Clinical and Molecular Differences between 4-Year-Old Monozygous Male Twins Mosaic for Normal, Premutation and Fragile X Full Mutation Alleles

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Sample Processing
2.3. CGG Sizing
2.4. FMR1 Methylation Analysis
2.5. Microarray SNP Testing
2.6. Follow-Up Neuropsychological Assessments
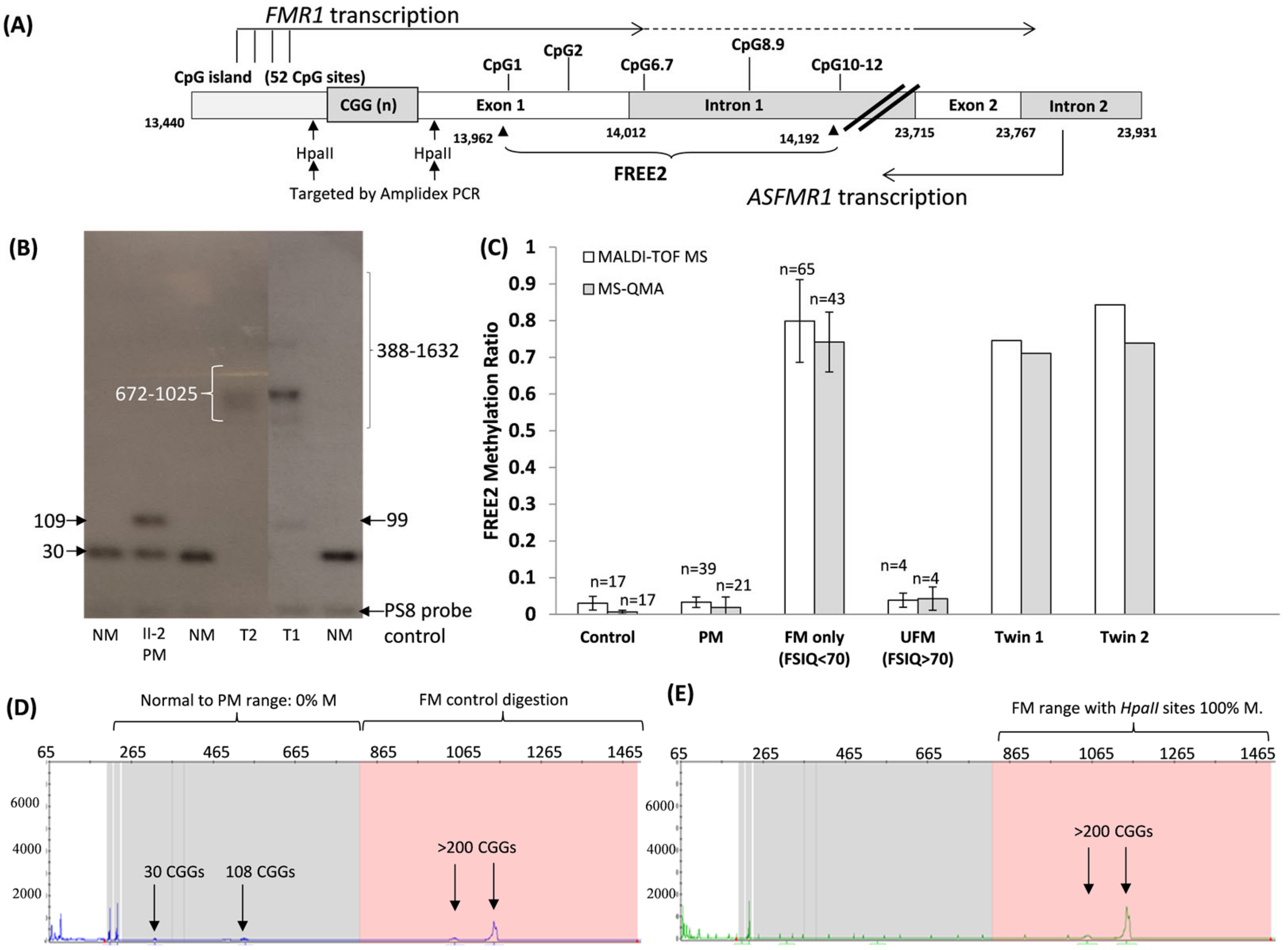
3. Results
3.1. Clinical History
3.2. Initial Diagostic Testing
3.3. Follow-Up Investigation of Similarities and Differences between Twin Phenotypes
3.4. DNA Methylation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootsak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Brylawski, B.P.; Chastain, P.D., 2nd; Cohen, S.M.; Cordeiro-Stone, M.; Kaufman, D.G. Mapping of an origin of DNA replication in the promoter of fragile X gene FMR1. Exp. Mol. Pathol. 2007, 82, 190–196. [Google Scholar] [CrossRef]
- Willemsen, R.; Bontekoe, C.J.; Severijnen, L.A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Tassone, F.; Loesch, D.Z.; Taylor, A.K.; Gehling, F.; Hagerman, R.J.; Burgess, T.; Ganesamoorthy, D.; Hennerich, D.; Gordon, L.; et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010, 19, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Revenga, L.; Madrigal, I.; Pagonabarraga, J.; Xuncla, M.; Badenas, C.; Kulisevsky, J.; Gomez, B.; Mila, M. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur. J. Hum. Genet. 2009, 17, 13591362. [Google Scholar] [CrossRef] [PubMed]
- Kraan, C.M.; Bui, Q.M.; Field, M.; Archibald, A.D.; Metcalfe, S.A.; Christie, L.M.; Bennetts, B.H.; Oertel, R.; Smith, M.J.; du Sart, D.; et al. FMR1 allele size distribution in 35,000 males and females: A comparison of developmental delay and general population cohorts. Genet. Med. 2018, 20, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, V.; Glaeser, D.; McQuaid, S.; Steinbach, P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur. J. Hum. Genet. 2015, 23, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; Francis, D.; Gaffney, L.; Greenberg, J.; Hills, L.; Li, X.; Godler, D.E.; Slater, H.R. Prenatal diagnosis of fragile X syndrome complicated by full mutation retraction. Am. J. Med. Genet. A 2015, 167A, 2485–2487. [Google Scholar] [CrossRef]
- Prawer, Y.; Hunter, M.; Cronin, S.; Ling, L.; Aliaga Vera, S.; Fahey, M.; Gelfand, N.; Oertel, R.; Bartlett, E.; Francis, D.; et al. Prenatal Diagnosis of Fragile X Syndrome in a Twin Pregnancy Complicated by a Complete Retraction. Genes 2018, 9, E287. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early Detection of Fragile X Syndrome: Applications of a Novel Approach for Improved Quantitative Methylation Analysis in Venous Blood and Newborn Blood Spots. Clin. Chem. 2014, 60, 963–973. [Google Scholar] [CrossRef]
- Khaniani, M.S.; Kalitsis, P.; Burgess, T.; Slater, H.R. An improved Diagnostic PCR Assay for identification of Cryptic Heterozygosity for CGG Triplet Repeat Alleles in the Fragile X Gene (FMR1). Mol. Cytogenet. 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Francis, D.; Burgess, T.; Mitchell, J.; Slater, H. Identification of small FRAXA premutations. Mol. Diagn. 2000, 5, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hadd, A.G.; Sah, S.; Houghton, J.F.; Filipovic-Sadic, S.; Zhang, W.; Hagerman, P.J.; Tassone, F.; Latham, G.J. High-resolution methylation polymerase chain reaction for fragile X analysis: Evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet. Med. 2011, 13, 528–538. [Google Scholar] [CrossRef]
- Weschler, D. Wechsler Preschool and Primary Scale of Intelligence—Third Edition Australian Standardised Edition; Pearson Clinical and Talent Assessment Australia and New Zealand: Sydney, NSW, Australia, 2004. [Google Scholar]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S.L. Autism Diagnostic Observation Schedule, 2nd Edition (ADOS-2); Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Hus, V.; Gotham, K.; Lord, C. Standardizing ADOS domain scores: separating severity of social affect and restricted and repetitive behaviors. J. Autism Dev. Disord. 2014, 44, 2400–2412. [Google Scholar] [CrossRef]
- Arpone, M.; Baker, E.K.; Bretherton, L.; Bui, M.; Li, X.; Whitaker, S.; Dissanayake, C.; Cohen, J.; Hickerton, C.; Rogers, C.; et al. Intragenic DNA methylation in buccal epithelial cells and intellectual functioning in a paediatric cohort of males with fragile X. Sci. Rep. 2018, 8, 3644. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.Y.; Aliaga, S.; Arpone, M.V.; Francis, D.; Li, X.; Chong, B.; Slater, H.R.; Rogers, C.; Bretherton, L.; Hunter, M.; et al. Partially Methylated Alleles, Microdeletion and Tissue Mosaicism in a Fragile X Male with Tremor and Ataxia at 30 Years of Age: A Case Report. Am. J. Med. Genet. 2016, 170, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.T.; Dudding, T.; Aliaga, S.M.; Arpone, M.; Francis, D.; Li, X.; Slater, H.R.; Rogers, C.; Bretherton, L.; du Sart, D.; et al. Molecular Inconsistencies in a Fragile X Male with Early Onset Ataxia. Genes 2016, 7, 68. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Houck, G.E., Jr.; Brown, W.T.; Dobkin, C.S. Mosaicism in fragile X affected males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Olmer, R.; De Diego Otero, Y.; Oostra, B.A. Twin sisters, monozygotic with the fragile X mutation, but with a different phenotype. J. Med. Genet. 2000, 37, 603–604. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, M.P.; Cohen, M.; Vnencak-Jones, C.L. Maternal FMR1 premutation allele expansion and contraction in fraternal twins. Am. J. Med. Genet. 2013, 161A, 2620–2625. [Google Scholar]
- Cornish, K.M.; Kraan, C.M.; Bui, Q.M.; Bellgrove, M.A.; Metcalfe, S.A.; Trollor, J.N.; Hocking, D.R.; Slater, H.R.; Inaba, Y.; Li, X.; et al. Novel methylation markers of the dysexecutive-psychiatric phenotype in FMR1 premutation women. Neurology 2015, 84, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, S.M.; Slater, H.R.; Francis, D.; Du Sart, D.; Li, X.; Amor, D.J.; Alliende, A.M.; Santa Maria, L.; Faundes, V.; Morales, P.; et al. Identification of Males with Cryptic Fragile X Alleles by Methylation-Specific Quantitative Melt Analysis. Clin. Chem. 2016, 62, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Shi, E.Z.; Li, X.; Herlihy, A.S.; Skinner, C.; Hagerman, R.J.; Francis, D.; et al. Detection of skewed X-chromosome inactivation in Fragile X syndrome and X chromosome aneuploidy using quantitative melt analysis. Exp. Rev. Mol. Med. 2015, 17, e13. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandelache, A.; Baker, E.K.; Aliaga, S.M.; Arpone, M.; Forbes, R.; Stark, Z.; Francis, D.; Godler, D.E. Clinical and Molecular Differences between 4-Year-Old Monozygous Male Twins Mosaic for Normal, Premutation and Fragile X Full Mutation Alleles. Genes 2019, 10, 279. https://doi.org/10.3390/genes10040279
Pandelache A, Baker EK, Aliaga SM, Arpone M, Forbes R, Stark Z, Francis D, Godler DE. Clinical and Molecular Differences between 4-Year-Old Monozygous Male Twins Mosaic for Normal, Premutation and Fragile X Full Mutation Alleles. Genes. 2019; 10(4):279. https://doi.org/10.3390/genes10040279
Chicago/Turabian StylePandelache, Alison, Emma K Baker, Solange M. Aliaga, Marta Arpone, Robin Forbes, Zornitza Stark, David Francis, and David E. Godler. 2019. "Clinical and Molecular Differences between 4-Year-Old Monozygous Male Twins Mosaic for Normal, Premutation and Fragile X Full Mutation Alleles" Genes 10, no. 4: 279. https://doi.org/10.3390/genes10040279