Pacbio Sequencing Reveals Identical Organelle Genomes between American Cranberry (Vaccinium macrocarpon Ait.) and a Wild Relative

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. PacBio Sequencing, Genome Assembly, and Annotation
2.3. Illumina Sequencing and Mapping
2.4. Phylogenetic Analysis
3. Results and Discussion
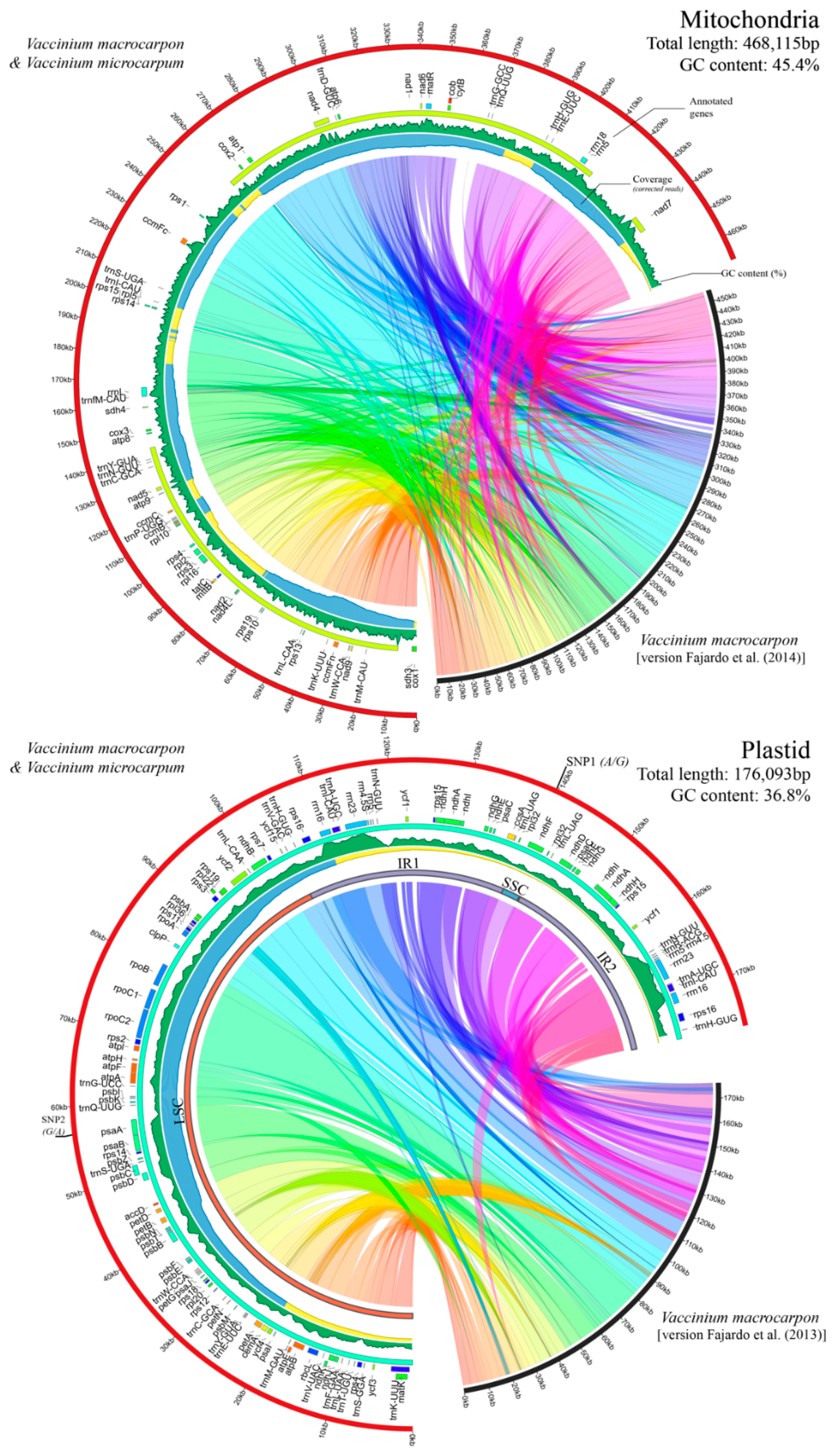
3.1. Cultivated and Wild Cranberries Share Identical Organelle Genomes
3.2. Genome Architecture and Gene Content
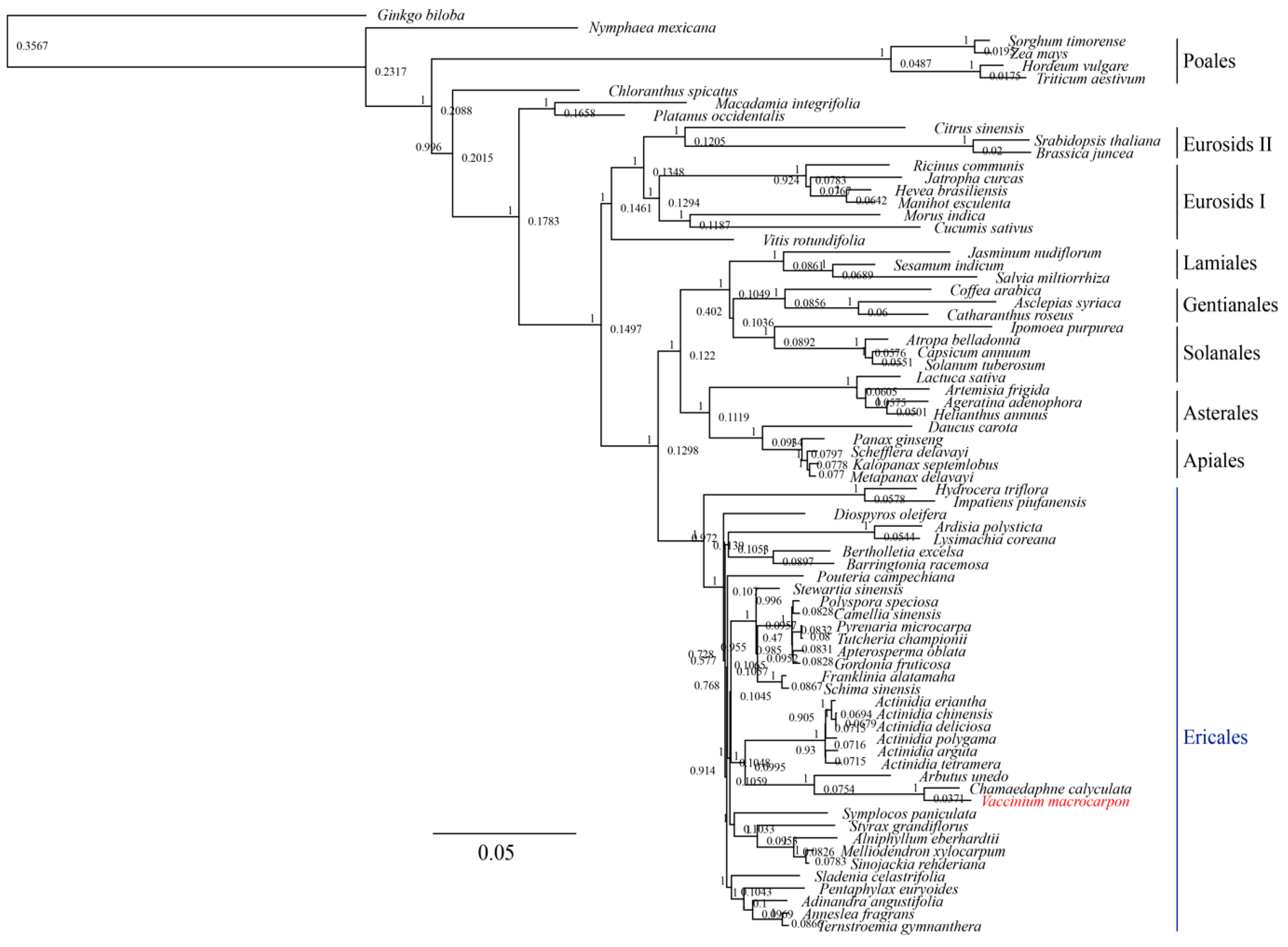
3.3. Phylogenetic Analysis of V. macrocarpon and Related Species
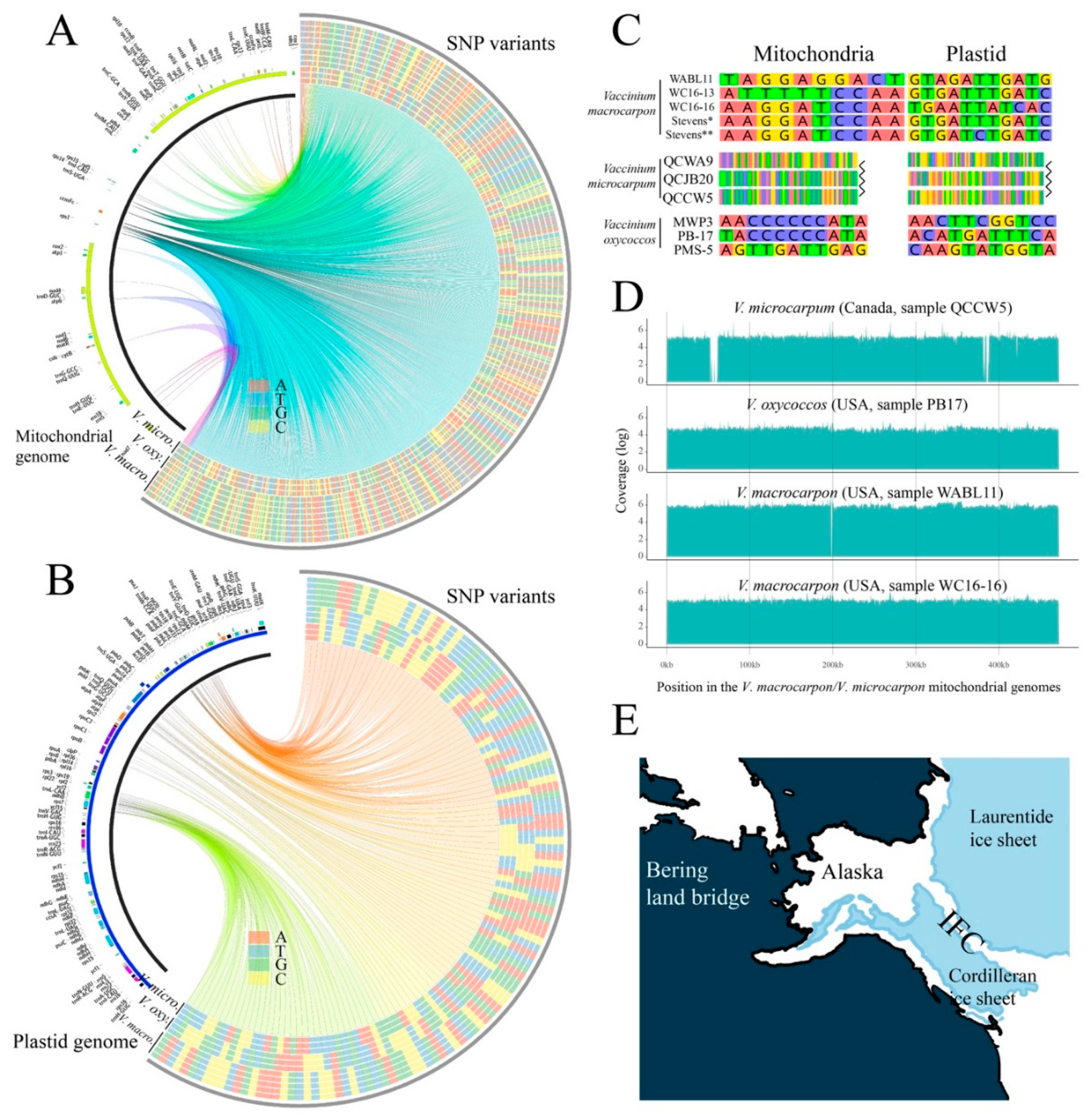
3.4. Divergent V. microcarpum Can Be Found in Eastern Canada
3.5. Relationships among Diploids, and the Link between Alaskan V. microcarpum and V. macrocarpon
3.6. The Origin of the Tetraploid V. oxycoccos
3.7. Conclusions and Further Directions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dempewolf, H.; Baute, G.; Anderson, J.; Kilian, B.; Smith, C.; Guarino, L. Past and Future Use of Wild Relatives in Crop Breeding. Crop Sci. 2017, 57, 1070–1082. [Google Scholar] [CrossRef]
- Hajjar, R.; Hodgkin, T. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 2007, 156, 1–13. [Google Scholar] [CrossRef]
- Jarvis, A.; Lane, A.; Hijmans, R.J. The effect of climate change on crop wild relatives. Agric. Ecosyst. Environ. 2008, 126, 13–23. [Google Scholar] [CrossRef]
- Maxted, N.; Kell, S.; Ford-Lloyd, B.; Dulloo, E.; Toledo, Á. Toward the Systematic Conservation of Global Crop Wild Relative Diversity. Crop Sci. 2012, 52, 774–785. [Google Scholar] [CrossRef]
- Vander Kloet, S.P. The taxonomy of Vaccinium section Oxycoccus. Rhodora 1983, 85, 1–43. [Google Scholar]
- Vander Kloet, S.P. Vaccinium Linnaeus. In Flora N. Am. North Mexico, Vol. 8: Magnoliophyta: Paeoniaceae to Ericaceae; Morin, N.R., Ed.; Oxford University Press: New York, NY, USA, 2009; pp. 515–530. [Google Scholar]
- Vorsa, N.; Johnson-Cicalese, J. American cranberry. Fruit Breed. 2012, 8, 191–223. [Google Scholar]
- Jacquemart, A.-L. Vaccinium Oxycoccos L. (Oxycoccus palustris Pers.) and Vaccinium microcarpum (Turcz. ex Rupr.) Schmalh. (Oxycoccus microcarpus Turcz. ex Rupr.). J. Ecol. 1997, 85, 381–396. [Google Scholar] [CrossRef]
- Vander Kloet, S.P. The Genus Vaccinium in North America; Agriculture Canada: Ottawa, ON, Canada, 1988. [Google Scholar]
- Bruederle, L.P.; Hugan, M.S.; Dignan, J.M.; Vorsa, N. Genetic Variation in Natural Populations of the Large Cranberry, Vaccinium macrocarpon Ait. (Ericaceae). Bull. Torrey Bot. Club 1996, 123, 41–47. [Google Scholar] [CrossRef]
- Smith, T.W.; Walinga, C.; Wang, S.; Kron, P.; Suda, J.; Zalapa, J. Evaluating the relationship between diploid and tetraploid Vaccinium oxycoccos (Ericaceae) in eastern Canada. Botany 2015, 93, 623–636. [Google Scholar] [CrossRef]
- Ballington, J.R. Collection, utilization, and preservation of genetic resources in Vaccinium. HortScience 2001, 36, 213–220. [Google Scholar] [CrossRef]
- Song, G.-Q.; Hancock, J.F. Vaccinium. In Wild Crop Relatives: Genomic and Breeding Resources: Temperate Fruits; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 197–221. ISBN 9783642160578. [Google Scholar]
- Eaton, G.W.; Mahrt, B.J. Cold hardiness testing of cranberry flower buds. Can. J. Plant Sci. 1977, 57, 461–465. [Google Scholar] [CrossRef]
- Ehlenfeldt, M.K.; Rowland, L.J.; Ogden, E.L.; Vinyard, B.T. Floral Bud Cold Hardiness of Vaccinium ashei, V. constablaei, and Hybrid Derivatives and the Potential for Producing Northern-adapted Rabbiteye Cultivars. HortScience 2007, 42, 1131–1134. [Google Scholar] [CrossRef]
- Asada, T. Distribution of two taxa in Vaccinium oxycoccus sensu lato (Ericaceae) along micro-scale environmental gradients in Bekanbeushi Peatland, northern Japan. Acta Phytotaxon. Geobot. 2001, 51, 169–176. [Google Scholar]
- Suda, J.; Lysák, M.A. A taxonomic study of the Vaccinium sect. Oxycoccus (Hill) WDJ Kock (Ericaceae) in the Czech Republic and adjacent territories. Folia Geobot. 2001, 36, 303–320. [Google Scholar] [CrossRef]
- Camp, W.H. A Preliminary Consideration of the Biosystematy of Oxycoccus. Bull. Torrey Bot. Club 1944, 71, 426–437. [Google Scholar] [CrossRef]
- Mahy, G.; Bruederle, L.P.; Connors, B.; Van Hofwegen, M.; Vorsa, N. Allozyme evidence for genetic autopolyploidy and high genetic diversity in tetraploid cranberry, Vaccinium oxycoccos (Ericaceae). Am. J. Bot. 2000, 87, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Soltis, P.S.; Rieseberg, L.H. Molecular Data and the Dynamic Nature of Polyploidy. CRC Crit. Rev. Plant Sci. 1993, 12, 243–273. [Google Scholar] [CrossRef]
- Georgi, L.; Johnson-Cicalese, J.; Honig, J.; Das, S.P.; Rajah, V.D.; Bhattacharya, D.; Bassil, N.; Rowland, L.J.; Polashock, J.; Vorsa, N. The first genetic map of the American cranberry: Exploration of synteny conservation and quantitative trait loci. Theor. Appl. Genet. 2013, 126, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pazaran, G.; Diaz-Garcia, L.; Schlautman, B.; Deutsch, J.; Salazar, W.; Hernandez-Ochoa, M.; Grygleski, E.; Steffan, S.; Iorizzo, M.; Polashock, J.; et al. Exploiting genotyping by sequencing to characterize the genomic structure of the American cranberry through high-density linkage mapping. BMC Genom. 2016, 17, 451. [Google Scholar] [CrossRef] [PubMed]
- Daverdin, G.; Johnson-Cicalese, J.; Zalapa, J.; Vorsa, N.; Polashock, J. Identification and mapping of fruit rot resistance QTL in American cranberry using GBS. Mol. Breed. 2017, 37, 38. [Google Scholar] [CrossRef]
- Schlautman, B.; Covarrubias-Pazaran, G. Development of a high-density cranberry SSR linkage map for comparative genetic analysis and trait detection. Mol. Breed. 2015, 35, 177. [Google Scholar] [CrossRef]
- Schlautman, B.; Covarrubias-Pazaran, G.; Diaz-Garcia, L.; Iorizzo, M.; Polashock, J.; Grygleski, E.; Vorsa, N.; Zalapa, J. Construction of a High-Density American Cranberry (Vaccinium macrocarpon Ait.) Composite Map Using Genotyping-by-Sequencing for Multi-pedigree Linkage Mapping. G3 2017, 7, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, L.; Covarrubias-Pazaran, G.; Schlautman, B.; Grygleski, E.; Zalapa, J. Image-based phenotyping for identification of QTL determining fruit shape and size in American cranberry (Vaccinium macrocarpon L.). PeerJ 2018, 6, e5461. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, L.; Schlautman, B.; Covarrubias-Pazaran, G.; Maule, A.; Johnson-Cicalese, J.; Grygleski, E.; Vorsa, N.; Zalapa, J. Massive phenotyping of multiple cranberry populations reveals novel QTLs for fruit anthocyanin content and other important chemical traits. Mol. Genet. Genom. 2018, 293, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Polashock, J.; Zelzion, E.; Fajardo, D.; Zalapa, J.; Georgi, L.; Bhattacharya, D.; Vorsa, N. The American cranberry: First insights into the whole genome of a species adapted to bog habitat. BMC Plant Biol. 2014, 14, 165. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, D.; Senalik, D.; Ames, M.; Zhu, H.; Steffan, S.A.; Harbut, R.; Polashock, J.; Vorsa, N.; Gillespie, E.; Kron, K.; et al. Complete plastid genome sequence of Vaccinium macrocarpon: Structure, gene content, and rearrangements revealed by next generation sequencing. Tree Genet. Genomes 2013, 9, 489–498. [Google Scholar] [CrossRef]
- Fajardo, D.; Schlautman, B.; Steffan, S.; Polashock, J.; Vorsa, N.; Zalapa, J. The American cranberry mitochondrial genome reveals the presence of selenocysteine (tRNA-Sec and SECIS) insertion machinery in land plants. Gene 2014, 536, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Senalik, D.; McCown, B.H.; Zeldin, E.L.; Speers, J.; Hyman, J.; Bassil, N.; Hummer, K.; Simon, P.W.; Zalapa, J.E. Mining and validation of pyrosequenced simple sequence repeats (SSRs) from American cranberry (Vaccinium macrocarpon Ait.). Theor. Appl. Genet. 2012, 124, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Bougie, T.C.; Bougie, T.A.; Schlautman, B.J.; Wiesman, E.; Guzman, A.; Fajardo, D.A.; Steffan, S.; Smith, T. Clonal diversity and genetic differentiation revealed by SSR markers in wild Vaccinium macrocarpon and Vaccinium oxycoccos. Ann. Appl. Biol. 2015, 166, 196–207. [Google Scholar] [CrossRef]
- Schlautman, B.; Bolivar-Medina, J.; Hodapp, S.; Zalapa, J. Cranberry SSR multiplexing panels for DNA horticultural fingerprinting and genetic studies. Sci. Hortic. 2017, 219, 280–286. [Google Scholar] [CrossRef]
- Schlautman, B.; Fajardo, D.; Bougie, T.; Wiesman, E.; Polashock, J.; Vorsa, N.; Steffan, S.; Zalapa, J. Development and validation of 697 novel polymorphic genomic and EST-SSR markers in the American cranberry (Vaccinium macrocarpon Ait.). Molecules 2015, 20, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Schlautman, B.; Covarrubias-Pazaran, G.; Fajardo, D.; Steffan, S.; Zalapa, J. Discriminating power of microsatellites in cranberry organelles for taxonomic studies in Vaccinium and Ericaceae. Genet. Resour. Crop Evol. 2017, 64, 451–466. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Silva, N.D.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Mayer, C. Phobos, a tandem repeat search tool for complete genomes. Version 2008, 3, 12. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003, 10. [Google Scholar] [CrossRef] [PubMed]
- Hisano, H.; Tsujimura, M.; Yoshida, H.; Terachi, T.; Sato, K. Mitochondrial genome sequences from wild and cultivated barley (Hordeum vulgare). BMC Genom. 2016, 17, 824. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, M.; Tsudzuki, T.; Sugiura, M. The chloroplast genome of Nicotiana sylvestris and Nicotiana tomentosiformis: Complete sequencing confirms that the Nicotiana sylvestris progenitor is the maternal genome donor of Nicotiana tabacum. Mol. Genet. Genom. 2006, 275, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the Complete Mitochondrial Genome Sequence of the Diploid Cotton Gossypium raimondii by Comparative Genomics Approaches. Biomed Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef] [PubMed]
- Handa, H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003, 31, 5907–5916. [Google Scholar] [CrossRef]
- Oda, K.; Yamato, K.; Ohta, E.; Nakamura, Y.; Takemura, M.; Nozato, N.; Akashi, K.; Ohyama, K. Transfer RNA genes in the mitochondrial genome from a liverwort, Marchantia polymorpha: The absence of chloroplast-like tRNAs. Nucleic Acids Res. 1992, 20, 3773–3777. [Google Scholar] [CrossRef] [PubMed]
- Ye, N.; Wang, X.; Li, J.; Bi, C.; Xu, Y.; Wu, D.; Ye, Q. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ 2017, 5, e3148. [Google Scholar] [CrossRef]
- Rose, J.P.; Kleist, T.J.; Löfstrand, S.D.; Drew, B.T.; Schönenberger, J.; Sytsma, K.J. Phylogeny, historical biogeography, and diversification of angiosperm order Ericales suggest ancient Neotropical and East Asian connections. Mol. Phylogenet. Evol. 2018, 122, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Szczecinska, M.; Gomolinska, A.; Szkudlarz, P.; Sawicki, J. Plastid and nuclear genomic resources of a relict and endangered plant species: Chamaedaphne calyculata (L.) Moench (Ericaceae). Turk. J. Bot. 2014, 38, 1229–1238. [Google Scholar] [CrossRef]
- Bartsch, I. Effects of fertilization on growth and nutrient use by Chamaedaphne calyculata in a raised bog. Can. J. Bot. 1994, 72, 323–329. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, P.D.; Froese, D.; Ives, J.W.; Soares, A.E.R.; Zazula, G.D.; Letts, B.; Andrews, T.D.; Driver, J.C.; Hall, E.; Hare, P.G.; et al. Bison phylogeography constrains dispersal and viability of the Ice Free Corridor in western Canada. Proc. Natl. Acad. Sci. USA 2016, 113, 8057–8063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porsild, A.E. Earth Mounds in Unglaciated Arctic Northwestern America. Geogr. Rev. 1938, 28, 46–58. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Species | Collected from | Sequencing Technology |
|---|---|---|---|
| V. macrocarpon (cultivar Stevens) | Madison, WI, USA | PacBio Sequel and Illumina | |
| V. microcarpum | Alaska, USA | ||
| WABL11 | V. macrocarpon | Vilas County, WI, USA | Illumina |
| WC16-13 | North-Central WI, USA | ||
| WC16-16 | NJ, USA | ||
| QCCW5 | V. microcarpum | Quebec, ON, Canada | |
| QCJB20 | |||
| QCWA9 | |||
| MWB3 | V. oxycoccos | Manitowish Waters, WI, USA | |
| PMS5 | |||
| PB17 | Pennington, MN, USA |
| Origin of the SNP | Mitochondria | Plastid | ||
|---|---|---|---|---|
| Counts | Percentage | Counts | Percentage | |
| V. macrocarpon | 827 | 99.3% | 33 | 26.4% |
| Private V. oxycoccos Alleles | 5 | 0.4% | 22 | 17.4% |
| V. microcarpum | 1 | 0.1% | 67 | 55.4% |
| Total | 833 | 100% | 122 | 100% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Garcia, L.; Rodriguez-Bonilla, L.; Rohde, J.; Smith, T.; Zalapa, J. Pacbio Sequencing Reveals Identical Organelle Genomes between American Cranberry (Vaccinium macrocarpon Ait.) and a Wild Relative. Genes 2019, 10, 291. https://doi.org/10.3390/genes10040291
Diaz-Garcia L, Rodriguez-Bonilla L, Rohde J, Smith T, Zalapa J. Pacbio Sequencing Reveals Identical Organelle Genomes between American Cranberry (Vaccinium macrocarpon Ait.) and a Wild Relative. Genes. 2019; 10(4):291. https://doi.org/10.3390/genes10040291
Chicago/Turabian StyleDiaz-Garcia, Luis, Lorraine Rodriguez-Bonilla, Jessica Rohde, Tyler Smith, and Juan Zalapa. 2019. "Pacbio Sequencing Reveals Identical Organelle Genomes between American Cranberry (Vaccinium macrocarpon Ait.) and a Wild Relative" Genes 10, no. 4: 291. https://doi.org/10.3390/genes10040291