-EM uncovers inchworm-like translocation of a viral IRES through the ribosome. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.; Savva, C.G.; Shin, B.S.; Dever, T.E.; Ramakrishnan, V.; Fernandez, I.S. Structural characterization of ribosome recruitment and translocation by type IV IRES. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Demeshkina, N.; Mancera-Martinez, E.; Melnikov, S.; Simonetti, A.; Myasnikov, A.; Yusupov, M.; Yusupova, G.; Hashem, Y. Structural insights into the role of diphthamide on elongation factor 2 in mRNA reading-frame maintenance. J. Mol. Biol. 2018, 430, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Hawer, H.; Utkur, K.; Arend, M.; Mayer, K.; Adrian, L.; Brinkmann, U.; Schaffrath, R. Importance of diphthamide modified EF2 for translational accuracy and competitive cell growth in yeast. PLoS ONE 2018, 13, e0205870. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, J.; Wu, F.; Xu, C.; Li, S.; Zhao, W.; Wu, Z.; Wu, J.; Zhou, C.Z.; Shi, Y. Solution structure of Kti11p from Saccharomyces cerevisiae reveals a novel zinc-binding module. Biochemistry 2005, 44, 8801–8809. [Google Scholar] [CrossRef]
- Abdel-Fattah, W.; Scheidt, V.; Uthman, S.; Stark, M.J.; Schaffrath, R. Insights into diphthamide, key diphtheria toxin effector. Toxins 2013, 5, 958–968. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, X.; Torelli, A.T.; Lee, M.; Dzikovski, B.; Koralewski, R.M.; Wang, E.; Freed, J.; Krebs, C.; Ealick, S.E.; et al. Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 2010, 465, 891–896. [Google Scholar] [CrossRef]
- Zhu, X.; Dzikovski, B.; Su, X.; Torelli, A.T.; Zhang, Y.; Ealick, S.E.; Freed, J.H.; Lin, H. Mechanistic understanding of Pyrococcus horikoshii Dph2, a [4Fe-4S] enzyme required for diphthamide biosynthesis. Mol. BioSyst. 2011, 7, 74–81. [Google Scholar] [CrossRef]
- Dong, M.; Horitani, M.; Dzikovski, B.; Freed, J.H.; Ealick, S.E.; Hoffman, B.M.; Lin, H. Substrate-dependent cleavage site selection by unconventional radical S-adenosylmethionine enzymes in diphthamide biosynthesis. J. Am. Chem. Soc. 2017, 139, 5680–5683. [Google Scholar] [CrossRef]
- Dong, M.; Kathiresan, V.; Fenwick, M.K.; Torelli, A.T.; Zhang, Y.; Caranto, J.D.; Dzikovski, B.; Sharma, A.; Lancaster, K.M.; Freed, J.H.; et al. Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis. Science 2018, 359, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Dong, M.; Zhang, Y.; Lee, E.A.; Lin, H. Cbr1 is a Dph3 reductase required for the tRNA wobble uridine modification. Nat. Chem. Biol. 2016, 12, 995–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, H.; Takahashi, T.; Kozaki, K.; Yatabe, Y.; Mitsudomi, T.; Fujii, Y.; Sugiura, T.; Matsuda, H.; Takahashi, T.; Takahashi, T. Detailed deletion mapping suggests the involvement of a tumor suppressor gene at 17p13.3, distal to p53, in the pathogenesis of lung cancers. Oncogene 1998, 17, 2095–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscia, D.S.; Morizio, R.; Venesio, T.; Palenzona, C.; Donadio, M.; Callahan, R. Prognostic significance of loss of heterozygosity at loci on chromosome 17p13.3-ter in sporadic breast cancer is evidence for a putative tumour suppressor gene. Br. J. Cancer 1999, 80, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Hoff, C.; Mollenhauer, J.; Waldau, B.; Hamann, U.; Poustka, A. Allelic imbalance and fine mapping of the 17p13.3 subregion in sporadic breast carcinomas. Cancer Genet. Cytogenet. 2001, 129, 145–149. [Google Scholar] [CrossRef]
- Saxena, A.; Clark, W.C.; Robertson, J.T.; Ikejiri, B.; Oldfield, E.H.; Ali, I.U. Evidence for the involvement of a potential second tumor suppressor gene on chromosome 17 distinct from p53 in malignant astrocytomas. Cancer Res. 1992, 52, 6716–6721. [Google Scholar] [PubMed]
- Schultz, D.C.; Vanderveer, L.; Berman, D.B.; Hamilton, T.C.; Wong, A.J.; Godwin, A.K. Identification of two candidate tumor suppressor genes on chromosome 17p13.3. Cancer Res. 1996, 56, 1997–2002. [Google Scholar] [PubMed]
- Phillips, N.J.; Ziegler, M.R.; Radford, D.M.; Fair, K.L.; Steinbrueck, T.; Xynos, F.P.; Donis-Keller, H. Allelic deletion on chromosome 17p13.3 in early ovarian cancer. Cancer Res. 1996, 56, 606–611. [Google Scholar]
- Liu, M.; Yin, K.; Guo, X.; Feng, H.; Yuan, M.; Liu, Y.; Zhang, J.; Guo, B.; Wang, C.; Zhou, G.; et al. Diphthamide biosynthesis 1 is a novel oncogene in colorectal cancer cells and is regulated by MiR-218-5p. Cell. Physiol. Biochem. 2017, 44, 505–514. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Nasiri, J.; Sedghi, M.; Salehi, M.; Hosseinzadeh, M.; Okamoto, N.; Mizuguchi, T.; Nakashima, M.; Miyatake, S.; Takata, A.; et al. A novel homozygous DPH1 mutation causes intellectual disability and unique craniofacial features. J. Hum. Genet. 2018, 63, 487–491. [Google Scholar] [CrossRef]
- Loucks, C.M.; Parboosingh, J.S.; Shaheen, R.; Bernier, F.P.; McLeod, D.R.; Seidahmed, M.Z.; Puffenberger, E.G.; Ober, C.; Hegele, R.A.; Boycott, K.M.; et al. Matching two independent cohorts validates DPH1 as a gene responsible for autosomal recessive intellectual disability with short stature, craniofacial, and ectodermal anomalies. Hum. Mutat. 2015, 36, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.R.; You, L.R.; Yan, Y.T.; Chen, C.M. Role of OVCA1/DPH1 in craniofacial abnormalities of Miller-Dieker syndrome. Hum. Mol. Genet. 2014, 23, 5579–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, J.; Oana, S.; Sakaguchi, T.; Nakashima, M.; Numabe, H.; Kawashima, H.; Matsumoto, N.; Miyake, N. Novel compound heterozygous DPH1 mutations in a patient with the unique clinical features of airway obstruction and external genital abnormalities. J. Hum. Genet. 2018, 63, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Behringer, R.R. Ovca1 regulates cell proliferation, embryonic development, and tumorigenesis. Genes Dev. 2004, 18, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, F.; Tsuda-Sakurai, K.; Yamazaki, T.; Nishio, R.; Nishimura, K.; Kimura, M.; Funakoshi, M.; Miura, M. Nutritional control of stem cell division through S-adenosylmethionine in Drosophila intestine. Dev. Cell 2018, 44, 741.e3–751.e3. [Google Scholar] [CrossRef] [PubMed]
- Arguelles, S.; Camandola, S.; Cutler, R.G.; Ayala, A.; Mattson, M.P. Elongation factor 2 diphthamide is critical for translation of two IRES-dependent protein targets, XIAP and FGF2, under oxidative stress conditions. Free Radic. Biol. Med. 2014, 67, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Bachran, C.; Gupta, P.; Miller-Randolph, S.; Wang, H.; Crown, D.; Zhang, Y.; Wein, A.N.; Singh, R.; Fattah, R.; et al. Diphthamide modification on eukaryotic elongation factor 2 is needed to assure fidelity of mRNA translation and mouse development. Proc. Natl. Acad. Sci. USA 2012, 109, 13817–13822. [Google Scholar] [CrossRef] [Green Version]
- Webb, T.R.; Cross, S.H.; McKie, L.; Edgar, R.; Vizor, L.; Harrison, J.; Peters, J.; Jackson, I.J. Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development. J. Cell Sci. 2008, 121, 3140–3145. [Google Scholar] [CrossRef] [Green Version]



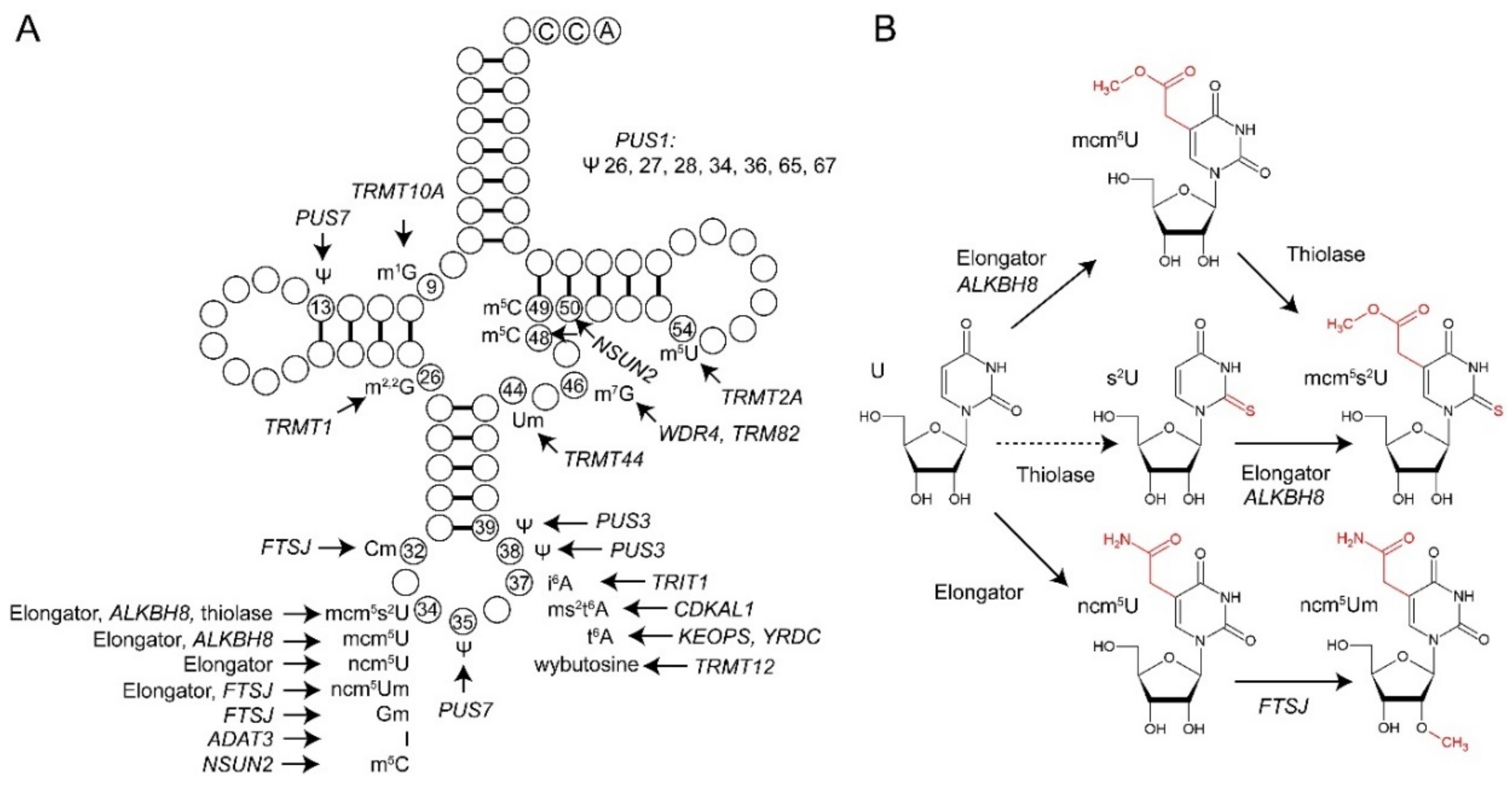{kind=link}
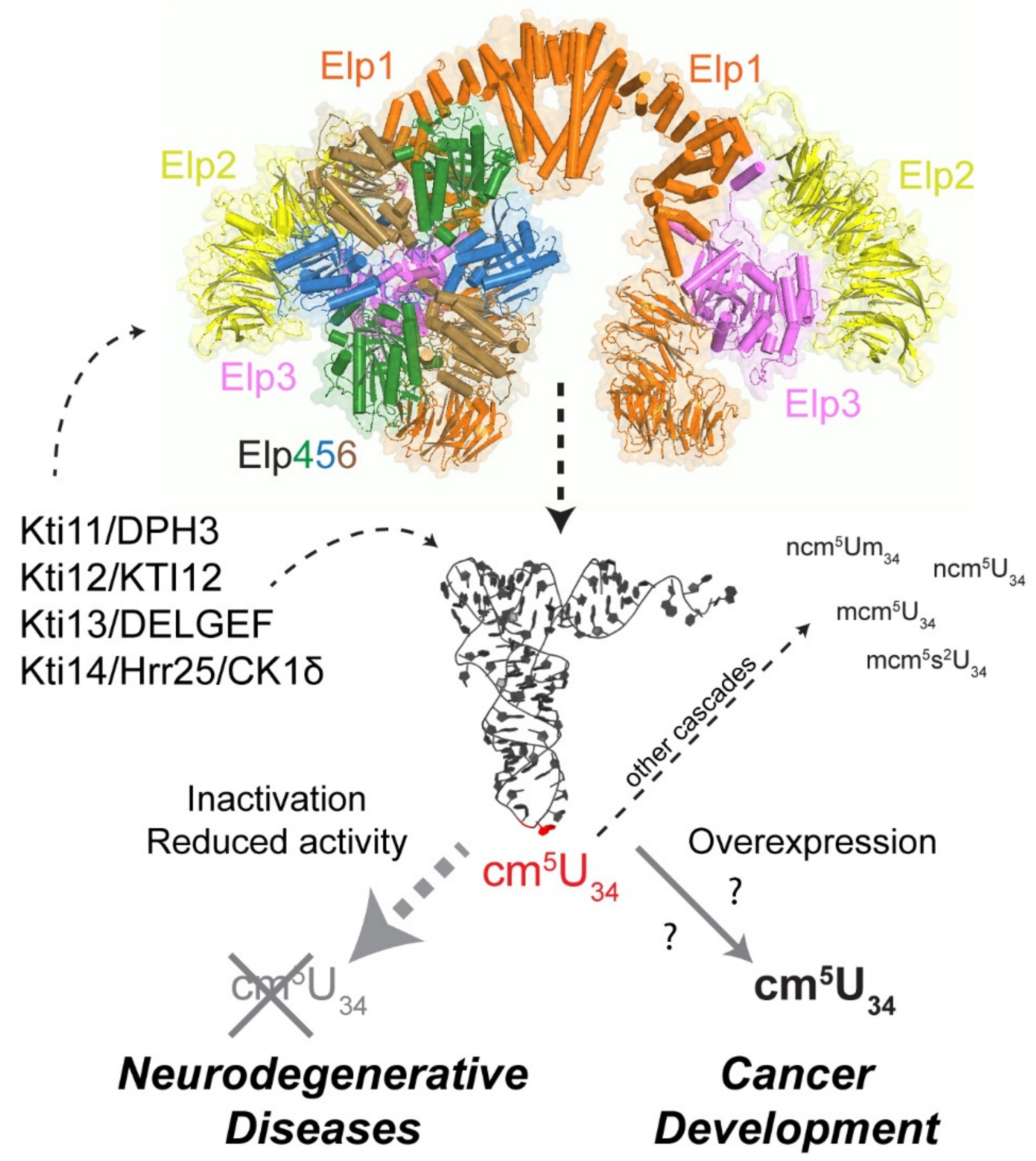{kind=link}
| Disease | Genes | Modification |
|---|---|---|
| Familial dysautonomia 1 [21,22,23] | IKBKAP | mcm5(s2)U34, ncm5U34, ncm5Um34 |
| Intellectual disability 1 [24,25] | ELP2 | mcm5(s2)U34, ncm5U34, ncm5Um34 |
| Amyotrophic lateral sclerosis 1 [26,27] | ELP3 | mcm5(s2)U34, ncm5U34, ncm5Um34 |
| Breast-, bladder-, colorectal-, cervix- and testicular cancer1 [28] | hTRM9L * | mcm5(s2)U34 (? *) |
| Urothelial cancer 2 [29] | ALKBH8 | mcm5(s2)U34 |
| Asthma 1 [30] | IKBKAP | mcm5(s2)U34, ncm5U34, ncm5Um34 |
| Melanoma 2,3 [31,32] | ELP1, 3, 5, 6, CTU1/2 | mcm5(s2)U34, ncm5U34, ncm5Um34 |
| Invasive breast cancer 2,3 [33] | ELP3, CTU1/2 | mcm5(s2)U34, ncm5U34, ncm5Um34 |
| X-linked mental retardation 1 [34] | FTSJ1 | Cm32, Gm34, yW37, ncm5Um34 |
| MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) 1 [11,35,36,37] | Mt-tRNA LeuUAA´, MTO1, GTPBP3 | τm5U34 (mito) |
| MERRF (myoclonus epilepsy with ragged-red fibers) 1 [11,38] | Mt-tRNA LysUUU, MTO1, GTPBP3, MTU1 | τm5s2U34 (mito) |
| Deafness associated with rRNA A1555G mutation 1 [39] | MTU1 | s2U (mito) |
| Acute infantile liver failure1 [40] | MTU1 | s2u (mito) |
| Neurodegeneration, Galloway-Mowat syndrome1 [41,42] | YRDC, KEOPS, OSGEPL1 | t6A37 |
| MERRF-like syndrome 1 [9] | YRDC, KEOPS, OSGEPL1 | t6A37 |
| Type 2 diabetes 1 [43,44] | CDKAL1 | ms2t6A37 |
| Breast cancer 2 [45] | TRMT12 | wybutosine37 |
| Intellectual disability 1 [46,47] | PUS3 | Ψ38/39 |
| Intellectual disability 1, Microcephaly 1, aggressive behavior 1 [48] | PUS7 | Ψ13/35 |
| Intellectual disability 1 [49] | NSUN2 | m5C34,48,49 |
| Dubowitz-like syndrome 1 [50] | NSUN2 | m5C34,48,49 |
| Noonan-like syndrome 1 [51] | NSUN2 | m5C34,48,49 |
| Skin-, breast- and colorectal cancer 2,3 [52,53,54] | NSUN2 | m5C34,48,49 |
| Intellectual disability 1 [55] | ADAT3 | I34 |
| Encephalopathy and myoclonic epilepsy 1 [56] | TRIT1 | i6A/ms2i6A37 |
| Lung- and breast cancer 1 [57,58,59] | TRIT1 | i6A/ms2i6A37 |
| Intellectual disability 1 [24] | TRMT1 | m2,2G26 |
| Primordial dwarfism 1 [60] | METTL1/WDR4, TRM82 | m7G46 |
| PEPS 1 (Partial epilepsy with pericentral spikes) [61] | TRMT44 | Um44 |
| Microcephaly 1 [62,63] | TRMT10A | m1G9 |
| Intellectual disability and early onset diabetes 1 [64,65], epilepsy 1 [65] | TRMT10A | m1G9 |
| Breast cancer 2 [66] | TRMT2A | m5U54 |
| Mitochondrial Myopathy and Sideroblastic Anemia 1 (MLASA) [67] | PUS1 | Ψmultiple (mito) |
| Yeast Gene | Human Orthologs/Synonym | Modifications |
|---|---|---|
| ELP1 | ELP1/IKAP | mcm5s2U; mcm5U; ncm5U; ncm5Um |
| ELP2 | ELP2 | mcm5s2U; mcm5U; ncm5U; ncm5Um |
| ELP3 | ELP3 | mcm5s2U; mcm5U; ncm5U; ncm5Um |
| ELP4 | ELP4 | mcm5s2U; mcm5U; ncm5U; ncm5Um |
| ELP5 | ELP5 | mcm5s2U; mcm5U; ncm5U; ncm5Um |
| ELP6 | ELP6 | mcm5s2U; mcm5U; ncm5U; ncm5Um |
| TRM9 | ALKBH8, hTRM9L * | mcm5s2U; mcm5U |
| TRM112 | TRMT112 | mcm5s2U; mcm5U |
| NFS1 | NFS1 | mcm5s2U |
| TUM1 | TUM1 | mcm5s 2U |
| URM1 | URM1 | mcm5s2U |
| UBA4 | UBA4 | mcm5s2U |
| NCS2 | CTU1 | mcm5s2U |
| NCS6 | CTU2 | mcm5s2U |
| Gene | Elongation factor 2 (EF2) Modification | xm 5U34 Modification | Species | Disease/Syndrome |
|---|---|---|---|---|
| DPH1 | absent | present | Human | Lung cancer [205] |
| Breast cancer [206,207] | ||||
| Brain tumors [208] | ||||
| Ovarian cancer [209,210] | ||||
| Colorectal cancer [211] | ||||
| Intellectual disability and craniofacial abnormalities [212,213] | ||||
| Miller-Dieker syndrome (MDS) [214] | ||||
| Airway obstruction and external genital abnormalities [215] | ||||
| Mouse | Embryonic lethal, cell proliferation defect, edema, polydactyly [216] Embryonic jaw shortening, cleft palate [214] | |||
| Fruit fly | Failure of intestinal stem cell proliferation [217] | |||
| KTI11/DPH3 | absent | absent | Chinese hamster | Reduced life span hunder [218] |
| Mouse | Necrosis, apoptosis and defects in development of placenta [219] | |||
| DPH4 | absent | present | Mouse | Neuronal underdevelopment, impaired growth and polydactyly [220] |
| DPH5 | absent | present | Fruit fly | Intestinal stem cell defect [217] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawer, H.; Hammermeister, A.; Ravichandran, K.E.; Glatt, S.; Schaffrath, R.; Klassen, R. Roles of Elongator Dependent tRNA Modification Pathways in Neurodegeneration and Cancer. Genes 2019, 10, 19. https://doi.org/10.3390/genes10010019
Hawer H, Hammermeister A, Ravichandran KE, Glatt S, Schaffrath R, Klassen R. Roles of Elongator Dependent tRNA Modification Pathways in Neurodegeneration and Cancer. Genes. 2019; 10(1):19. https://doi.org/10.3390/genes10010019
Chicago/Turabian StyleHawer, Harmen, Alexander Hammermeister, Keerthiraju Ethiraju Ravichandran, Sebastian Glatt, Raffael Schaffrath, and Roland Klassen. 2019. "Roles of Elongator Dependent tRNA Modification Pathways in Neurodegeneration and Cancer" Genes 10, no. 1: 19. https://doi.org/10.3390/genes10010019