A “NOTCH” Deeper into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer

 , , ,
, , ,
Abstract
:1. Introduction
2. Epithelial–Mesenchymal Transition
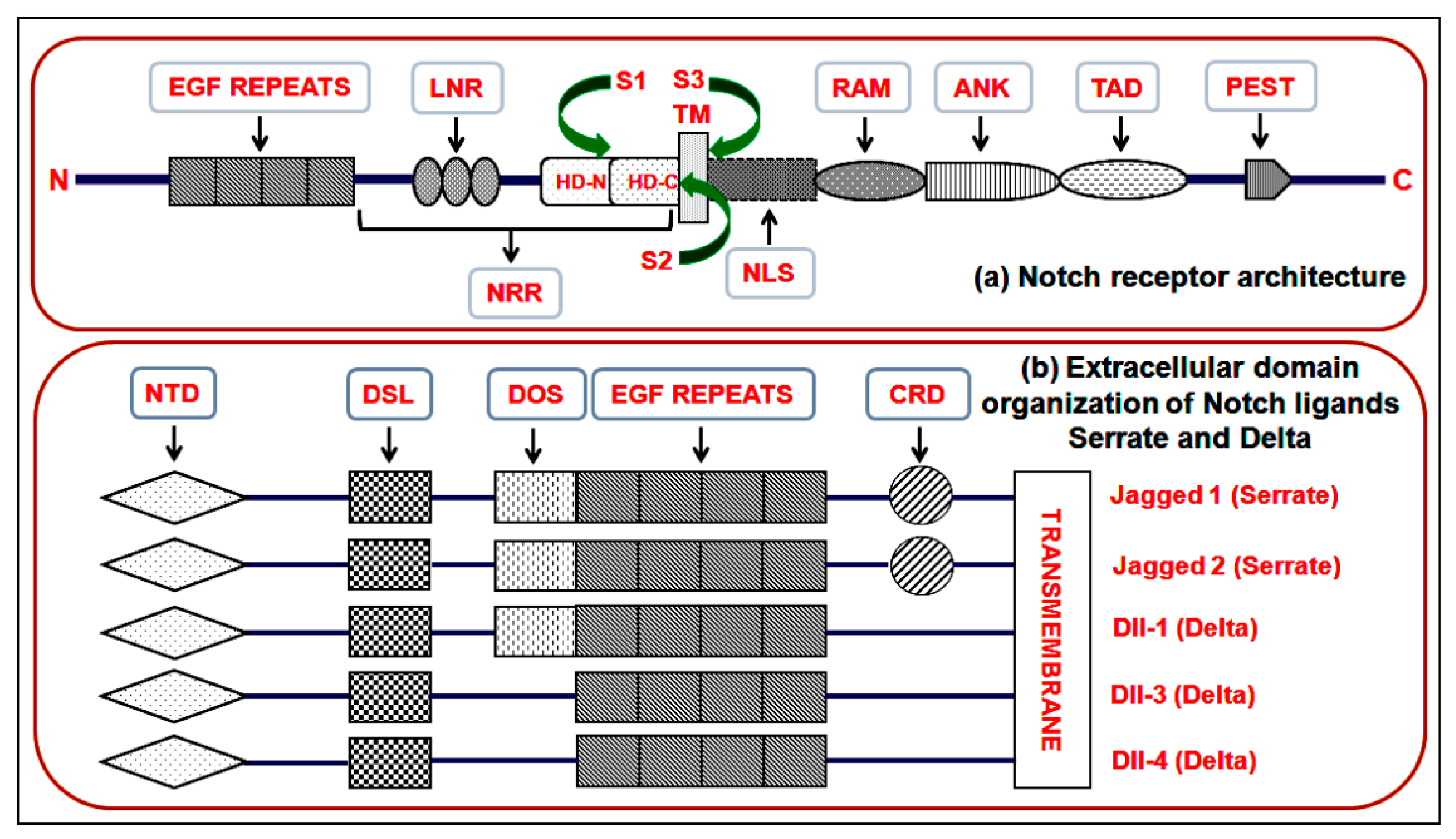
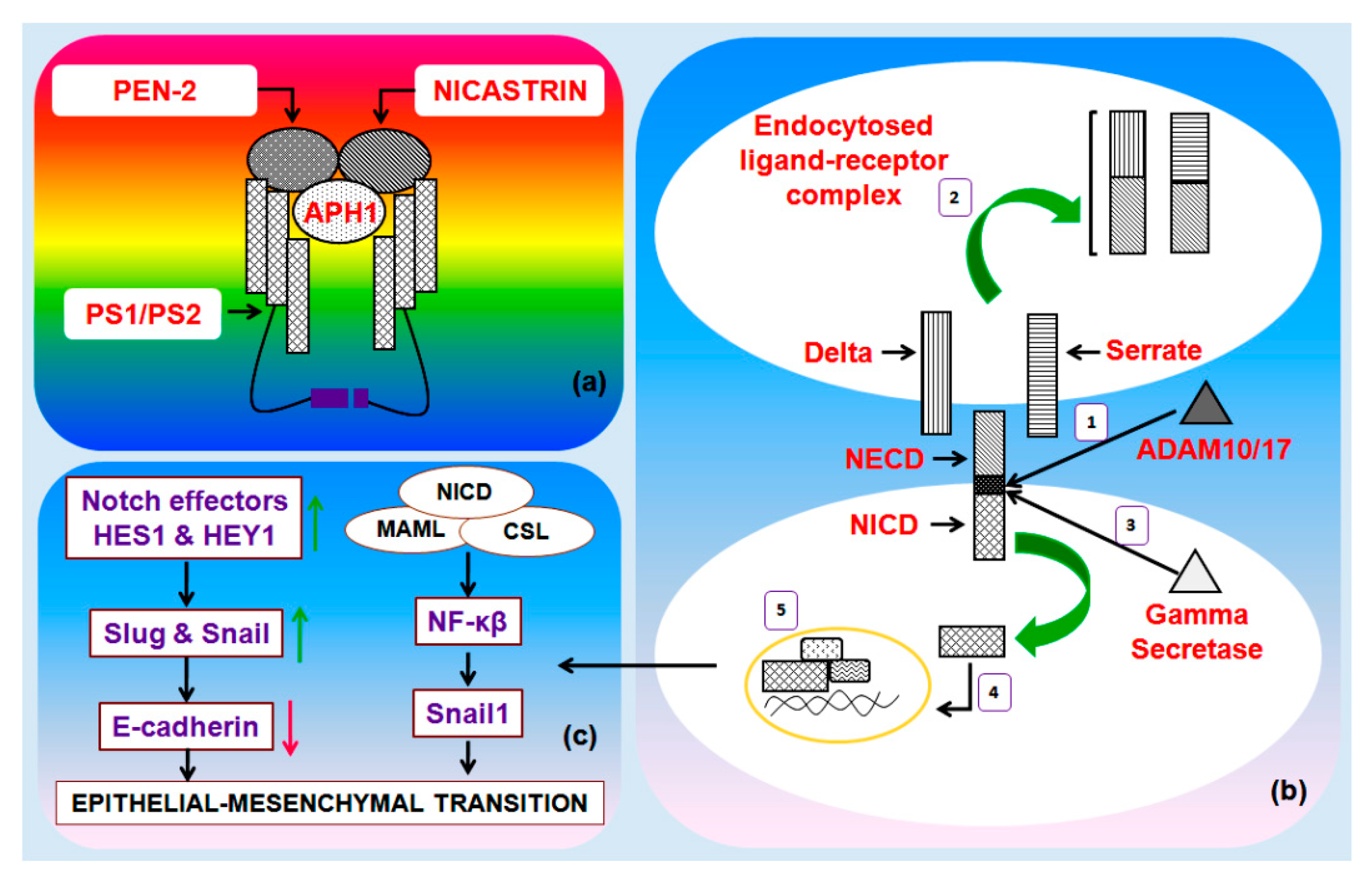
3. The Notch Pathway
4. Notch-Mediated EMT in Breast Cancer
4.1. The Notch/Akt Module
4.2. The Notch/Cytokine Module
4.3. The Notch/Hypoxia Module
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dai, X.; Li, Y.; Bai, Z.; Tang, X.Q. Molecular portraits revealing the heterogeneity of breast tumor subtypes defined using immunohistochemistry markers. Sci. Rep. 2015, 5, 14499. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Chu, P.Y.; Yiang, G.T.; Wu, M.Y. The Molecular Mechanism of Epithelial-Mesenchymal Transition for Breast Carcinogenesis. Biomolecules 2019, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Epithelial-mesenchymal transition: Untangling EMT’s functions. Nat. Rev. Cancer 2016, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal Transition—A Hallmark of Breast Cancer Metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Chiappetta, G. The Epithelial-to-Mesenchymal Transition in Breast Cancer: Focus on Basal-Like Carcinomas. Cancers 2017, 9, 134. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wen, X.F.; Guo, Y.X.; Zhang, G.J. Targeting Notch degradation system provides promise for breast cancer therapeutics. Crit. Rev. Oncol. Hematol. 2016, 104, 21–29. [Google Scholar] [CrossRef]
- Rangel, M.C.; Bertolette, D.; Castro, N.P.; Klauzinska, M.; Cuttitta, F.; Salomon, D.S. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 156, 211–226. [Google Scholar] [CrossRef]
- Wang, Q.; Shi, Y.; Butler, H.J.; Xue, J.; Wang, G.; Duan, P.; Zheng, H. Role of delta-like ligand-4 in chemoresistance against docetaxel in MCF-7 cells. Hum. Exp. Toxicol. 2017, 36, 328–338. [Google Scholar] [CrossRef]
- Baker, A.T.; Zlobin, A.; Osipo, C. Notch-EGFR/HER2 Bidirectional Crosstalk in Breast Cancer. Front. Oncol. 2014, 4, 360. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Škovierová, H.; Okajčeková, T.; Strnádel, J.; Vidomanová, E.; Halašová, E. Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review). Int. J. Mol. Med. 2018, 41, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Ashaie, M.A.; Chowdhury, E.H. Cadherins: The Superfamily Critically Involved in Breast Cancer. Curr. Pharm. Des. 2016, 22, 616–638. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-Mesenchymal Transition and Breast Cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef]
- Acar, A.; Simões, B.M.; Clarke, R.B.; Brennan, K. A Role for Notch Signalling in Breast Cancer and Endocrine Resistance. Stem Cells Int. 2016, 2016, 2498764. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhai, W.J.; Teng, C.B. Notch Signaling in Pancreatic Development. Int. J. Mol. Sci. 2016, 17, 48. [Google Scholar] [CrossRef]
- Yahyanejad, S.; Theys, J.; Vooijs, M. Targeting Notch to overcome radiation resistance. Oncotarget 2016, 7, 7610–7628. [Google Scholar] [CrossRef]
- Crabtree, J.S.; Singleton, C.S.; Miele, L. Notch Signaling in Neuroendocrine Tumors. Front. Oncol. 2016, 6, 94. [Google Scholar] [CrossRef]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef]
- Allenspach, E.J.; Maillard, I.; Aster, J.C.; Pear, W.S. Notch signaling in cancer. Cancer Biol. Ther. 2002, 1, 466–476. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Kotiyal, S.; Bhattacharya, S. Breast cancer stem cells, EMT and therapeutic targets. Biochem. Biophys. Res. Commun. 2014, 453, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Elzamly, S.; Badri, N.; Padilla, O.; Dwivedi, A.K.; Alvarado, L.A.; Hamilton, M.; Diab, N.; Rock, C.; Elfar, A.; Teleb, M.; et al. Epithelial-Mesenchymal Transition Markers in Breast Cancer and Pathological Response after Neoadjuvant Chemotherapy. Breast Cancer 2018, 12, 1178223418788074. [Google Scholar]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The Role of Notch Signaling Pathway in Epithelial-Mesenchymal Transition (EMT) During Development and Tumor Aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial–mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef]
- Katz, Y.; Li, F.; Lambert, N.J.; Sokol, E.S.; Tam, W.L.; Cheng, A.W.; Airoldi, E.M.; Lengner, C.J.; Gupta, P.B.; Yu, Z.; et al. Musashi proteins are post-transcriptional regulators of the epithelial-luminal cell state. eLife 2014, 3, e03915. [Google Scholar] [CrossRef]
- Calaf, G.M.; Balajee, A.S.; Montalvo-Villagra, M.T.; Leon, M.; Daniela, N.M.; Alvarez, R.G.; Roy, D.; Narayan, G.; Abarca-Quinones, J. Vimentin and Notch as biomarkers for breast cancer progression. Oncol. Lett. 2014, 7, 721–727. [Google Scholar] [CrossRef]
- Sadeghi, N.; Gerber, D.E. Targeting the PI3K pathway for cancer therapy. Future Med. Chem. 2012, 4, 1153–1169. [Google Scholar] [CrossRef] [PubMed]
- Sangphech, N.; Osborne, B.A.; Palaga, T. Notch signaling regulates the phosphorylation of Akt and survival of lipopolysaccharide-activated macrophages via regulator of G protein signaling 19 (RGS19). Immunobiol. 2014, 219, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.M.; Yao, M.; Liu, S.X.; Hao, J.; Liu, Q.J.; Gao, F. Interplay between the Notch and PI3K/Akt pathways in high glucose-induced podocyte apoptosis. Am. J. Physiol. Ren. Physiol. 2014, 306, F205–F213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhang, J.; Xiong, N.; Li, S.; Chen, Y.; Yang, H.; Wu, C.; Zeng, H.; Liu, Y. Notch-1 signaling activates NF-κB in human breast carcinoma MDA-MB-231 cells via PP2A-dependent AKT pathway. Med. Oncol. 2016, 33, 33. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Shimizu, M.; Izrailit, J.; Ng, N.F.; Buchman, Y.; Pan, J.G.; Dering, J.; Reedijk, M. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res. Treat. 2010, 123, 113–124. [Google Scholar] [CrossRef]
- Chen, Y.; Li, D.; Liu, H.; Xu, H.; Zheng, H.; Qian, F.; Li, W.; Zhao, C.; Wang, Z.; Wang, X. Notch-1 signaling facilitates survivin expression in human non-small cell lung cancer cells. Cancer Biol. Ther. 2011, 11, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhao, F.; Lu, J.; Li, T.; Yang, H.; Wu, C.; Liu, Y. Notch-1 Signaling Promotes the Malignant Features of Human Breast Cancer through NF-κB Activation. PLoS ONE 2014, 9, e95912. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Li, Y.Y.; Zhang, H.Y.; Wang, F.; He, H.L.; Yao, J.C.; Liu, L.; Li, S.S. Role of matrix metalloproteinase-9 in transforming growth factor-ß1-induced epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Oncol. Targets Ther. 2017, 10, 2837–2847. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.K.; Kaushik, N.; Suh, Y.; Yoo, K.C.; Cui, Y.H.; Kim, M.J.; Lee, H.J.; Kim, I.G.; Lee, S.J. Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget 2016, 7, 53430–53442. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchyme transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.; Shao, S.; Zuo, X.; Ning, Q.; Luo, M.; Gu, S.; Zhao, X. Notch1 induces epithelial-mesenchymal transition and the cancer stem cell phenotype in breast cancer cells and STAT3 plays a key role. Int. J. Oncol. 2015, 46, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133hi cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.C.; Kulp, S.K.; Huang, H.L.; Tu, H.J.; Salunke, S.B.; Sullivan, N.J.; Sun, D.; Wicha, M.S.; Shapiro, C.L.; Chen, C.S. Function of Integrin-Linked Kinase in Modulating the Stemness of IL-6–Abundant Breast Cancer Cells by Regulating γ-Secretase–Mediated Notch1 Activation in Caveolae. Neoplasia 2015, 17, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Barnawi, R.; Al-Khaldi, S.; Majed Sleiman, G.; Sarkar, A.; Al-Dhfyan, A.; Al-Mohanna, F.; Ghebeh, H.; Al-Alwan, M. Fascin is Critical for the Maintenance of Breast Cancer Stem Cell Pool Predominantly via the Activation of the Notch Self-Renewal Pathway. Stem Cells 2016, 34, 2799–2813. [Google Scholar] [CrossRef]
- Dinicola, S.; Fabrizi, G.; Masiello, M.G.; Proietti, S.; Palombo, A.; Minini, M.; Harrath, A.H.; Alwasel, S.H.; Ricci, G.; Catizone, A.; et al. Inositol induces mesenchymal-epithelial reversion in breast cancer cells through cytoskeleton rearrangement. Exp. Cell Res. 2016, 345, 37–50. [Google Scholar] [CrossRef]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Imanaka, N.; Chen, J.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [Green Version]
- Bocca, C.; Ievolella, M.; Autelli, R.; Motta, M.; Mosso, L.; Torchio, B.; Bozzo, F.; Cannito, S.; Paternostro, C.; Colombatto, S.; et al. Expression of Cox-2 in human breast cancer cells as a critical determinant of epithelial-to-mesenchymal transition and invasiveness. Expert Opin. Ther. Targets 2014, 18, 121–135. [Google Scholar] [CrossRef]
- Hugo, H.J.; Saunders, C.; Ramsay, R.G.; Thompson, E.W. New Insights on COX-2 in Chronic Inflammation Driving Breast Cancer Growth and Metastasis. J. Mammary Gland Biol. Neoplasia 2015, 20, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.M.; Hess, D.; Lala, P.K. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Nandi, P.; Omar, A.; Ugwuagbo, K.C.; Lala, P.K. EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.H.; Liang, L.Z.; Liu, X.L.; Wu, J.N.; Su, K.; Chen, J.Y.; Zheng, Q.Y. LncRNA UCA1/miR-124 axis modulates TGFß1-induced epithelial-mesenchymal transition and invasion of tongue cancer cells through JAG1/Notch signaling. J. Cell. Biochem. 2019, 120, 10495–10504. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Cheng, S.H.; Wu, C.H.; Li, W.Y.; Wang, J.S.; Kung, M.L.; Chu, T.H.; Huang, S.T.; Feng, C.T.; Huang, S.C.; et al. Delta-like 1 homologue promotes tumorigenesis and epithelial-mesenchymal transition of ovarian high-grade serous carcinoma through activation of Notch signaling. Oncogene 2019, 38, 3201–3215. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, X.; Song, J.; Wang, Y.; Zhao, Y.; Meng, J. MicroRNA-539 inhibits the progression of Wilms’ Tumor through downregulation of JAG1 and Notch1/3. Cancer Biomark. 2018, 24, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wang, Z.; Xiong, X.; Zhong, Y.; Zhang, W.; Dong, Y.; Li, J.; Zhu, Z.; Zhang, W.; Wu, H.; et al. Membrane-tethered Notch1 exhibits oncogenic property via activation of EGFR-PI3K-AKT pathway in oral squamous cell carcinoma. J. Cell. Physiol. 2019, 234, 5940–5952. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Fan, S.; Lin, H.; Lian, W. STAT3-induced upregulation of long noncoding RNA HNF1A-AS1 promotes the progression of oral squamous cell carcinoma via activating Notch signaling pathway. Cancer Biol. Ther. 2018, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zheng, G.; Zhou, L.; Li, P.; Yun, M.; Shi, Q.; Wang, T.; Wu, X. Notch signalling induces epithelial-mesenchymal transition to promote metastasis in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 42, 2276–2284. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Chen, M.; Lu, M.; Xin-Xiang, J.; Meng-Xiong, P.; Jun-Wu, M. Glutaredoxin 3 promotes migration and invasion via the Notch signalling pathway in oral squamous cell carcinoma. Free Radic Res. 2018, 52, 390–401. [Google Scholar] [CrossRef]
- Yang, X.; Xia, W.; Chen, L.; Wu, C.X.; Zhang, C.C.; Olson, P.; Wang, X.Q. Synergistic antitumor effect of a γ-secretase inhibitor PF-03084014 and sorafenib in hepatocellular carcinoma. Oncotarget 2018, 9, 34996–35007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Song, W.; Kan, P.; Huang, C.; Ma, Z.; Wu, Q.; Yao, X.; Zhang, B. Overexpression of Epsin 3 enhances migration and invasion of glioma cells by inducing epithelial-mesenchymal transition. Oncol. Rep. 2018, 40, 3049–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, Q.; Lin, L.; Wang, R.; Chen, L.; Du, W.; Jiang, C.; Li, R. Targeting the Notch1 oncogene by miR-139-5p inhibits glioma metastasis and epithelial-mesenchymal transition (EMT). BMC Neurol. 2018, 18, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.B.; Li, W.; Chu, A.X. MicroRNA-133a inhibits gastric cancer cells growth, migration, and epithelial-mesenchymal transition process by targeting presenilin 1. J. Cell Biochem. 2019, 120, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, L.; He, X.; Li, S.; Sun, R.; Xiang, Q.; Ren, G.; Xiang, T. KRAB zinc-finger protein 382 regulates epithelial-mesenchymal transition and functions as a tumor suppressor, but is silenced by CpG methylation in gastric cancer. Int. J. Oncol. 2018, 53, 961–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, G.; Cheng, Z.; Dai, L.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA-XIST Suppresses Proliferation and TGF-ß1-Induced EMT in NSCLC Through the Notch-1 Pathway by Regulation of miR-137. Genet. Test. Mol. Biomark. 2018, 22, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Jolly, M.K.; Tripathi, S.C.; Aguilar, M.; Hanash, S.M.; Levine, H.; Onuchic, J.N. Numb prevents a complete epithelial-mesenchymal transition by modulating Notch signalling. J. R. Soc. Interface 2017, 14, 20170512. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.W.; Wang, H.; Bao, Y.F.; Xie, K. Notch signaling molecule is involved in the invasion of MiaPaCa2 cells induced by CoCl2 via regulating epithelial-mesenchymal transition. Mol. Med. Rep. 2018, 17, 4965–4972. [Google Scholar] [CrossRef] [Green Version]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Que, J.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1758. [Google Scholar] [CrossRef] [Green Version]
- Fukusumi, T.; Guo, T.W.; Sakai, A.; Ando, M.; Ren, S.; Haft, S.; Liu, C.; Amornphimoltham, P.; Gutkind, J.S.; Califano, J.A. The NOTCH4-HEY1 Pathway Induces Epithelial-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 619–633. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jin, X.; Wang, Z.; Zhang, X.; Liu, S.; Liu, G. Downregulation of SNHG1 suppresses cell proliferation and invasion by regulating Notch signaling pathway in esophageal squamous cell cancer. Cancer Biomark. 2017, 21, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| S. No. | Cancer Type | Associated Factors | Interactions | References |
|---|---|---|---|---|
| 1 | Tongue Cancer | JAG1/Notch, LncRNA UCA1/miRNA-124, TGFβ1 | UCA1 knockdown increases, whereas miR-124 inhibition decreases TGFβ1-induced EMT and invasion in tongue cancer cells through miR-124 downstream JAG1/Notch signaling | [54] |
| 2 | Ovarian Cancer | Notch1, DLK1 | Delta-like 1 homolog (DLK1) over-expression promotes ovarian carcinogenesis through Notch activation and EMT induction | [55] |
| 3 | Wilms’ Tumor (WT) | JAG1/Notch 1/3, miRNA-539 | miR-539 inhibits EMT in WT by inhibiting the expression of JAG1/Notch 1/3 cascade | [56] |
| 4 | Oral Squamous Cell Carcinoma (OSCC) | Notch1, EGFR/PI3K/Akt | Notch1 modulates EMT by the activation of EGFR/PI3K/Akt pathway in OSCC | [57] |
| 5 | Oral Squamous Cell Carcinoma | Notch1, HNF1A-SA1, STAT3 | The transcription factor STAT3 upregulates HNF1A-SA1 and facilitates OSCC progression by activation of the Notch signaling cascade | [58,59] |
| 6 | Oral Squamous Cell Carcinoma | Notch1, GLRX3 | Glutaredoxin 3 (GLRX3) knock-down limits Notch activity in OSCC by reversing EMT | [60] |
| 7 | Hepatocellular Carcinoma | Notch1, Snail-1, N-cadherin, ABCG2, Nanog, Oct4 | Notch1-Snail1 signaling pathway contributes to sorafenib resistance by promoting EMT and EMT-mediated CSC features, such as upregulated expression of Snail-1, N-cadherin, ABCG2, Nanog and Oct4, and reduced expression of E-cadherin | [61] |
| 8 | Glioblastoma | Notch1/2/3/4, EPN3, WNT/β-catenin | EPN3 may be involved in the Notch and WNT/β-catenin signaling pathways that in turn promotes EMT in glioblastoma cells by activating Slug, Twist and ZEB1, but not Snail-1 or ZEB2 | [62] |
| 9 | Glioblastoma | Notch1, miRNA-139-5p | miR-139-5p inhibits Notch1 and prevents glioma metastasis and EMT | [63] |
| 10 | Gastric Cancer | PS1, miRNA-133a | miR-133a prevents EMT in gastric cells by targeting PS1, a key component in the Notch signaling pathway | [64] |
| 11 | Gastric Cancer | Notch1, ZNF-382 | KRAB zinc finger protein 382 (ZNF-382) is frequently methylated in gastric cancer and can reverse the EMT program in gastric cancer cells through Notch signaling | [65] |
| 12 | Non-Small Cell Lung Cancer (NSCLC) | Notch1, XIST, miR-137 | XIST suppresses TGF-β1-induced EMT in NSCLC by regulating the Notch1 pathway | [66] |
| 13 | Lung Cancer | Notch1, Numb | Numb functions as a suppressor for a full EMT and thus behaves as a ‘phenotypic stability factor’ by regulating Notch-driven EMT | [67] |
| 14 | Pancreatic Cancer | Notch1, HIF-1α | HIF-1α and Notch1 may be involved in regulating the EMT program in MiaPaCa2 cells | [68] |
| 15 | Squamous Cell Carcinoma (SCC) | Notch1, Notch3, TGFβ, ZEB1 | Notch1 activation and EMT are coupled to trigger SCC tumor initiation in association with TGF-β located in the tumor microenvironment. In response, TGFβ activates ZEB1 that represses Notch3, thereby preventing terminal differentiation | [69] |
| 16 | Head and Neck Squamous Cell Carcinoma (HNSCC) | Notch4, HEY1 | Notch4 and HEY1 associate to induce cisplatin resistance and promote EMT in HNSCC | [70] |
| 17 | Esophageal Squamous Cell Cancer | Notch1, SNHG1, HES1 | Small nucleolar RNA host gene 1 (SNHG1) suppresses Notch1 and HES1 and inhibits EMT | [71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kar, R.; Jha, N.K.; Jha, S.K.; Sharma, A.; Dholpuria, S.; Asthana, N.; Chaurasiya, K.; Singh, V.K.; Burgee, S.; Nand, P. A “NOTCH” Deeper into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer. Genes 2019, 10, 961. https://doi.org/10.3390/genes10120961
Kar R, Jha NK, Jha SK, Sharma A, Dholpuria S, Asthana N, Chaurasiya K, Singh VK, Burgee S, Nand P. A “NOTCH” Deeper into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer. Genes. 2019; 10(12):961. https://doi.org/10.3390/genes10120961
Chicago/Turabian StyleKar, Rohan, Niraj Kumar Jha, Saurabh Kumar Jha, Ankur Sharma, Sunny Dholpuria, Nidhi Asthana, Kundan Chaurasiya, Vivek Kumar Singh, Shuaib Burgee, and Parma Nand. 2019. "A “NOTCH” Deeper into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer" Genes 10, no. 12: 961. https://doi.org/10.3390/genes10120961