
Set-Based Rare Variant Expression Quantitative Trait Loci in Blood and Brain from Alzheimer Disease Study Participants

 ,
,
Abstract
:1. Introduction
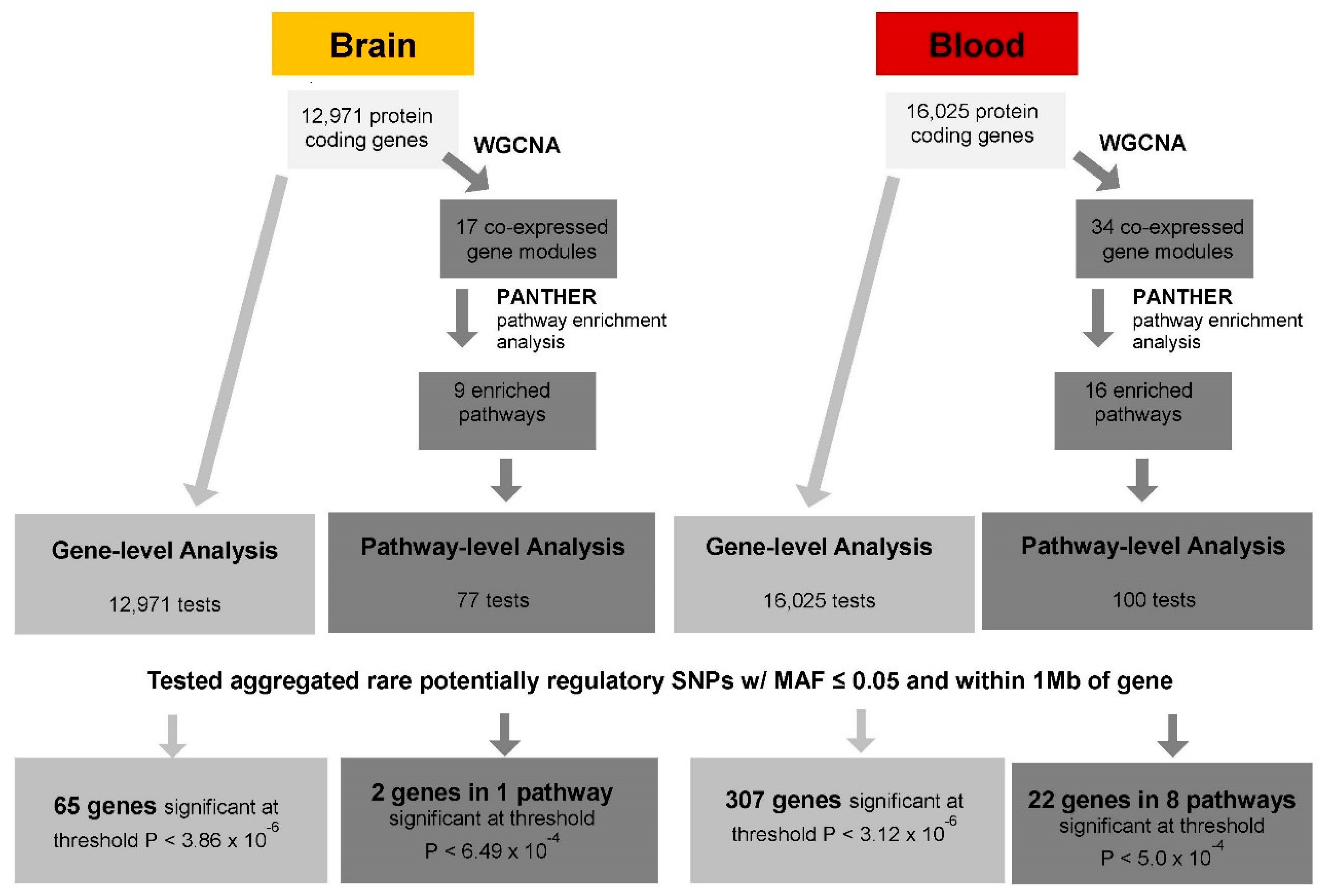
2. Materials and Methods
2.1. Study Cohorts
2.2. Data Processing
2.3. Functional Annotation of Variants
2.4. Set-Based eQTL Analysis
2.4.1. Gene-Level cis-eQTL Analysis
2.4.2. Pathway-Level cis-eQTL Analysis
2.4.3. Comparison of Rare and Common eQTLs
3. Results
3.1. Gene-Level eQTL Associations
3.2. Variant-Level eQTL Associations
3.3. Pathways Enriched in the Brain and Blood
3.4. Gene Targets of eQTLs in the Brain and Blood
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| AD | Alzheimer disease |
| ADNI | Alzheimer’s Disease Neuroimaging Initiative |
| eQTL | Expression quantitative trait locus |
| CADD | Combined annotation-dependent depletion |
| GWAVA | Genome-wide annotation of variants |
| MAF | Minor allele frequency |
| MCI | Mild cognitive impairment |
| ME | module eigengene |
| PANTHER | Protein Analysis Through Evolutionary Relationships |
| ROSMAP | Religious Orders Study/Memory and Aging Project |
| SNP | Single nucleotide polymorphism |
| SVA | Surrogate variable analysis |
| WCGNA | Weighted gene co-expression network analysis |
| WGS | Whole-genome sequence |
References
- Alzheimer’s Association. Facts and Figures. 2020. Available online: https://alz.org/alzheimers-dementia/facts-figures (accessed on 20 June 2020).
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fisk, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Sims, R.; van der Lee, S.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Farrell, J.; Bennett, D.A.; Buxbaum, J.D.; Byrd, G.S.; Ertekin-Taner, N.; Evans, D.; Foroud, T.; et al. Two rare AKAP9 variants are associated with Alzheimer disease in African Americans. Alzheimers Dement. 2014, 10, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Wetzel-Smith, M.K.; Hunkapiller, J.; Bhangale, T.R.; Srinivasan, K.; Maloney, J.A.; Atwal, J.K.; Sa, S.M.; Yaylaoglu, M.B.; Foreman, O.; Ortmann, W.; et al. A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat. Med. 2014, 20, 1452–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bis, J.; Jian, X.; Kunkle, B.; Chen, Y.; Hamilton-Nelson, K.; Bush, W.S.; Salerno, W.J.; Lancour, D.; Ma, Y.; Renton, A.E.; et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 2020, 25, 1901–1903. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, C.; Beecham, G.; Vardarajan, B.N.; Ma, Y.; Lancour, D.; Farrell, J.J.; Chung, J. A rare missense variant of CASP7 is associated with familial late-onset Alzheimer’s disease. Alzheimers Dement. 2020, 15, 441–452. [Google Scholar] [CrossRef]
- Patel, D.; Mez, J.; Vardarajan, B.N.; Staley, L.; Chung, J.; Zhang, X.; Farrell, J.J.; Rynkiewicz, M.J.; Cannon-Albright, L.A.; Teerlink, C.C.; et al. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw. Open 2019, 2, e191350. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Ghani, M.; Guo, Z.; Deming, Y.; Wang, K.; Sims, R.; Mao, C.; Yao, Y.; Cruchaga, C.; Stephan, D.A.; et al. An APOE-independent cis-eSNP on chromosome 19q13.32 influences tau levels and late-onset Alzheimer’s disease risk. Neurobiol. Aging 2018, 66, 178.e1–178.e8. [Google Scholar] [CrossRef]
- Zou, F.; Carrasquillo, M.M.; Pankratz, V.S.; Belbin, O.; Morgan, K.; Allen, M.; Wilcox, S.L.; Ma, L.; Walker, L.P.; Kouri, N.; et al. Gene expression levels as endophenotypes in genome-wide association studies of Alzheimer disease. Neurology 2010, 74, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kim, Y.; Tsang, E.K.; Davis, J.R.; Damani, F.N.; Chiang, C.; Hess, G.T.; Zappala, Z.; Strober, B.J.; Scott, A.J.; et al. The impact of rare variation on gene expression across tissues. Nature 2017, 550, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Akinsanmi, I.; Arafat, D.; Cradick, T.J.; Lee, C.M.; Banskota, S.; Marigorta, U.M.; Bao, G.; Gibson, G. A burden of rare variants associated with extremes of gene expression in human peripheral blood. Am. J. Hum. Genet. 2016, 98, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.B.; Lappalainen, T.; Gutierrez-Arcelus, M.; Dermitzakis, E.T. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011, 7, e1002144. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, G.; Yang, E.; Ji, G.; Brinkmeyer-Langford, C.L.; Cai, J.J. Aberrant gene expression in humans. PLoS Genet. 2015, 11, e1004942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daye, Z.J.; Chen, J.; Li, H. High-dimensional heteroscedastic regression with an application to eQTL data analysis. Biometrics 2012, 68, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ibrahim, J.G.; Zou, F. Genomewide multiple-loci mapping in experimental crosses by iterative adaptive penalized regression. Genetics 2010, 185, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lin, C.; Chen, C.; Chen, J.J. Applying genome-wide gene-based expression quantitative trait locus mapping to study population ancestry and pharmacogenetics. BMC Genom. 2014, 15, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.Q.; Li, D.; Yang, W.; Zhang, Y.; Liu, J.; Tong, W. A gene module-based eQTL analysis prioritizing disease genes and pathways in kidney cancer. Comput. Struct. Biotechnol. J. 2017, 15, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.M.; Thwing, A.; Fingerlin, T. eQTL mapping of rare variant associations using RNA-seq data: An evaluation of approaches. PLoS ONE 2019, 14, e0223273. [Google Scholar] [CrossRef]
- Alzheimer’s Disease Neuroimaging Initiative. ADNI—Alzheimer’s Disease Neuroimaging Initiative. Available online: http://adni.loni.usc.edu/ (accessed on 11 December 2018).
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Wilson, R.S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 2012, 9, 628–645. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.A.; Schneider, J.A.; Buchman, A.S.; Barnes, L.L.; Wilson, R.S.; Boyle, P.A.; Wilson, R.S. Overview and findings from the Rush Memory and Aging Project. Curr. Alzheimer Res. 2012, 9, 646–663. [Google Scholar] [CrossRef]
- AMP-AD Knowledge Portal. 2018. Available online: https://www.synapse.org/ (accessed on 1 July 2018).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Patel, D.; Zhang, X.; Farrell, J.; Chung, J.; Stein, T.D.; Lunetta, K.L.; Farrer, L.A. Cell-type specific expression quantitative trait loci associated with Alzheimer disease in blood and brain tissue. MedRxiv 2020. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, G.R.S.; Dunham, I.; Zeggini, E.; Flicek, P. Functional annotation of non-coding sequence variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; Christiani, D.C.; Wurfel, M.H.; Lin, X.; NHLBI GO Exome Sequencing Project—ESP Lung Project Team; et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 8 April 2020).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Hirayama, A.; Ikeda, S.; Shirahata, A.; Shoji, F.; Maruyama, M.; Kayano, M.; Bundo, M.; Hattori, K.; Yoshida, S.; et al. Comparative analysis of cerebrospinal fluid metabolites in Alzheimer’s disease and idiopathic normal pressure hydrocephalus in a Japanese cohort. Biomark. Res. 2018, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Xin, J.; Hu, Y.; Zhang, L.; Wang, J. Analyzing the genes related to Alzheimer’s disease via a network and pathway-based approach. Alzheimers Res. Ther. 2017, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.L.; Tang, N.L.S.; Tam, C.W.C.; Lui, V.W.C.; Suen, E.W.C.; Chiu, H.F.K.; Lam, L.C.W. Association between HLA-A alleles and Alzheimer’s disease in a southern Chinese community. Dement. Geriatr. Cogn. Disord. 2008, 26, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.; Wang, Y.; Wang, H.; Zhang, W.; Hu, N.; Tan, L.; Sun, L.; Tan, M.-S.; Zhu, X.-C.; et al. The genetic variation of ARRB2 is associated with late-onset Alzheimer’s disease in Han Chinese. Curr. Alzheimer Res. 2014, 11, 408–412. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Šerý, O.; Janoutová, J.; Ewerlingová, L.; Hálová, A.; Lochman, J.; Janout, V.; Khan, N.A.; Balcar, V.J. CD36 gene polymorphism is associated with Alzheimer’s disease. Biochimie 2017, 135, 46–53. [Google Scholar] [CrossRef]
- Nho, K.; Nudelman, K.; Allen, M.; Hodges, A.; Kim, S.; Risacher, S.L.; Apostolova, L.G.; Lin, K.; Lunnon, K.; Wang, X.; et al. Genome-wide transcriptome analysis identifies novel dysregulated genes implicated in Alzheimer’s pathology. Alzheimers Dement. 2020, 16, 1213–1223. [Google Scholar] [CrossRef]
- Song, I.; Kim, Y.; Chung, S.; Cho, H. Association between serum haptoglobin and the pathogenesis of Alzheimer’s disease. Intern. Med. 2015, 54, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Pruunsild, P.; Greenberg, M.E.; Bading, H. Lineage divergence of activity-driven transcription and evolution of cognitive ability. Nat. Rev. Neurosci. 2018, 19, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Tian, M.; Zheng, Y.; Gong, F.; Fu, A.K.Y.; Ip, N.Y. Stimulation of the hippocampal POMC/MC4R circuit alleviates synaptic plasticity impairment in an Alzheimer’s disease model. Cell Rep. 2016, 17, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Smith, R.G.; Lunnon, K. DNA modifications and Alzheimer’s disease. In Neuroepigenomics in Aging and Disease; Delgado-Morales, R., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 303–319. [Google Scholar]
- Blalock, E.M.; Geddes, J.W.; Chen, K.C.; Porter, N.M.; Markesbery, W.R.; Landfield, P.W. Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA 2004, 101, 2173–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreft, K.L.; van Meurs, M.; Wierenga-Wolf, A.F.; Melief, M.; van Strien, M.E.; Hol, E.M.; Oostra, B.A.; Laman, J.D.; Hintzen, R.Q. Abundant kif21b is associated with accelerated progression in neurodegenerative diseases. Acta Neuropathol. Commun. 2014, 2, 144. [Google Scholar] [CrossRef]
- Hares, K.; Miners, J.S.; Cook, A.J.; Rice, C.; Scolding, N.; Love, S.; Wilkins, A. Overexpression of kinesin superfamily motor proteins in Alzheimer’s disease. J. Alzheimers Dis. 2017, 60, 1511–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, N.S.; Vardarajan, B.; Mayeux, R. Genomic variation in educational attainment modifies Alzheimer disease risk. Neurol. Genet. 2019, 5, e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, M.; Kato, T. Increased expression of PAD2 after repeated intracerebroventricular infusions of soluble Abeta(25-35) in the Alzheimer’s disease model rat brain: Effect of memantine. Cell Mol. Biol. Lett. 2009, 14, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Herrick, S.; Evers, D.M.; Lee, J.; Udagawa, N.; Pak, D.T.S. Postsynaptic PDLIM5 / Enigma Homolog binds SPAR and causes dendritic spine shrinkage. Mol. Cell Neurosci. 2010, 43, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, C.E.; Goyette, J.; Utter, V.; Rahimi, F.; Yang, Z.; Geczy, C.L.; Halliday, G.M. Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- El-Battari, A.; Mathieu, S.; Sigaud, R.; Prorok-Hamon, M.; Ouafik, L.; Jeanneau, C. Elucidating the roles of Alzheimer disease-associated proteases and the signal-peptide peptidase-like 3 (SPPL3) in the shedding of glycosyltransferases. BioRxiv 2018, 317214. [Google Scholar]
- Zhang, S.; Qin, C.; Cao, G.; Guo, L.; Feng, C.; Zhang, W. Genome-wide analysis of DNA methylation profiles in a senescence-accelerated mouse prone 8 brain using whole-genome bisulfite sequencing. Bioinformatics 2017, 33, 1591–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cotto, M.; Guo, L.; Karwan, M.; Sen, S.K.; Barb, J.; Collado, C.J.; Elloumi, F.; Palmieri, E.M.; Boelte, K.; Kolodgie, F.D.; et al. TREML4 promotes inflammatory programs in human and murine macrophages and alters atherosclerosis lesion composition in the apolipoprotein E deficient mouse. Front. Immunol. 2020, 11, 397. [Google Scholar] [CrossRef]
- Gireud-Goss, M.; Reyes, S.; Tewari, R.; Patrizz, A.; Howe, M.D.; Kofler, J.; Waxham, M.N.; McCullough, L.D.; Bean, A.J. The ubiquitin ligase UBE4B regulates amyloid precursor protein ubiquitination, endosomal trafficking, and amyloid β42 generation and secretion. Mol. Cell. Neurosci. 2020, 108, 103542. [Google Scholar] [CrossRef]
- Moradifard, S.; Hoseinbeyki, M.; Ganji, S.M.; Minuchehr, Z. Analysis of microRNA and Gene Expression Profiles in Alzheimer’s Disease: A Meta-Analysis Approach. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef]
- Gleichmann, M.; Zhang, Y.; Wood, W.H.; Becker, K.G.; Mughal, M.R.; Pazin, M.J.; van Praag, H.; Kobilo, T.; Zonderman, A.B.; Troncoso, J.C.; et al. Molecular changes in brain aging and Alzheimer’s disease are mirrored in experimentally silenced cortical neuron networks. Neurobiol. Aging 2012, 33, 205.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.A.; Davies, P.; Dobrenis, K.; Huang, M. Tomoregulin-2 is found extensively in plaques in Alzheimer’s disease brain. J. Neurochem. 2006, 98, 34–44. [Google Scholar] [CrossRef]
- Esteve, P.; Rueda-Carrasco, J.; Mateo, M.I.; Martin-Bermejo, M.J.; Draffin, J.; Pereyra, G.; Sandonís, A.; Crespo, I.; Moreno, I.; Aso, E.; et al. Elevated levels of secreted-frizzled-related-protein 1 contribute to Alzheimer’s disease pathogenesis. Nat. Neurosci. 2019, 22, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Vélez, J.I.; Lopera, F.; Sepulveda-Falla, D.; Patel, H.R.; Johar, A.S.; Chuah, A.; Tobón, C.; Rivera, D.; Villegas, A.; Cai, Y.; et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol. Psychiatry 2016, 21, 916–924. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, M.; Rabaneda, L.G.; Murillo-Carretero, M.; Ortega-Martínez, S.; Martínez-Chantar, M.L.; Woodhoo, A.; Luka, Z.; Wagner, C.; Lu, S.C.; Mato, J.M.; et al. Glycine N-methyltransferase expression in the hippocampus and its role in neurogenesis and cognitive performance. Hippocampus 2014, 24, 840–852. [Google Scholar] [CrossRef] [Green Version]
- Lardenoije, R.; Roubroeks, J.A.Y.; Pishva, E.; Leber, M.; Wagner, H.; Iatrou, A.; Smith, A.R.; Smith, R.G.; Eijssen, L.M.T.; Kleineidam, L.; et al. Alzheimer’s disease-associated (hydroxy)methylomic changes in the brain and blood. Clin. Epigenetics 2019, 11, 164. [Google Scholar] [CrossRef]
- Chen, X.; Ji, B.; Hao, X.; Li, X.; Eisele, F.; Nyström, T.; Petranovic, D. FMN reduces Amyloid-β toxicity in yeast by regulating redox status and cellular metabolism. Nat. Commun. 2020, 11, 867. [Google Scholar] [CrossRef]
- Zhu, C.; Jiang, G.; Chen, J.; Zhou, Z.; Cheng, Q. Serum haptoglobin in Chinese patients with Alzheimer’s disease and mild cognitive impairment: A case-control study. Brain Res. Bull. 2018, 137, 301–305. [Google Scholar] [CrossRef]
- Ayton, S.; Janelidze, S.; Roberts, B.; Palmqvist, S.; Kalinowski, P.; Diouf, I.; Belaidi, A.A.; Stomrud, E.; Bush, A.I.; Hansson, O. Acute phase markers in CSF reveal inflammatory changes in Alzheimer’s disease that intersect with pathology, APOE ε4, sex and age. Prog. Neurobiol. 2021, 198, 101904. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Gómez, J.L.; Pelletier, A.; Rougier, M.; Korobelnik, J.; Schweitzer, C.; Delyfer, M.; Catheline, G.; Monfermé, S.; Dartigues, J.-F.; Delcourt, C.; et al. Association of retinal nerve fiber layer thickness with brain alterations in the visual and limbic networks in elderly adults without dementia. JAMA Netw. Open 2018, 1, e184406. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Zhuang, Y.; Ying, Z.; Agrawal, R.; Yang, X.; Gomez-Pinilla, F. Traumatic brain injury induces genome-wide transcriptomic, methylomic, and network perturbations in brain and blood predicting neurological disorders. EBioMedicine 2017, 16, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Page, A.; Dupuis, G.; Frost, E.H.; Larbi, A.; Pawelec, G.; Witkowski, J.M.; Fulop, T. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp. Gerontol. 2018, 107, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Khan, A.F.; Adewale, Q.; Shirazi, A.H. Blood and brain gene expression trajectories mirror neuropathology and clinical deterioration in neurodegeneration. Brain 2020, 143, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Iijima, R.; Ogishima, S.; Kikuchi, M.; Matsuoka, Y.; Ghosh, S.; Miyamoto, T.; Miyashita, A.; Kuwano, R.; Tanaka, H. AlzPathway: A comprehensive map of signaling pathways of Alzheimer’s disease. BMC Syst. Biol. 2012, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef]
- Hsu, W.-C.; Wang, H.-K.; Lee, L.-C.; Fung, H.-C.; Lin, J.-C.; Hsu, H.-P.; Wu, Y.-R.; Ro, L.-S.; Hu, F.-J.; Chang, Y.-T.; et al. Promoter polymorphisms modulating HSPA5 expression may increase susceptibility to Taiwanese Alzheimer’s disease. J. Neural. Transm. 2008, 115, 1537–1543. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s disease: Chemokines produced by astrocytes and chemokine receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar] [PubMed]
- Zuena, A.R.; Casolini, P.; Lattanzi, R.; Maftei, D. Chemokines in Alzheimer’s disease: New insights into prokineticins, chemokine-like proteins. Front. Pharmacol. 2019, 10, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, Z.; Mobilio, F.; Walker, F.R.; Taylor, J.M.; Crack, P.J. Abrogation of type-I interferon signalling alters the microglial response to Aβ 1–42. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, D.; Gong, P.; Lin, A.; Zhang, Y.; Ye, R.D.; Tang, Y. Formyl peptide receptor 2 deficiency improves cognition and attenuates tau hyperphosphorylation and astrogliosis in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Li, C.; Zhang, L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: Implications for cerebral ischemia and neurodegenerative diseases. Neurobiol. Aging 2019, 87, 1–10. [Google Scholar] [CrossRef]
- Chu, J.; Lauretti, E.; Di Meco, A.; Praticò, D. FLAP pharmacological blockade modulates metabolism of endogenous tau in vivo. Transl. Psychiatry 2013, 3, e333. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, M.; Roy, K.; Das, S.; Mukhopadhyay, D. The N-terminal SH3 domain of Grb2 is required for endosomal localization of AβPP. J. Alzheimers Dis. 2012, 32, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Blair, J.A.; Wang, C.; Hernandez, D.; Siedlak, S.L.; Rodgers, M.S.; Achar, R.K.; Fahmy, L.M.; Torres, S.L.; Petersen, R.B.; Zhu, X.; et al. Individual case analysis of postmortem interval time on brain tissue preservation. PLoS ONE 2016, 11, e015615. [Google Scholar]



{kind=link}
{kind=link}
| Dataset | Race | N | AD Cases | MCI Cases | Controls | Female | Age * |
|---|---|---|---|---|---|---|---|
| ROSMAP (Brain) | NHW 98% AA 2% Other <0.01% | 475 | 281 | 0 | 194 | 63% | 85.9 (4.8) |
| ADNI (Blood) | NHW 93% AA 4% Other 3% | 713 | 207 | 284 | 222 | 44% | 76.3 (8.1) |
| Chr | Begin Position | End Position | Gene | Brain | Blood | ||||
|---|---|---|---|---|---|---|---|---|---|
| CVar + | Unique Var ^ | p-Value | CVar + | Unique Var ^ | p-Value | ||||
| 6 | 41,942,338 | 43,929,364 | GNMT | 671 | 437 | 1.85 × 10−6 | 1006 | 640 | 2.87 × 10−7 |
| 11 | 17,434,230 | 19,468,040 | LDHC | 429 | 273 | 2.07 × 10−7 | 762 | 473 | 2.25 × 10−10 |
| 15 | 64,039,999 | 66,063,761 | RBPMS2 | 404 | 249 | 9.90 × 10−8 | 648 | 417 | 1.69 × 10−36 |
| 16 | 67,034,867 | 69,106,452 | DUS2 | 714 | 482 | 1.98 × 10−6 | 1085 | 723 | 6.41 × 10−08 |
| 16 | 71,090,452 | 73,094,829 | HP | 741 | 461 | 2.28 × 10−9 | 1206 | 750 | 2.43 × 10−11 |
| Pathway | # Genes in Pathway | Gene Module | # Module Genes in Pathway | Module Genes | Uncorrected p-Value | FDR | ||
|---|---|---|---|---|---|---|---|---|
| Expected # of Genes * | Fold Enrichment † | +/− | ||||||
| BRAIN | ||||||||
| Apoptosis signaling | 77 | 7 | 12 | 1.64 | 7.3 | + | 3.09 × 10−7 | 5.01 × 10−5 |
| Toll receptor signaling | 32 | 8 | 6 | 0.46 | 12.97 | + | 1.45 × 10−5 | 2.36 × 10−3 |
| Wnt signaling | 235 | 4 | 21 | 7.35 | 2.86 | + | 3.49 × 10−5 | 5.65 × 10−3 |
| Cadherin signaling | 127 | 4 | 14 | 3.97 | 3.53 | + | 9.59 × 10−5 | 7.77 × 10−3 |
| CCKR signaling map | 111 | 7 | 10 | 2.37 | 4.22 | + | 2.22 × 10−4 | 1.20 × 10−2 |
| Gonadotropin-releasing hormone receptor | 152 | 4 | 14 | 4.75 | 2.95 | + | 5.28 × 10−4 | 2.14 × 10−2 |
| p53 | 62 | 7 | 7 | 1.32 | 5.29 | + | 5.76 × 10−4 | 2.33 × 10−2 |
| Inflammation mediated by chemokine and cytokine signaling | 173 | 16 | 5 | 0.51 | 9.89 | + | 1.54 × 10−4 | 2.50 × 10−2 |
| Angiogenesis | 126 | 4 | 12 | 3.94 | 3.05 | + | 9.95 × 10−4 | 3.22 × 10−2 |
| BLOOD | ||||||||
| Blood coagulation | 43 | 24 | 8 | 0.27 | 29.9 | + | 8.22 × 10−10 | 1.34 × 10−7 |
| Parkinson’s disease | 85 | 15 | 7 | 0.79 | 8.82 | + | 2.28 × 10−5 | 3.72 × 10−3 |
| Inflammation mediated by chemokine and cytokine signaling | 237 | 14 | 11 | 2.27 | 4.84 | + | 2.50 × 10−5 | 4.08 × 10−3 |
| T-cell activation | 73 | 32 | 4 | 0.18 | 22.02 | + | 3.87 × 10−5 | 6.30 × 10−3 |
| B-cell activation | 66 | 12 | 6 | 0.66 | 9.08 | + | 7.89 × 10−5 | 1.29 × 10−2 |
| PDGF signaling | 127 | 12 | 7 | 1.27 | 5.5 | + | 3.79 × 10−4 | 2.06 × 10−2 |
| Apoptosis signaling | 112 | 5 | 12 | 3.15 | 3.81 | + | 1.51 × 10−4 | 2.47 × 10−2 |
| JAK/STAT signaling | 17 | 7 | 4 | 0.28 | 14.33 | + | 3.21 × 10−4 | 2.62 × 10−2 |
| Ras | 64 | 5 | 8 | 1.8 | 4.44 | + | 7.61 × 10−4 | 3.10 × 10−2 |
| CCKR signaling map | 164 | 5 | 14 | 4.61 | 3.04 | + | 3.94 × 10−4 | 3.21 × 10−2 |
| Angiotensin II-stimulated signaling through G proteins and β-arrestin | 33 | 5 | 6 | 0.93 | 6.47 | + | 6.14 × 10−4 | 3.34 × 10−2 |
| Histamine H2 receptor-mediated signaling | 24 | 5 | 5 | 0.67 | 7.41 | + | 1.03 × 10−3 | 3.37 × 10−2 |
| Inflammation mediated by chemokine and cytokine signaling | 237 | 24 | 7 | 1.47 | 4.75 | + | 7.94 × 10−4 | 4.32 × 10−2 |
| Heme biosynthesis | 11 | 6 | 4 | 0.25 | 15.93 | + | 2.74 × 10−4 | 4.47 × 10−2 |
| Integrin signalling | 180 | 7 | 11 | 2.96 | 3.72 | + | 2.84 × 10−4 | 4.62 × 10−2 |
| Inflammation mediated by chemokine and cytokine signaling | 237 | 20 | 8 | 1.78 | 4.48 | + | 5.07 × 10−4 | 8.26 × 10−2 |
| CHR | Begin Position | End Position | Gene | CVAR + | Unique VAR ^ | p-Value | Gene Module | Pathway |
|---|---|---|---|---|---|---|---|---|
| 17 | 31,600,172 | 33,592,552 | CCL7 * | 340 | 206 | 1.84 × 10−5 | 16 | Inflammation mediated by chemokine and cytokine signaling |
| 17 | 31,648,819 | 33,621,655 | CCL8 * | 319 | 195 | 4.50 × 10−4 | 16 | Inflammation mediated by chemokine and cytokine signaling |
| 17 | 72,322,351 | 74,401,630 | GRB2 | 1108 | 717 | 6.04 × 10−7 | 14 | Inflammation mediated by chemokine and cytokine signaling |
| 2 | 217,992,496 | 220,001,949 | CXCR2 | 943 | 564 | 1.53 × 10−6 | 14 | Inflammation mediated by chemokine and cytokine signaling |
| 5 | 174,085,268 | 176,108,976 | HRH2 | 335 | 196 | 9.07 × 10−6 | 5 | Histamine H2 receptor mediated signaling |
| 1 | 25,859,096 | 27,901,441 | RPS6KA1 | 1208 | 790 | 1.01 × 10−5 | 5 | Ras Pathway, CCKR signaling map |
| 11 | 76,033,278 | 78,180,311 | PAK1 | 565 | 355 | 1.83 × 10−5 | 5 | Ras Pathway, CCKR Signaling map |
| 21 | 33,696,834 | 35,718,581 | IFNAR1 | 525 | 332 | 1.98 × 10−5 | 14 | Inflammation mediated by chemokine and cytokine signaling |
| 3 | 11,628,812 | 13,702,170 | RAF1 | 431 | 255 | 2.11 × 10−5 | 14 | Inflammation mediated by chemokine and cytokine signaling |
| 1 | 83,964,144 | 85,961,982 | GNG5 | 589 | 336 | 3.43 × 10−5 | 5 | Histamine H2 receptor mediated signaling |
| 9 | 115,150,150 | 117,160,754 | ALAD | 620 | 425 | 4.97 × 10−5 | 6 | Heme biosynthesis |
| 1 | 44,478,672 | 46,476,606 | UROD | 1061 | 649 | 5.92 × 10−5 | 6 | Heme biosynthesis |
| 19 | 13,202,507 | 15,228,794 | PRKACA | 767 | 501 | 7.60 × 10−5 | 5 | Histamine H2 receptor mediated signaling, CCKR signaling map |
| 9 | 127,005,465 | 128998618 | HSPA5 | 1235 | 692 | 7.91 × 10−5 | 15 | Parkinson disease |
| 8 | 21,946,761 | 23968794 | TNFRSF10C | 817 | 520 | 8.77 × 10−5 | 5 | Apoptosis signaling |
| 19 | 51,273,985 | 53272173 | FPR2 | 440 | 280 | 1.23 × 10−4 | 14 | Inflammation mediated by chemokine and cytokine signaling |
| 13 | 30,317,837 | 32,332,540 | ALOX5AP | 297 | 197 | 1.26 × 10−4 | 14 | Inflammation mediated by chemokine and cytokine signaling |
| 2 | 200,984,212 | 203,030,077 | CFLAR | 637 | 404 | 2.42 × 10−4 | 5 | Apoptosis signaling |
| 17 | 39,458,200 | 41,463,831 | STAT5A | 1093 | 708 | 3.08 × 10−4 | 12 | PDGF signaling |
| 14 | 50,190,597 | 52,294,891 | NIN | 516 | 323 | 3.65 × 10−4 | 12 | PDGF signaling |
| 12 | 49,108,257 | 51,158,233 | TMBIM6 | 1082 | 716 | 4.48 × 10−4 | 5 | Apoptosis signaling |
| eQTLs in Blood | eQTLs in Brain | ||
|---|---|---|---|
| eGene | Reference | eGene | Reference |
| ABCA7 * | [36] | ACOT1 | [37] |
| ADAMTSL4 | [38] | HLA-A | [39] |
| ARRB2 | [40] | HLA-DOB * | [26] |
| ATG7 | [41] | HLA-DRB1 * | [35,41] |
| CD36 | [42] | HLA-DRB5 * | [36] |
| CREB5 | [43] | HP | [44] |
| CTNNAL1 | [45] | POMC | [46] |
| ECHDC3 * | [35] | RNF39 | [47,48] |
| HP | [44] | ZNF253 | [49] |
| KF1B | [50,51] | ||
| LRRC2 | [52] | ||
| MS4A6A * | [36] | ||
| PADI2 | [53] | ||
| PDLIM5 | [54] | ||
| S100A12 | [55] | ||
| SPPL3 | [56] | ||
| TMEM51 | [57] | ||
| TREML4 | [58] | ||
| UBE4B | [59] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, D.; Zhang, X.; Farrell, J.J.; Lunetta, K.L.; Farrer, L.A. Set-Based Rare Variant Expression Quantitative Trait Loci in Blood and Brain from Alzheimer Disease Study Participants. Genes 2021, 12, 419. https://doi.org/10.3390/genes12030419
Patel D, Zhang X, Farrell JJ, Lunetta KL, Farrer LA. Set-Based Rare Variant Expression Quantitative Trait Loci in Blood and Brain from Alzheimer Disease Study Participants. Genes. 2021; 12(3):419. https://doi.org/10.3390/genes12030419
Chicago/Turabian StylePatel, Devanshi, Xiaoling Zhang, John J. Farrell, Kathryn L. Lunetta, and Lindsay A. Farrer. 2021. "Set-Based Rare Variant Expression Quantitative Trait Loci in Blood and Brain from Alzheimer Disease Study Participants" Genes 12, no. 3: 419. https://doi.org/10.3390/genes12030419