
Methanogenesis and Salt Tolerance Genes of a Novel Halophilic Methanosarcinaceae Metagenome-Assembled Genome from a Former Solar Saltern

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Metagenome Information
2.2. MAG Assembly
2.3. Comparative Genomics
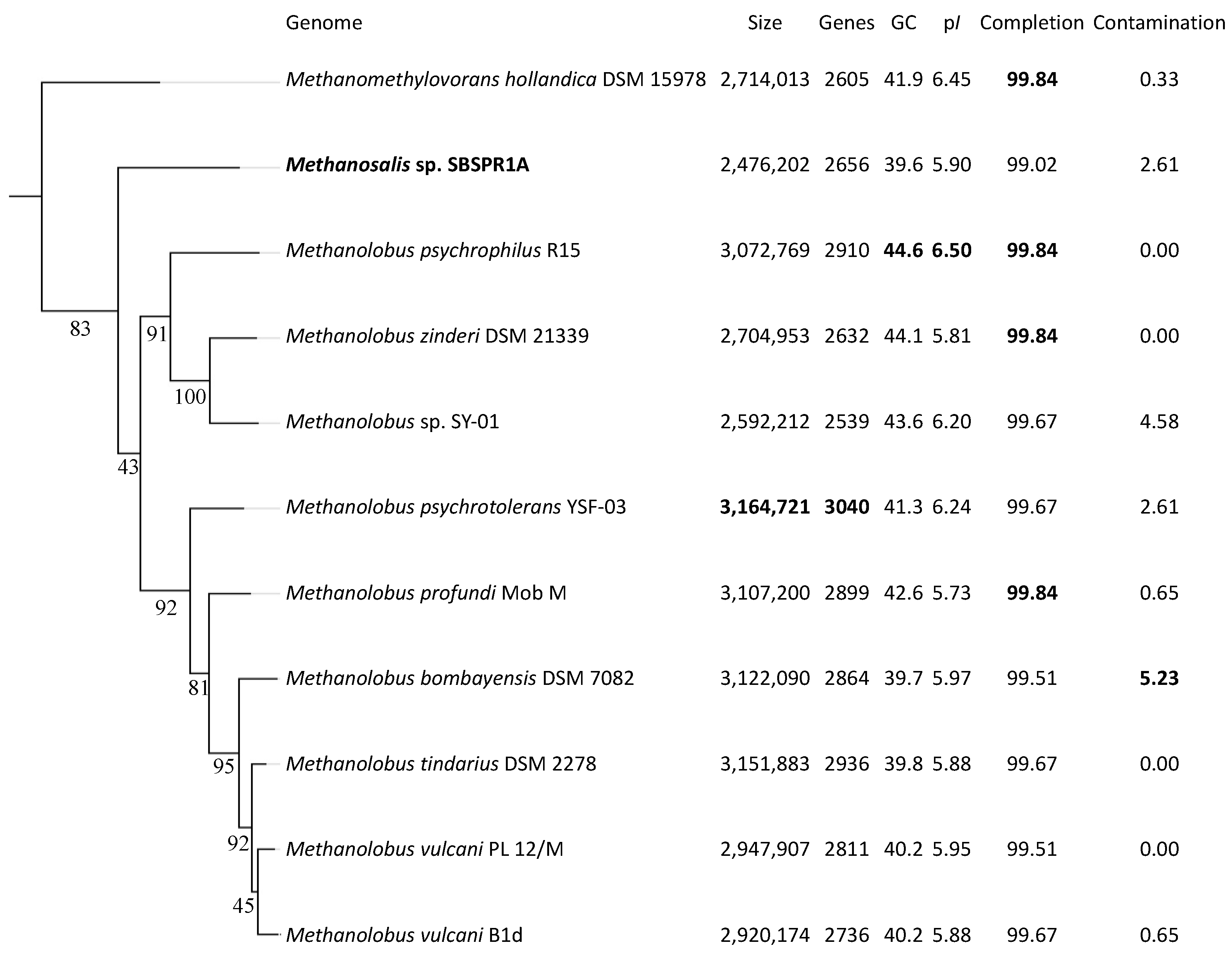
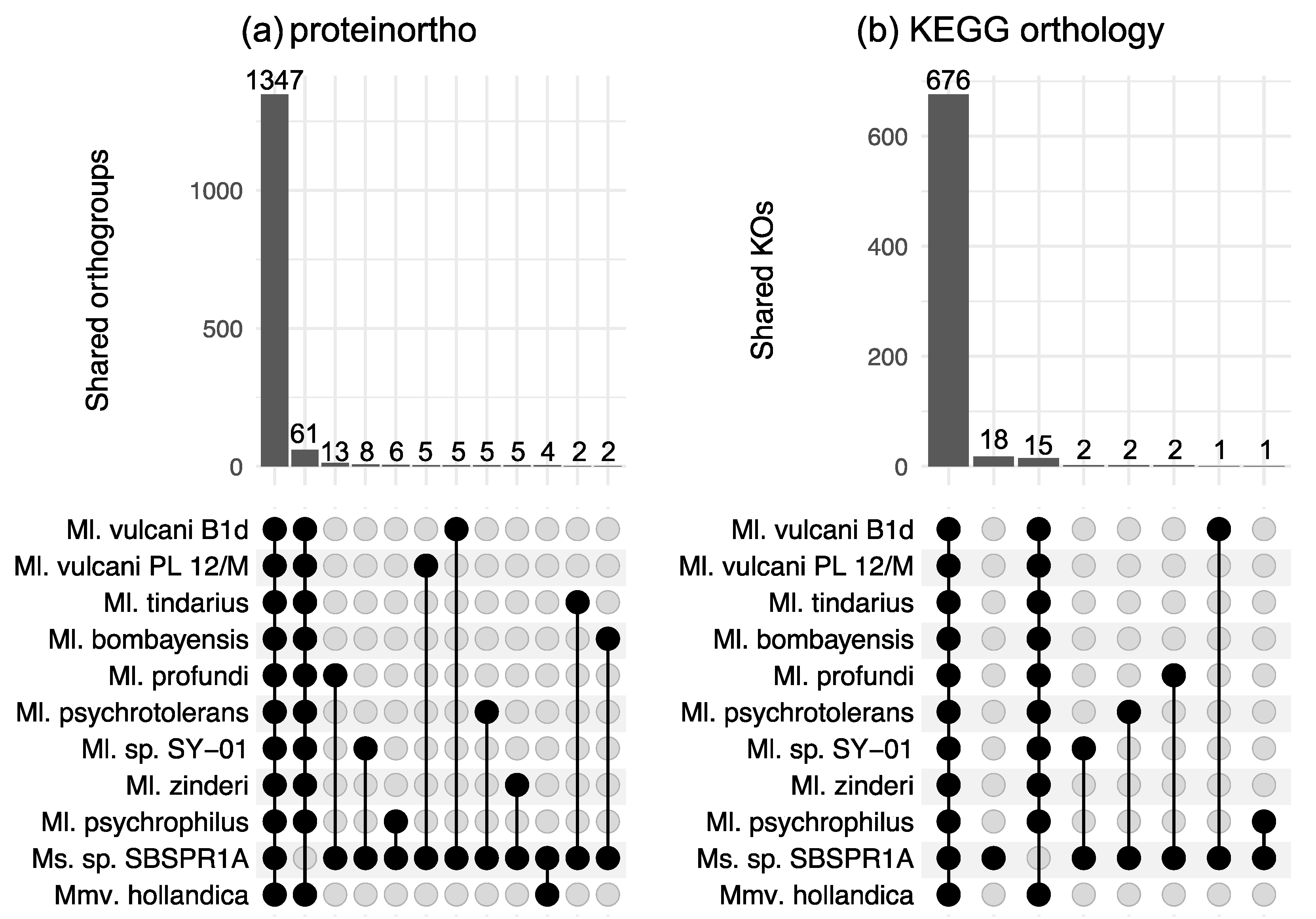
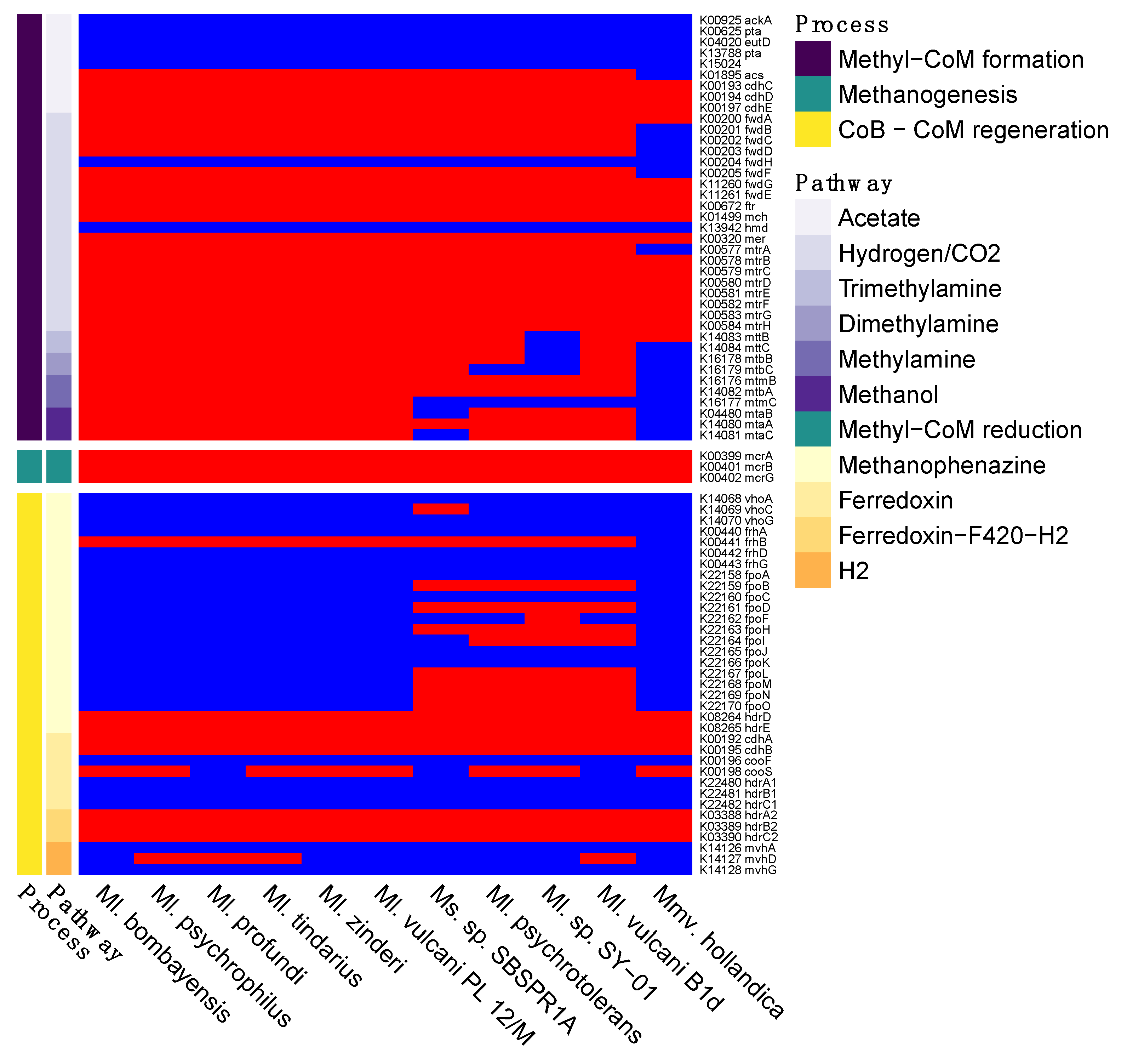
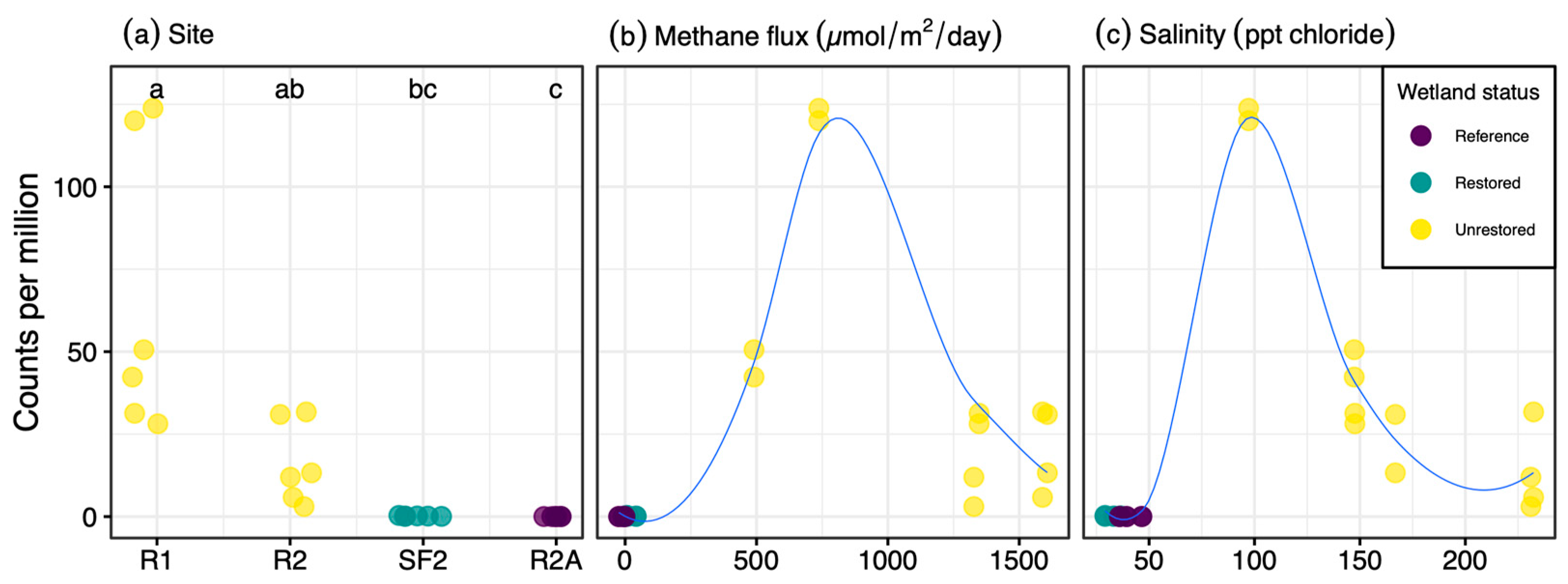
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Conrad, R. Importance of Hydrogenotrophic, Aceticlastic and Methylotrophic Methanogenesis for Methane Production in Terrestrial, Aquatic and Other Anoxic Environments: A Mini Review. Pedosphere 2020, 30, 25–39. [Google Scholar] [CrossRef]
- Liu, Y.; Whitman, W. Metabolic, Phylogenetic, and Ecological Diversity of the Methanogenic Archaea. Ann. N. Y. Acad. Sci. 2008, 1125, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. The Family Methanosarcinaceae. In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 259–281. ISBN 978-3-642-38954-2. [Google Scholar]
- König, H.; Stetter, K.O. Isolation and Characterization of Methanolobus Tindarius, Sp. Nov., a Coccoid Methanogen Growing Only on Methanol and Methylamines. Zent. Für Bakteriol. Mikrobiol. Hyg. Abt Orig. C Allg. Angew. Ökol. Mikrobiol. 1982, 3, 478–490. [Google Scholar] [CrossRef]
- Doerfert, S.N.; Reichlen, M.; Iyer, P.; Wang, M.; Ferry, J.G. Methanolobus Zinderi Sp. Nov., a Methylotrophic Methanogen Isolated from a Deep Subsurface Coal Seam. Int. J. Syst. Evol. Microbiol. 2009, 59, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Ni, S.; Boone, D.R. Isolation and Characterization of a Dimethyl Sulfide-Degrading Methanogen, Methanolobus Siciliae HI350, from an Oil Well, Characterization of M. Siciliae T4/MT, and Emendation of M. Siciliae. Int. J. Syst. Evol. Microbiol. 1991, 41, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Mochimaru, H.; Tamaki, H.; Hanada, S.; Imachi, H.; Nakamura, K.; Sakata, S.; Kamagata, Y. Methanolobus Profundi Sp. Nov., a Methylotrophic Methanogen Isolated from Deep Subsurface Sediments in a Natural Gas Field. Int. J. Syst. Evol. Microbiol. 2009, 59, 714–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, P.C.; Ranade, D.R.; Mandelco, L.; Boone, D.R. Isolation and Characterization of Methanolobus Bombayensis Sp. Nov., a Methylotrophic Methanogen That Requires High Concentrations of Divalent Cations. Int. J. Syst. Evol. Microbiol. 1994, 44, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Lomans, B.P.; Maas, R.; Luderer, R.; Op den Camp, H.J.M.; Pol, A.; van der Drift, C.; Vogels, G.D. Isolation and Characterization of Methanomethylovorans Hollandica Gen. Nov., Sp. Nov., Isolated from Freshwater Sediment, a Methylotrophic Methanogen Able To Grow on Dimethyl Sulfide and Methanethiol. Appl. Environ. Microbiol. 1999, 65, 3641–3650. [Google Scholar] [CrossRef] [Green Version]
- Cha, I.-T.; Min, U.-G.; Kim, S.-J.; Yim, K.J.; Roh, S.W.; Rhee, S.-K. Methanomethylovorans Uponensis Sp. Nov., a Methylotrophic Methanogen Isolated from Wetland Sediment. Antonie Van Leeuwenhoek 2013, 104, 1005–1012. [Google Scholar] [CrossRef]
- Jiang, B.; Parshina, S.N.; van Doesburg, W.; Lomans, B.P.; Stams, A.J.M.Y. 2005 Methanomethylovorans Thermophila Sp. Nov., a Thermophilic, Methylotrophic Methanogen from an Anaerobic Reactor Fed with Methanol. Int. J. Syst. Evol. Microbiol. 2005, 55, 2465–2470. [Google Scholar] [CrossRef] [Green Version]
- Simankova, M.V.; Kotsyurbenko, O.R.; Lueders, T.; Nozhevnikova, A.N.; Wagner, B.; Conrad, R.; Friedrich, M.W. Isolation and Characterization of New Strains of Methanogens from Cold Terrestrial Habitats. Syst. Appl. Microbiol. 2003, 26, 312–318. [Google Scholar] [CrossRef]
- Oremland, R.S.; Boone, D.R. Methanolobus Taylorii Sp. Nov., a New Methylotrophic, Estuarine Methanogen. Int. J. Syst. Bacteriol. 1994, 44, 573–575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Jiang, N.; Liu, X.; Dong, X. Methanogenesis from Methanol at Low Temperatures by a Novel Psychrophilic Methanogen, “Methanolobus Psychrophilus” Sp. Nov., Prevalent in Zoige Wetland of the Tibetan Plateau. Appl. Environ. Microbiol. 2008, 74, 6114–6120. [Google Scholar] [CrossRef] [Green Version]
- Oren, A. Taxonomy of Halophilic Archaea: Current Status and Future Challenges. Extremophiles 2014, 18, 825–834. [Google Scholar] [CrossRef]
- Ventosa, A.; Mellado, E.; Sanchez-Porro, C.; Marquez, M.C. Halophilic and Halotolerant Micro-Organisms from Soils. In Microbiology of Extreme Soils; Dion, P., Nautiyal, C.S., Eds.; Soil Biology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 87–115. ISBN 978-3-540-74231-9. [Google Scholar]
- Vavourakis, C.D.; Andrei, A.-S.; Mehrshad, M.; Ghai, R.; Sorokin, D.Y.; Muyzer, G. A Metagenomics Roadmap to the Uncultured Genome Diversity in Hypersaline Soda Lake Sediments. Microbiome 2018, 6, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirete, S.; Mora-Ruiz, M.R.; Lamprecht-Grandío, M.; de Figueras, C.G.; Rosselló-Móra, R.; González-Pastor, J.E. Salt Resistance Genes Revealed by Functional Metagenomics from Brines and Moderate-Salinity Rhizosphere within a Hypersaline Environment. Front. Microbiol. 2015, 6, 1121. [Google Scholar] [CrossRef] [PubMed]
- Muyodi, F.J. Microbiological analysis of the waters of Lake Victoria in relation to the invasion of the water hyacinth, Eichhornia crassipes (Mart.) Solms. In A Case Study of the Lakeshores of Mwanza Municipality; University of Dar es Salaam: Dar es Salaam, Tanzania, 2000; pp. 158–173. [Google Scholar]
- Chen, S.-C.; Huang, H.-H.; Lai, M.-C.; Weng, C.-Y.; Chiu, H.-H.; Tang, S.-L.; Rogozin, D.Y.; Degermendzhy, A.G. Methanolobus Psychrotolerans Sp. Nov., a Psychrotolerant Methanoarchaeon Isolated from a Saline Meromictic Lake in Siberia. Int. J. Syst. Evol. Microbiol. 2018, 68, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Vreeland, R.H. Mechanisms of Halotolerance in Microorganisms. Crit. Rev. Microbiol. 2009, 14, 311–356. [Google Scholar] [CrossRef]
- Oremland, R.S.; Polcin, S. Methanogenesis and Sulfate Reduction: Competitive and Noncompetitive Substrates in Estuarine Sediments. Appl. Environ. Microbiol. 1982, 44, 1270–1276. [Google Scholar] [CrossRef] [Green Version]
- Oremland, R.S.; Marsh, L.M.; Polcin, S. Methane Production and Simultaneous Sulphate Reduction in Anoxic, Salt Marsh Sediments. Nature 1982, 296, 143–145. [Google Scholar] [CrossRef]
- Mcgenity, T.; Sorokin, D. Methanogens and Methanogenesis in Hypersaline Environments. In Biogenesis of Hydrocarbons; Springer International Publishing: New York, NY, USA, 2018; pp. 1–27. ISBN 978-3-319-53114-4. [Google Scholar]
- Matarredona, L.; Camacho, M.; Zafrilla, B.; Bonete, M.-J.; Esclapez, J. The Role of Stress Proteins in Haloarchaea and Their Adaptive Response to Environmental Shifts. Biomolecules 2020, 10, 1390. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of Adaptation of Microorganisms of the Three Domains of Life to High Salt Concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef]
- Vera-Gargallo, B.; Ventosa, A. Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain). Genes 2018, 9, 152. [Google Scholar] [CrossRef] [Green Version]
- Youssef, N.H.; Savage-Ashlock, K.N.; McCully, A.L.; Luedtke, B.; Shaw, E.I.; Hoff, W.D.; Elshahed, M.S. Trehalose/2-Sulfotrehalose Biosynthesis and Glycine-Betaine Uptake Are Widely Spread Mechanisms for Osmoadaptation in the Halobacteriales. ISME J. 2014, 8, 636–649. [Google Scholar] [CrossRef]
- Zhou, J.; Theroux, S.M.; Bueno de Mesquita, C.P.; Hartman, W.H.; Tringe, S.G. Microbial Drivers of Methane Emissions from Unrestored Industrial Salt Ponds. ISME J. 2021. [Google Scholar] [CrossRef] [PubMed]
- von Meijenfeldt, F.A.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust Taxonomic Classification of Uncharted Microbial Sequences and Bins with CAT and BAT. Genome Biol. 2019, 20, 217. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an Efficient Tool for Accurately Reconstructing Single Genomes from Complex Microbial Communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-W.; Tang, Y.-H.; Tringe, S.G.; Simmons, B.A.; Singer, S.W. MaxBin: An Automated Binning Method to Recover Individual Genomes from Metagenomes Using an Expectation-Maximization Algorithm. Microbiome 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-H.; Liao, Y.-C. Accurate Binning of Metagenomic Contigs via Automated Clustering Sequences Using Information of Genomic Signatures and Marker Genes. Sci. Rep. 2016, 6, 24175. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of Genomes from Metagenomes via a Dereplication, Aggregation and Scoring Strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Pohlert, T. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR). 2014. Available online: https://CRAN.R-project.org/package=PMCMR (accessed on 28 July 2021).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Chen, L.-X.; Anantharaman, K.; Shaiber, A.; Eren, A.M.; Banfield, J.F. Accurate and Complete Genomes from Metagenomes. Genome Res. 2020, 30, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Paulino, D.; Warren, R.L.; Vandervalk, B.P.; Raymond, A.; Jackman, S.D.; Birol, I. Sealer: A Scalable Gap-Closing Application for Finishing Draft Genomes. BMC Bioinform. 2015, 16, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. GigaScience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R.; et al. IMG/M v.5.0: An Integrated Data Management and Comparative Analysis System for Microbial Genomes and Microbiomes. Nucleic Acids Res. 2019, 47, D666–D677. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Seshadri, R.; Varghese, N.J.; Eloe-Fadrosh, E.A.; Meier-Kolthoff, J.P.; Göker, M.; Coates, R.C.; Hadjithomas, M.; Pavlopoulos, G.A.; Paez-Espino, D.; et al. 1,003 Reference Genomes of Bacterial and Archaeal Isolates Expand Coverage of the Tree of Life. Nat. Biotechnol. 2017, 35, 676–683. [Google Scholar] [CrossRef]
- Ticak, T.; Hariraju, D.; Arcelay, M.B.; Arivett, B.A.; Fiester, S.E.; Ferguson, D.J. Isolation and Characterization of a Tetramethylammonium-Degrading Methanococcoides Strain and a Novel Glycine Betaine-Utilizing Methanolobus Strain. Arch. Microbiol. 2015, 197, 197–209. [Google Scholar] [CrossRef]
- Kadam, P.C.; Boone, D.R. Physiological Characterization and Emended Description of Methanolobus Vulcani. Int. J. Syst. Bacteriol. 1995, 45, 400–402. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Matheus Carnevali, P.B.; Cheng, J.-F.; Ivanova, N.N.; et al. Cryptic Inoviruses Revealed as Pervasive in Bacteria and Archaea across Earth’s Biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Chen, S.-C.; Lai, M.-C.; Huang, H.-H.; Chiu, H.-H.; Tang, S.-L.; Rogozin, D.Y.; Degermendzhy, A.G. Methanolobus Halotolerans Sp. Nov., Isolated from the Saline Lake Tus in Siberia. Int. J. Syst. Evol. Microbiol. 2020, 70, 5586–5593. [Google Scholar] [CrossRef]
- Göker, M.; Klenk, H.-P. Phylogeny-Driven Target Selection for Largescale Genome-Sequencing (and Other) Projects. Stand. Genom. Sci. 2013, 8, 360–374. [Google Scholar] [CrossRef] [Green Version]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast Selection of Best-Fit Models of Protein Evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Chalita, M.; Ha, S.-M.; Na, S.-I.; Yoon, S.-H.; Chun, J. 2017 ContEst16S: An Algorithm That Identifies Contaminated Prokaryotic Genomes Using 16S RNA Gene Sequences. Int. J. Syst. Evol. Microbiol. 2017, 67, 2053–2057. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Blankenberg, D.; Taylor, J.; Schenck, I.; He, J.; Zhang, Y.; Ghent, M.; Veeraraghavan, N.; Albert, I.; Miller, W.; Makova, K.D.; et al. A Framework for Collaborative Analysis of ENCODE Data: Making Large-Scale Analyses Biologist-Friendly. Genome Res. 2007, 17, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)Orthologs in Large-Scale Analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krassowski, M. Krassowski/complex-upset. GitHub 2020. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the Integration of Metabolic Annotations and the Reconstruction, Comparison and Analysis of Metabolic Models for Plants, Fungi and Microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc Collection of Microbial Genomes and Metabolic Pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically Driven Sequencing of Extremely Halophilic Archaea Reveals Strategies for Static and Dynamic Osmo-Response. PLoS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps, R Package Version 1.0.12; 2019. Available online: http://CRAN.R-project.org/package=pheatmap (accessed on 28 July 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Deppenmeier, U. The unique biochemistry of methanogenesis. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2002; Volume 71, pp. 223–283. [Google Scholar]
- Oren, A. Life at High Salt Concentrations, Intracellular KCl Concentrations, and Acidic Proteomes. Front. Microbiol. 2013, 4, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, C.P.; Murrell, J.C.; Shouche, Y.S. Molecular Diversity of Methanogens and Identification of Methanolobus Sp. as Active Methylotrophic Archaea in Lonar Crater Lake Sediments. FEMS Microbiol. Ecol. 2012, 81, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Schink, B.; Zeikus, J.G.Y. 1982 Microbial Ecology of Pectin Decomposition in Anoxic Lake Sediments. Microbiology 1982, 128, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Lovley, D.R.; Klug, M.J. Methanogenesis from Methanol and Methylamines and Acetogenesis from Hydrogen and Carbon Dioxide in the Sediments of a Eutrophic Lake. Appl. Environ. Microbiol. 1983, 45, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Orphan, V.J.; Jahnke, L.L.; Embaye, T.; Turk, K.A.; Pernthaler, A.; Summons, R.E.; Marais, D.J.D. Characterization and Spatial Distribution of Methanogens and Methanogenic Biosignatures in Hypersaline Microbial Mats of Baja California. Geobiology 2008, 6, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.-C.; Elling, F.J.; Nigro, L.M.; Samarkin, V.; Joye, S.B.; Teske, A.; Hinrichs, K.-U. Multiple Evidence for Methylotrophic Methanogenesis as the Dominant Methanogenic Pathway in Hypersaline Sediments from the Orca Basin, Gulf of Mexico. Geochim. Cosmochim. Acta 2016, 187, 1–20. [Google Scholar] [CrossRef]
- Zhuang, G.-C.; Heuer, V.B.; Lazar, C.S.; Goldhammer, T.; Wendt, J.; Samarkin, V.A.; Elvert, M.; Teske, A.P.; Joye, S.B.; Hinrichs, K.-U. Relative Importance of Methylotrophic Methanogenesis in Sediments of the Western Mediterranean Sea. Geochim. Cosmochim. Acta 2018, 224, 171–186. [Google Scholar] [CrossRef]
- Weston, N.B.; Dixon, R.E.; Joye, S.B. Ramifications of Increased Salinity in Tidal Freshwater Sediments: Geochemistry and Microbial Pathways of Organic Matter Mineralization. J. Geophys. Res. Biogeosciences 2006, 111. [Google Scholar] [CrossRef] [Green Version]
- Kristjansson, J.K.; Schönheit, P. Why Do Sulfate-Reducing Bacteria Outcompete Methanogenic Bacteria for Substrates? Oecologia 1983, 60, 264–266. [Google Scholar] [CrossRef]
- Oren, A. Formation and Breakdown of Glycine Betaine and Trimethylamine in Hypersaline Environments. Antonie Van Leeuwenhoek 1990, 58, 291–298. [Google Scholar] [CrossRef]
- Kiene, R.P.; Visscher, P.T. Production and Fate of Methylated Sulfur Compounds from Methionine and Dimethylsulfoniopropionate in Anoxic Salt Marsh Sediments. Appl. Environ. Microbiol. 1987, 53, 2426–2434. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Priscu, J.C.; Xiong, J.; Conrad, R.; Vick-Majors, T.; Chu, H.; Hou, J. Salinity Drives Archaeal Distribution Patterns in High Altitude Lake Sediments on the Tibetan Plateau. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef]
- Smith, J.M.; Green, S.J.; Kelley, C.A.; Prufert-Bebout, L.; Bebout, B.M. Shifts in Methanogen Community Structure and Function Associated with Long-Term Manipulation of Sulfate and Salinity in a Hypersaline Microbial Mat. Environ. Microbiol. 2008, 10, 386–394. [Google Scholar] [CrossRef]
- Deole, R.; Challacombe, J.; Raiford, D.W.; Hoff, W.D. An Extremely Halophilic Proteobacterium Combines a Highly Acidic Proteome with a Low Cytoplasmic Potassium Content. J. Biol. Chem. 2013, 288, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, M.E.; Fitz-Gibbon, S.T.; Oren, A.; House, C.H. Amino Acid Signatures of Salinity on an Environmental Scale with a Focus on the Dead Sea. Environ. Microbiol. 2010, 12, 2613–2623. [Google Scholar] [CrossRef]
- Paul, S.; Bag, S.K.; Das, S.; Harvill, E.T.; Dutta, C. Molecular Signature of Hypersaline Adaptation: Insights from Genome and Proteome Composition of Halophilic Prokaryotes. Genome Biol. 2008, 9, R70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elevi Bardavid, R.; Oren, A. Acid-Shifted Isoelectric Point Profiles of the Proteins in a Hypersaline Microbial Mat: An Adaptation to Life at High Salt Concentrations? Extremophiles 2012, 16, 787–792. [Google Scholar] [CrossRef]
- Sur, S.; Bothra, A.K.; Sen, A. Proteome Analysis Reveals the Influence of Isoelectric Point and Amino Acid Usages on the Lifestyle of Nitrogen Fixing Microorganisms. Indian J. Biotechnol. 2013, 12, 88–97. [Google Scholar]
- Schwartz, R.; Ting, C.S.; King, J. Whole Proteome PI Values Correlate with Subcellular Localizations of Proteins for Organisms within the Three Domains of Life. Genome Res. 2001, 11, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueno de Mesquita, C.P.; Zhou, J.; Theroux, S.M.; Tringe, S.G. Methanogenesis and Salt Tolerance Genes of a Novel Halophilic Methanosarcinaceae Metagenome-Assembled Genome from a Former Solar Saltern. Genes 2021, 12, 1609. https://doi.org/10.3390/genes12101609
Bueno de Mesquita CP, Zhou J, Theroux SM, Tringe SG. Methanogenesis and Salt Tolerance Genes of a Novel Halophilic Methanosarcinaceae Metagenome-Assembled Genome from a Former Solar Saltern. Genes. 2021; 12(10):1609. https://doi.org/10.3390/genes12101609
Chicago/Turabian StyleBueno de Mesquita, Clifton P., Jinglie Zhou, Susanna M. Theroux, and Susannah G. Tringe. 2021. "Methanogenesis and Salt Tolerance Genes of a Novel Halophilic Methanosarcinaceae Metagenome-Assembled Genome from a Former Solar Saltern" Genes 12, no. 10: 1609. https://doi.org/10.3390/genes12101609