Effect of CYP3A4*22 and PPAR-α Genetic Variants on Platelet Reactivity in Patients Treated with Clopidogrel and Lipid-Lowering Drugs Undergoing Elective Percutaneous Coronary Intervention

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Exposure and Outcome
2.3. Validation Cohorts
2.4. Statistical Analysis
3. Results
3.1. Study Cohort
3.2. Validation Cohorts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mehta, S.R.; Yusuf, S.; Peters, R.J.; Bertrand, M.E.; Lewis, B.S.; Natarajan, M.K.; Malmberg, K.; Rupprecht, H.-J.; Zhao, F.; Chrolavicius, S.; et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001, 358, 527–533. [Google Scholar] [CrossRef]
- Angiolillo, D.J.; Fernández-Ortiz, A.; Bernardo, E.; Ramírez, C.; Cavallari, U.; Trabetti, E.; Sabaté, M.; Hernández, R.; Moreno, R.; Escaned, J.; et al. Contribution of gene sequence variations of the hepatic cytochrome P450 3A4 enzyme to variability in individual responsiveness to clopidogrel. Arter. Thromb. Vasc. Biol. 2006, 26, 1895–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhindi, R.; Ormerod, O.; Newton, J.; Banning, A.P.; Testa, L. Interaction between statins and clopidogrel: Is there anything clinically relevant? QJM Int. J. Med. 2008, 101, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliden, K.P.; DiChiara, J.; Lawal, L.; Singla, A.; Antonino, M.J.; Baker, B.A.; Bailey, W.L.; Tantry, U.S.; Gurbel, P.A. The association of cigarette smoking with enhanced platelet inhibition by clopidogrel. J. Am. Coll. Cardiol. 2008, 52, 531–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremmel, T.; Steiner, S.; Seidinger, D.; Koppensteiner, R.; Panzer, S.; Kopp, C.W. Calcium-channel blockers decrease clopidogrel-mediated platelet inhibition. Heart 2009, 96, 186–189. [Google Scholar] [CrossRef]
- Harmsze, A.M.; Van Werkum, J.W.; Berg, J.M.T.; Zwart, B.; Bouman, H.J.; Breet, N.J.; Hof, A.W.V.; Ruven, H.J.T.; Hackeng, C.M.; Klungel, O.H.; et al. CYP2C19*2 and CYP2C9*3 alleles are associated with stent thrombosis: A case-control study. Eur. Heart J. 2010, 31, 3046–3053. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.M.; Maddox, T.M.; Wang, L.; Fihn, S.D.; Jesse, R.L.; Peterson, E.D.; Rumsfeld, J.S. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009, 301, 937. [Google Scholar] [CrossRef]
- Hochholzer, W.; Trenk, D.; Fromm, M.F.; Valina, C.M.; Stratz, C.; Bestehorn, H.-P.; Büttner, H.J.; Neumann, F.-J. Impact of cytochrome P450 2C19 loss-of-function polymorphism and of major demographic characteristics on residual platelet function after loading and maintenance treatment with clopidogrel in patients undergoing elective coronary stent placement. J. Am. Coll. Cardiol. 2010, 55, 2427–2434. [Google Scholar] [CrossRef] [Green Version]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Kazui, M.; Nishiya, Y.; Ishizuka, T.; Hagihara, K.; Farid, N.A.; Okazaki, O.; Ikeda, T.; Kurihara, A. Identification of the human cytochrome p450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2009, 38, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Yasmina, A.; De Boer, A.; Klungel, O.H.; Deneer, V.H. Pharmacogenomics of oral antiplatelet drugs. Pharmacogenomics 2014, 15, 509–528. [Google Scholar] [CrossRef]
- Park, J.J.; Park, K.W.; Kang, J.; Jeon, K.-H.; Kang, S.-H.; Ahn, H.S.; Han, J.-K.; Koh, J.-S.; Lee, S.-E.; Yang, H.-M.; et al. Genetic determinants of clopidogrel responsiveness in Koreans treated with drug-eluting stents. Int. J. Cardiol. 2013, 163, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Simon, T.; Collet, J.P.; Anderson, J.L.; Antman, E.M.; Bliden, K.; Cannon, C.P.; Danchin, N.; Giusti, B.; Gurbel, P.; et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: A meta-analysis. JAMA 2010, 304, 1821–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuldiner, A.R.; O’Connell, J.R.; Bliden, K.P.; Gandhi, A.; Ryan, K.; Horenstein, R.B.; Damcott, C.M.; Pakyz, R.; Tantry, U.S.; Gibson, Q.; et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 2009, 302, 849. [Google Scholar] [CrossRef] [PubMed]
- NIH, National Library of Medicine, National Center for Biotechnology Information, dbSNP Database. Available online: https://ncbi.nlm.nih.gov/snp/ (accessed on 27 August 2020).
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharm. J. 2010, 11, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, K.; Thomas, M.; Winter, S.; Nussler, A.K.; Niemi, M.; Schwab, M.; Zanger, U.M. PPARA: A novel genetic determinant of CYP3A4 in vitro and in vivo. Clin. Pharmacol. Ther. 2012, 91, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.Y.; Armstrong, P.C.; Dhanji, A.-R.A.; Tucker, A.T.; Paul-Clark, M.J.; Mitchell, J.A.; Warner, T.D. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arter. Thromb. Vasc. Biol. 2009, 29, 706–711. [Google Scholar] [CrossRef] [Green Version]
- Breet, N.J.; Van Werkum, J.W.; Bouman, H.J.; Kelder, J.C.; Ruven, H.J.T.; Bal, E.T.; Deneer, V.H.; Harmsze, A.M.; Van Der Heyden, J.A.S.; Rensing, B.J.W.M.; et al. Comparison of platelet function tests in predicting clinical outcome in patients undergoing coronary stent implantation. JAMA 2010, 303, 754. [Google Scholar] [CrossRef]
- Brar, S.S.; Berg, J.T.; Marcucci, R.; Price, M.J.; Valgimigli, M.; Kim, H.-S.; Patti, G.; Breet, N.J.; DiSciascio, G.; Cuisset, T.; et al. Impact of platelet reactivity on clinical outcomes after percutaneous coronary intervention. J. Am. Coll. Cardiol. 2011, 58, 1945–1954. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.W.A.; Bergmeijer, T.O.; Vos, G.-J.A.; Kelder, J.C.; Qaderdan, K.; Godschalk, T.C.; Breet, N.J.; Deneer, V.H.M.; Hackeng, C.M.; Berg, J.M.T. Tailored P2Y12 inhibitor treatment in patients undergoing non-urgent PCI—The POPular Risk Score study. Eur. J. Clin. Pharmacol. 2019, 75, 1201–1210. [Google Scholar] [CrossRef]
- Tantry, U.S.; Bonello, L.; Aradi, D.; Price, M.J.; Jeong, Y.-H.; Angiolillo, D.J.; Stone, G.W.; Curzen, N.; Geisler, T.; Berg, J.T.; et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J. Am. Coll. Cardiol. 2013, 62, 2261–2273. [Google Scholar] [CrossRef] [PubMed]
- Bergmeijer, T.O.; Reny, J.-L.; Pakyz, R.E.; Gong, L.; Lewis, J.P.; Kim, E.-Y.; Aradi, D.; Fernández-Cadenas, I.; Horenstein, R.B.; Lee, M.T.M.; et al. Genome-wide and candidate gene approaches of clopidogrel efficacy using pharmacodynamic and clinical end points—Rationale and design of the International Clopidogrel Pharmacogenomics Consortium (ICPC). Am. Heart J. 2018, 198, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.S.; Bergmeijer, T.O.; Gong, L.; Reny, J.; Lewis, J.P.; Mitchell, B.D.; Alexopoulos, D.; Aradi, D.; Altman, R.B.; Bliden, K.; et al. Genome-wide association study of platelet reactivity and cardiovascular response in patients treated with clopidogrel: A study by the International Clopidogrel Pharmacogenomics Consortium (ICPC). Clin. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
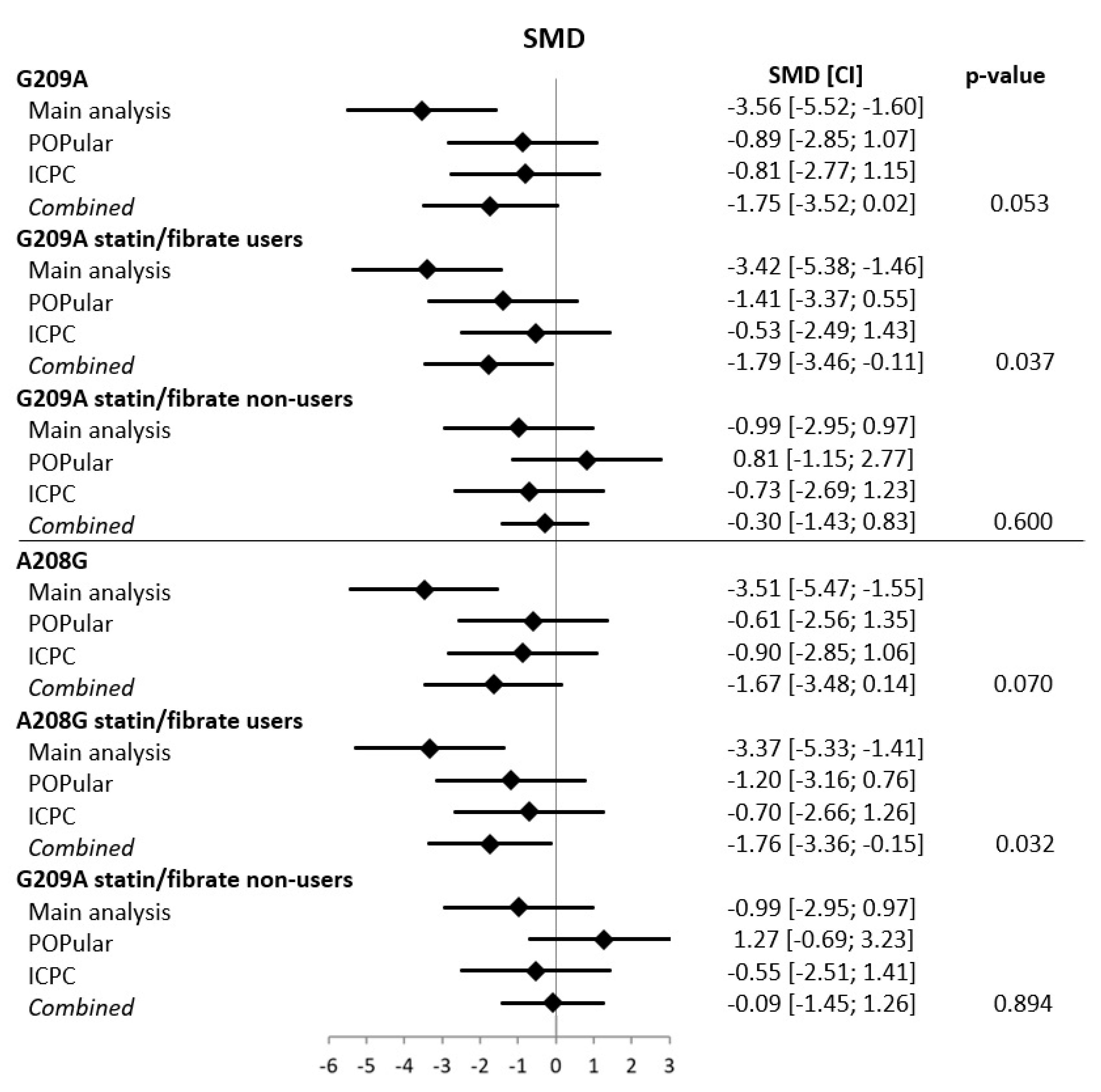
- Kreutz, R.P.; Owens, J.; Jin, Y.; Nystrom, P.; Desta, Z.; Kreutz, Y.; Breall, J.A.; Li, L.; Chiang, C.; Kovacs, R.J.; et al. Cytochrome P450 3A4*22, PPAR-α, and ARNT polymorphisms and clopidogrel response. Clin. Pharmacol. Adv. Appl. 2013, 5, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbiyik, F.; Ray, D.M.; Gettings, K.F.; Blumberg, N.; Francis, C.W.; Phipps, R.P. Human bone marrow megakaryocytes and platelets express PPARγ, and PPARγ agonists blunt platelet release of CD40 ligand and thromboxanes. Blood 2004, 104, 1361–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, F.Y.; Davidson, S.J.; Moraes, L.A.; Traves, S.L.; Paul-Clark, M.; Bishop-Bailey, D.; Warner, T.D.; Mitchell, J.A. Role of nuclear receptor signaling in platelets: Antithrombotic effects of PPARβ. FASEB J. 2005, 20, 326–328. [Google Scholar] [CrossRef]
- Fuentes, E.; Palomo, I. Mechanism of antiplatelet action of hypolipidemic, antidiabetic and antihypertensive drugs by PPAR activation. Vasc. Pharmacol. 2014, 62, 162–166. [Google Scholar] [CrossRef]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From Orphan Receptors to Drug Discovery†. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef]
- Paumelle, R.; Blanquart, C.; Briand, O.; Barbier, O.; Duhem, C.; Woerly, G.; Percevault, F.; Fruchart, J.C.; Dombrowicz, D.; Glineur, C.; et al. Acute antiinflammatory properties of statins involve peroxisome proliferator-activated receptor-alpha via inhibition of the protein kinase C signaling pathway. Circ. Res. 2006, 98, 361–369. [Google Scholar] [CrossRef]
- Elens, L.; Bouamar, R.; Hesselink, D.A.; Haufroid, V.; Van Der Heiden, I.P.; Van Gelder, T.; Van Schaik, R.H.N. A new functional CYP3A4 intron 6 polymorphism significantly affects tacrolimus pharmacokinetics in kidney transplant recipients. Clin. Chem. 2011, 57, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Stone, G.W.; Witzenbichler, B.; Weisz, G.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; Cox, D.A.; Duffy, P.L.; Mazzaferri, E.; et al. Platelet reactivity and clinical outcomes after coronary artery implantation of drug-eluting stents (ADAPT-DES): A prospective multicentre registry study. Lancet 2013, 382, 614–623. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| All Patients (n = 1124) | |
|---|---|
| Characteristics | n (%) |
| Age, years (mean, SD) | 63.9 ± 10.8 |
| Sex (male) | 848 (75.4) |
| BMI, kg/m2 (mean, SD) | 27.5 ± 4.2 |
| Smoking habit | |
| No | 632 (56.1) |
| Current smoker or stopped < 6 months | 247 (22.0) |
| Past smoker | 238 (21.2) |
| Unknown | 7 (0.6) |
| Disease history | |
| Hypertension | 936 (83.3) |
| Diabetes | 235 (20.9) |
| Myocardial infarction | 349 (31.0) |
| Co-medication at baseline | |
| Beta-blocker | 900 (80.1) |
| ACE inhibitor | 422 (37.5) |
| Calcium channel blocker | 314 (27.9) |
| ARB | 204 (18.1) |
| Diuretic | 313 (27.8) |
| Statin or fibrate | 996 (88.6) |
| Simvastatin | 647 (67.7) |
| Atorvastatin | 193 (19.4) |
| Rosuvastatin | 81 (8.1) |
| Pravastatin | 58 (5.8) |
| Fluvastatin | 7 (0.7) |
| Fibrate | 3 (0.3) |
| Unknown/missing | 9 (0.9) |
| Proton pump inhibitor | 454 (40.4) |
| Impaired renal function ‡ | 9 (0.8) |
| Clopidogrel loading † | 395 (35.1) |
| CYP2C19 metabolizer | |
| Normal | 821 (73.0) |
| Intermediate | 274 (24.4) |
| Poor | 29 (2.6) |
| CYP3A4*22 | |
| MAF | 6.3% |
| *1/*1 | 987 (87.8) |
| *1/*22 | 132 (11.7) |
| *22/*22 | 5 (0.4) |
| PPAR-α G209A | |
| MAF | 25.4% |
| GG | 630 (56.0) |
| GA | 417 (37.1) |
| AA | 77 (6.9) |
| PPAR-α A208G | |
| MAF | 27.0% |
| AA | 597 (53.1) |
| AG | 448 (39.9) |
| GG | 79 (7.0) |
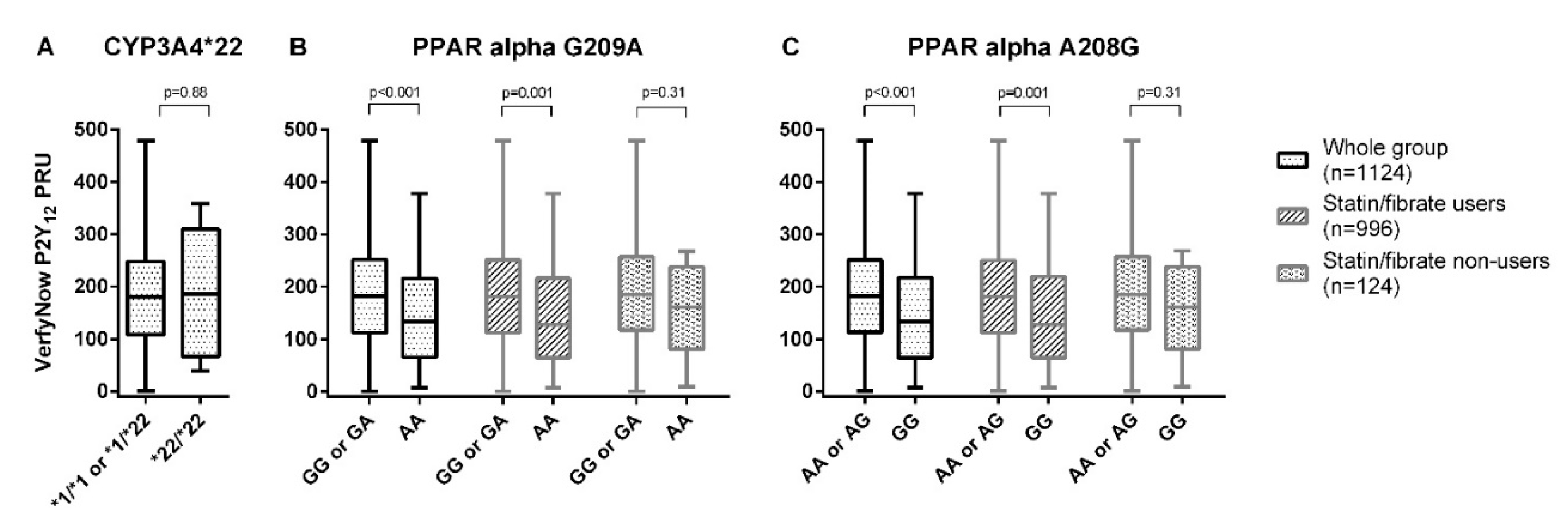
| Genetic Variant | Mean ± SD | Coefficient (95% CI) | |
|---|---|---|---|
| Crude | Adjusted | ||
| All patients (n = 1124) | |||
| CYP3A4*22 | |||
| *1/*1 or *1/*22 (n = 1119) | 181 ± 96 | Ref | Ref |
| *22/*22 (n = 5) | 188 ± 128 | 6.6 (−77.6, 90.8), p = 0.88 | NA ‡ |
| PPAR-α G209A | |||
| GG or GA (n = 1047) | 184 ± 95 | Ref | Ref |
| AA (n = 77) | 144.0 ± 94 | −40.1 (−62.1, −18.0), p < 0.001 | −24.6 (−44.7, −4.6), p = 0.016 † |
| PPAR-α A208G | |||
| AA or AG (n = 1045) | 184 ± 95 | Ref | Ref |
| GG (n = 79) | 145 ± 95 | −39.1 (−60.9, −17.3), p < 0.001 | −24.56 (−44.3, −4.8), p = 0.015 † |
| In statin/fibrate users (n = 996) | |||
| PPAR-α G209A | |||
| GG or GA (n = 928) | 184 ± 95 | Ref | Ref |
| AA (n = 68) | 143 ± 95 | −40.8 (−64.2, −17.4), p = 0.001 | −26.7 (−47.9, −5.4), p = 0.014 § |
| PPAR-α A208G | |||
| AA or AG (n = 926) | 184 ± 95 | Ref | Ref |
| GG (n = 70) | 144 ± 96 | −39.7 (−62.8, −16.6), p = 0.001 | −26.5 (−47.5, −5.6), p = 0.013 § |
| In statin/fibrate non-users (n = 124) | |||
| PPAR-α G209A | |||
| GG or GA (n = 115) | 190 ± 100 | Ref | Ref |
| AA (n = 9) | 154 ± 90 | −34.3 (−103.2, 32.7), p = 0.31 | NA ‡ |
| PPAR-α A208G | |||
| GG or GA (n = 115) | 190 ± 100 | Ref | Ref |
| AA (n = 9) | 154 ± 90 | −34.3 (−103.2, 32.7), p = 0.31 | NA ‡ |
| Genetic Variant | HPR+ (n = 436) | HPR− (n = 688) | Crude OR (95% CI) | Adjusted OR (95% CI) |
|---|---|---|---|---|
| All patients (n = 1124) | ||||
| CYP3A4*22 | ||||
| *1/*1 or *1/*22 (n = 1119) | 434 (38.8) | 685 (61.2) | Ref | Ref |
| *22/*22 (n = 5) | 2 (40.0) | 3 (60.0) | 1.05 (0.18, 6.32) | NA ‡ |
| PPAR-α G209A | ||||
| GG or GA (n = 1047) | 414 (39.5) | 633 (60.5) | Ref | Ref |
| AA (n = 77) | 22 (28.6) | 55 (71.4) | 0.61 (0.37, 1.02) | 0.82 (0.47, 1.43) * |
| PPAR-α A208G | ||||
| AA or AG (n = 1045) | 413 (39.5) | 632 (60.5) | Ref | Ref |
| GG (n = 79) | 23 (29.1) | 56 (70.9) | 0.63 (0.38, 1.04) | 0.83 (0.48, 1.43) * |
| In statin/fibrate users (n = 996) | ||||
| PPAR-α G209A | ||||
| GG or GA (n = 928) | 371 (39.2) | 576 (60.8) | Ref | Ref |
| AA (n = 68) | 19 (27.1) | 51 (72.9) | 0.60 (0.35, 1.03) | 0.77 (0.43, 1.38) § |
| PPAR-α A208G | ||||
| AA or AG (n = 926) | 370 (39.2) | 575 (60.8) | Ref | Ref |
| GG (n = 70) | 20 (27.8) | 52 (72.2) | 0.62 (0.36, 1.05) | 0.78 (0.43, 1.38) § |
| In statin/fibrate non-users (n = 124) | ||||
| PPAR-α G209A | ||||
| GG or GA (n = 115) | 42 (43.8) | 54 (56.3) | Ref | Ref |
| AA (n = 9) | 3 (42.9) | 4 (57.1) | 0.66 (0.17, 3.04) | NA ‡ |
| PPAR-α A208G | ||||
| GG or GA (n = 115) | 42 (43.8) | 54 (56.3) | Ref | Ref |
| AA (n = 9) | 3 (42.9) | 4 (57.1) | 0.66 (0.17, 3.04) | NA ‡ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergmeijer, T.O.; Yasmina, A.; Vos, G.J.A.; Janssen, P.W.A.; Hackeng, C.M.; Kelder, J.C.; Verma, S.S.; Ritchie, M.D.; Gong, L.; Klein, T.E.; et al. Effect of CYP3A4*22 and PPAR-α Genetic Variants on Platelet Reactivity in Patients Treated with Clopidogrel and Lipid-Lowering Drugs Undergoing Elective Percutaneous Coronary Intervention. Genes 2020, 11, 1068. https://doi.org/10.3390/genes11091068
Bergmeijer TO, Yasmina A, Vos GJA, Janssen PWA, Hackeng CM, Kelder JC, Verma SS, Ritchie MD, Gong L, Klein TE, et al. Effect of CYP3A4*22 and PPAR-α Genetic Variants on Platelet Reactivity in Patients Treated with Clopidogrel and Lipid-Lowering Drugs Undergoing Elective Percutaneous Coronary Intervention. Genes. 2020; 11(9):1068. https://doi.org/10.3390/genes11091068
Chicago/Turabian StyleBergmeijer, Thomas O., Alfi Yasmina, Gerrit J. A. Vos, Paul W. A. Janssen, Christian M. Hackeng, Johannes C. Kelder, Shefali S. Verma, Marylyn D. Ritchie, Li Gong, Teri E. Klein, and et al. 2020. "Effect of CYP3A4*22 and PPAR-α Genetic Variants on Platelet Reactivity in Patients Treated with Clopidogrel and Lipid-Lowering Drugs Undergoing Elective Percutaneous Coronary Intervention" Genes 11, no. 9: 1068. https://doi.org/10.3390/genes11091068