
Methylphosphonate Degradation and Salt-Tolerance Genes of Two Novel Halophilic Marivita Metagenome-Assembled Genomes from Unrestored Solar Salterns


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
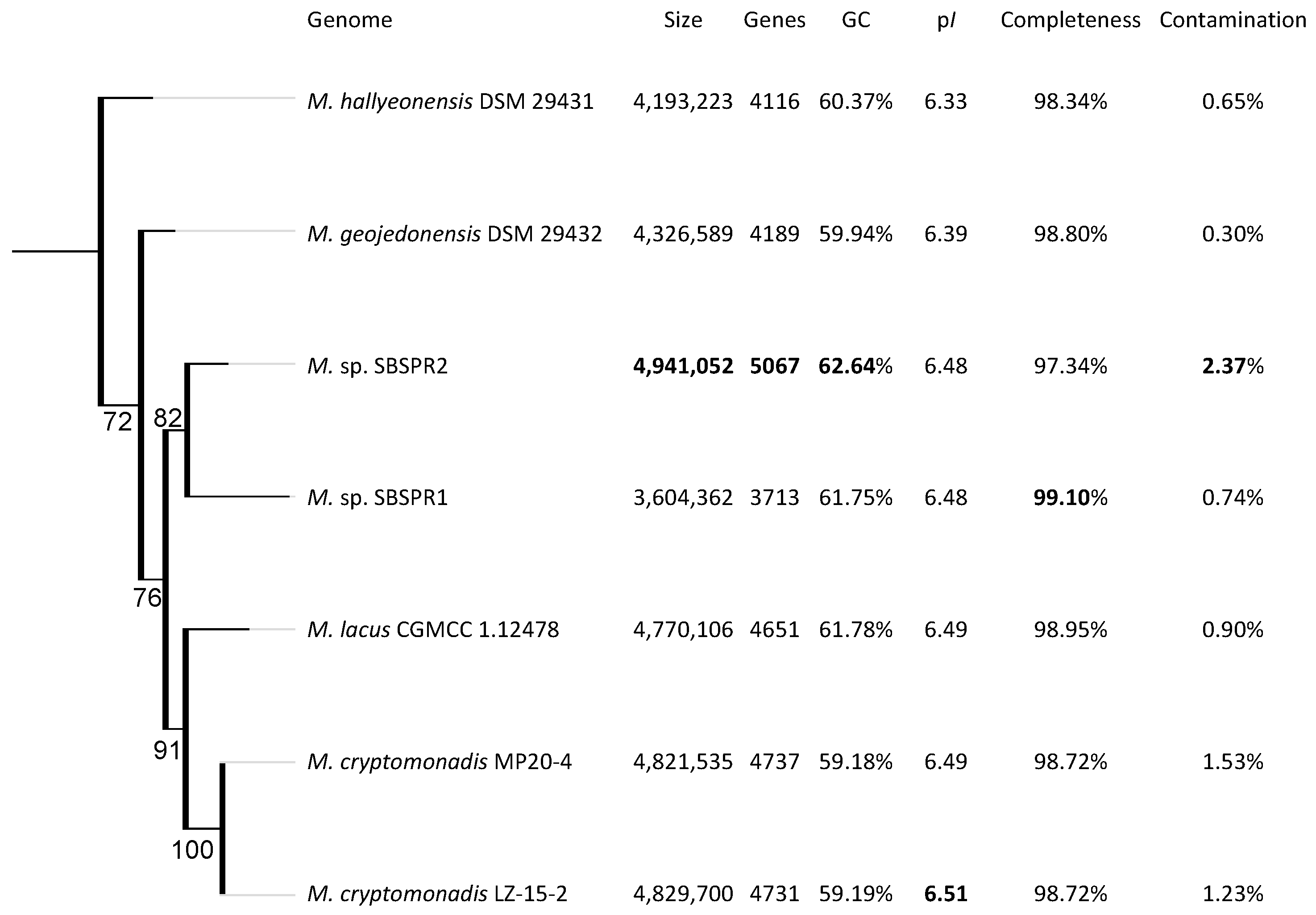
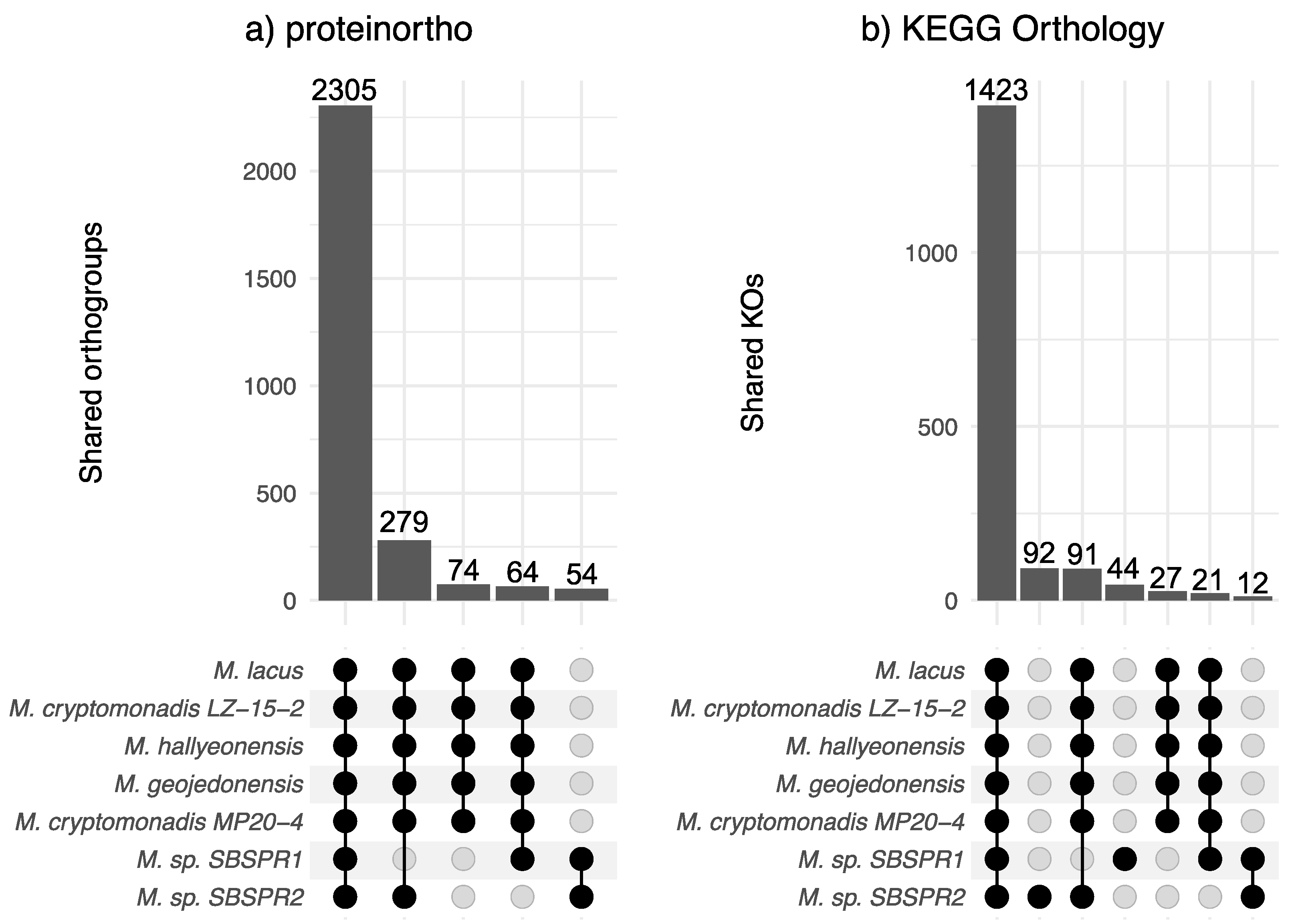
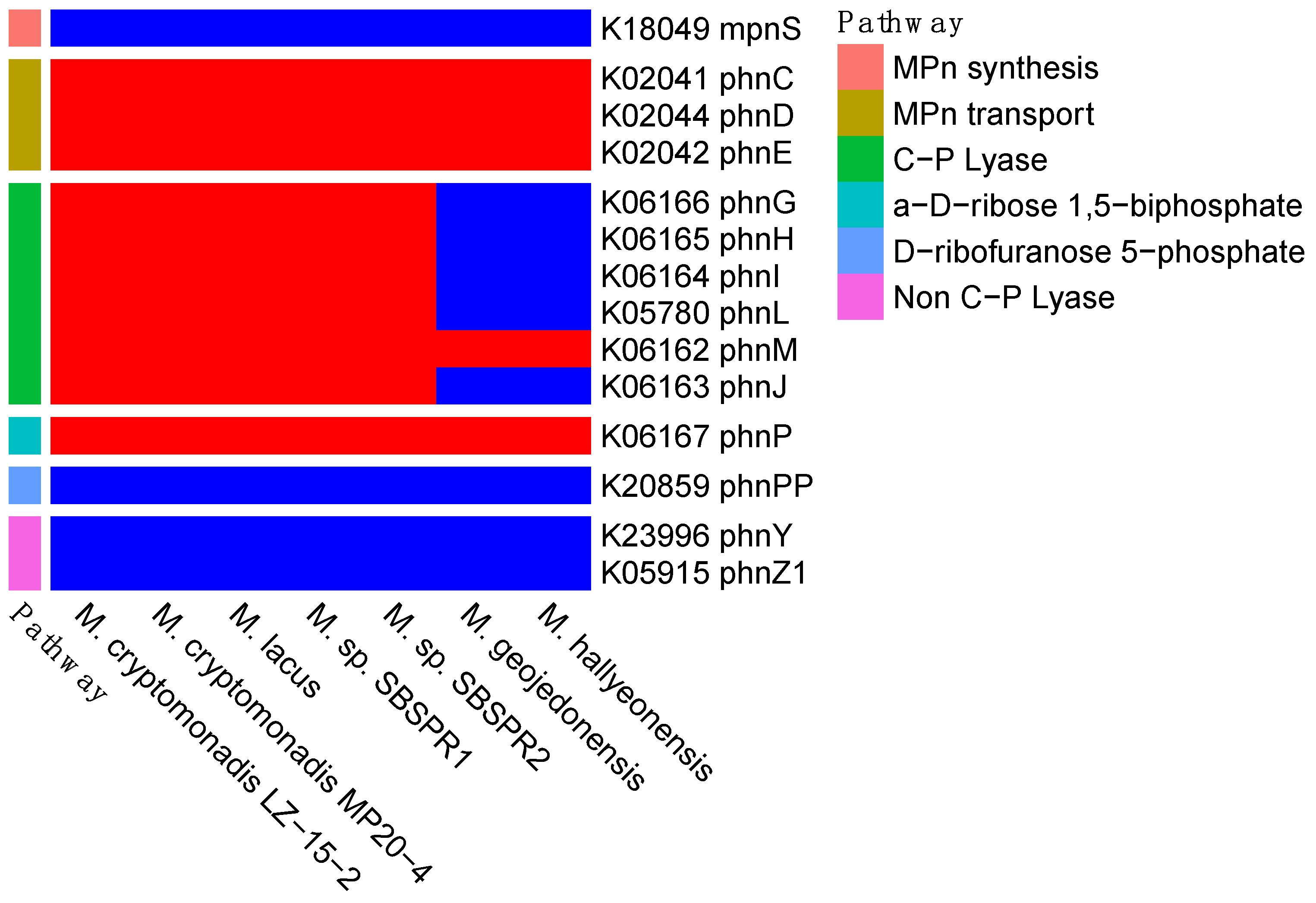
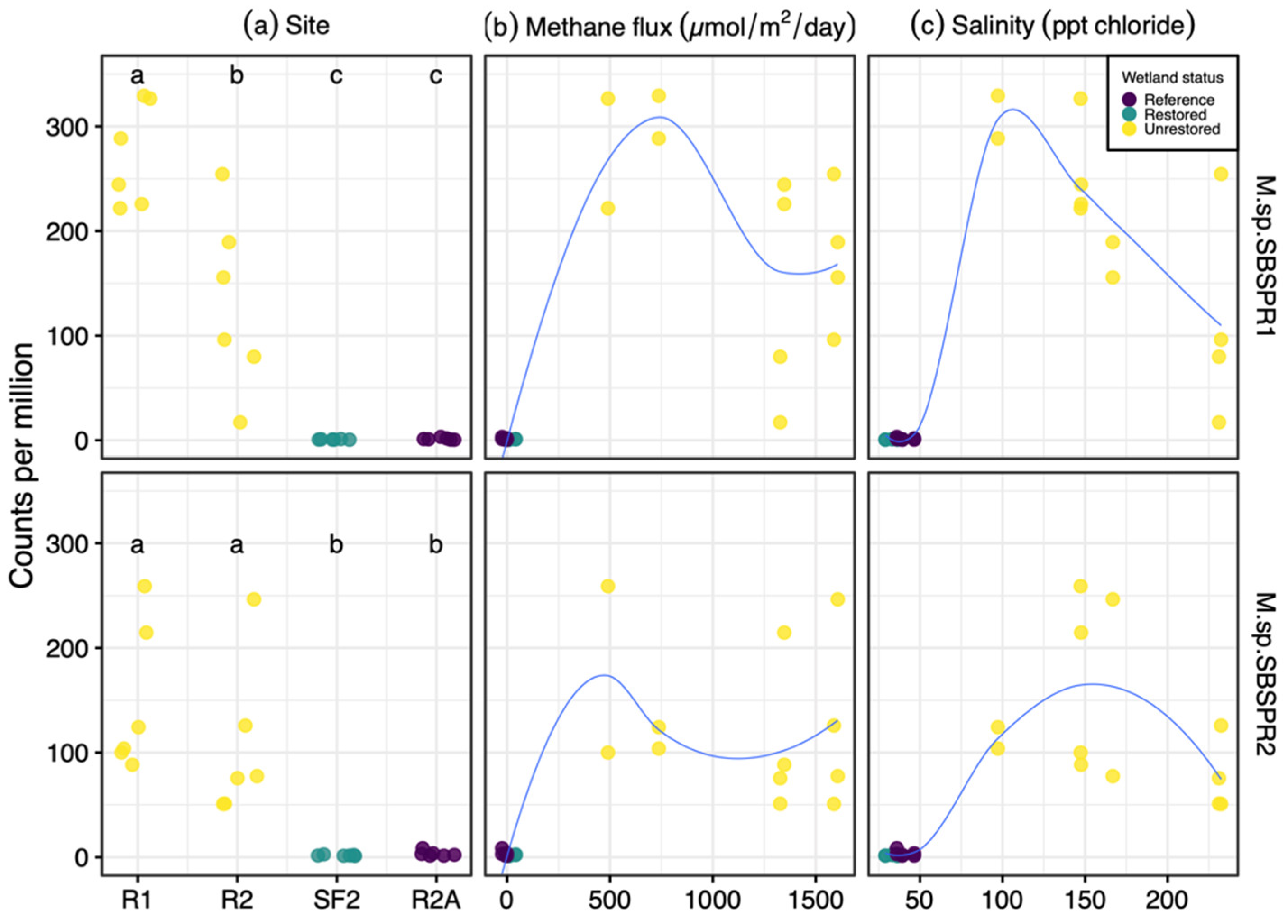
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karl, D.M.; Beversdorf, L.; Björkman, K.M.; Church, M.J.; Martinez, A.; Delong, E.F. Aerobic Production of Methane in the Sea. Nat. Geosci. 2008, 1, 473–478. [Google Scholar] [CrossRef]
- Del Valle, D.; Karl, D. Aerobic Production of Methane from Dissolved Water-Column Methylphosphonate and Sinking Particles in the North Pacific Subtropical Gyre. Aquat. Microb. Ecol. 2014, 73, 93–105. [Google Scholar] [CrossRef]
- Repeta, D.J.; Ferrón, S.; Sosa, O.A.; Johnson, C.G.; Repeta, L.D.; Acker, M.; DeLong, E.F.; Karl, D.M. Marine Methane Paradox Explained by Bacterial Degradation of Dissolved Organic Matter. Nat. Geosci. 2016, 9, 884–887. [Google Scholar] [CrossRef]
- Martínez, A.; Ventouras, L.-A.; Wilson, S.T.; Karl, D.M.; Delong, E.F. Metatranscriptomic and Functional Metagenomic Analysis of Methylphosphonate Utilization by Marine Bacteria. Front. Microbiol. 2013, 4, 340. [Google Scholar] [CrossRef]
- Yao, M.; Henny, C.; Maresca, J.A. Freshwater Bacteria Release Methane as a By-Product of Phosphorus Acquisition. Appl. Environ. Microbiol. 2016, 82, 6994–7003. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Alowaifeer, A.; Kerner, P.; Balasubramanian, N.; Patterson, A.; Christian, W.; Tarver, A.; Dore, J.E.; Hatzenpichler, R.; Bothner, B.; et al. Aerobic Bacterial Methane Synthesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2019229118. [Google Scholar] [CrossRef] [PubMed]
- Kamat, S.S.; Williams, H.J.; Raushel, F.M. Intermediates in the Transformation of Phosphonates to Phosphate by Bacteria. Nature 2011, 480, 570–573. [Google Scholar] [CrossRef]
- Kamat, S.S.; Williams, H.J.; Dangott, L.J.; Chakrabarti, M.; Raushel, F.M. The Catalytic Mechanism for Aerobic Formation of Methane by Bacteria. Nature 2013, 497, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, W.W.; Griffin, B.M.; Cicchillo, R.M.; Gao, J.; Janga, S.C.; Cooke, H.A.; Circello, B.T.; Evans, B.S.; Martens-Habbena, W.; Stahl, D.A.; et al. Synthesis of Methylphosphonic Acid by Marine Microbes: A Source for Methane in the Aerobic Ocean. Science 2012, 337, 1104–1107. [Google Scholar] [CrossRef]
- Born, D.A.; Ulrich, E.C.; Ju, K.-S.; Peck, S.C.; van der Donk, W.A.; Drennan, C.L. Structural Basis for Methylphosphonate Biosynthesis. Science 2017, 358, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Sosa, O.A.; Repeta, D.J.; DeLong, E.F.; Ashkezari, M.D.; Karl, D.M. Phosphate-Limited Ocean Regions Select for Bacterial Populations Enriched in the Carbon-Phosphorus Lyase Pathway for Phosphonate Degradation. Environ. Microbiol. 2019, 21, 2402–2414. [Google Scholar] [CrossRef]
- Oliverio, A.M.; Bissett, A.; McGuire, K.; Saltonstall, K.; Turner, B.L.; Fierer, N. The Role of Phosphorus Limitation in Shaping Soil Bacterial Communities and Their Metabolic Capabilities. mBio 2020, 11, e01718-20. [Google Scholar] [CrossRef]
- Carini, P.; White, A.E.; Campbell, E.O.; Giovannoni, S.J. Methane Production by Phosphate-Starved SAR11 Chemoheterotrophic Marine Bacteria. Nat. Commun. 2014, 5, 4346. [Google Scholar] [CrossRef] [PubMed]
- Teikari, J.E.; Fewer, D.P.; Shrestha, R.; Hou, S.; Leikoski, N.; Mäkelä, M.; Simojoki, A.; Hess, W.R.; Sivonen, K. Strains of the Toxic and Bloom-Forming Nodularia Spumigena (Cyanobacteria) Can Degrade Methylphosphonate and Release Methane. ISME J. 2018, 12, 1619–1630. [Google Scholar] [CrossRef]
- Zhou, J.; Theroux, S.M.; Bueno de Mesquita, C.P.; Hartman, W.H.; Tringe, S.G. Microbial Drivers of Methane Emissions from Unrestored Industrial Salt Ponds. ISME J. 2021, 16, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R.; et al. IMG/M v.5.0: An Integrated Data Management and Comparative Analysis System for Microbial Genomes and Microbiomes. Nucleic Acids Res. 2019, 47, D666–D677. [Google Scholar] [CrossRef]
- Hwang, C.Y.; Bae, G.D.; Yih, W.; Cho, B.C.Y. Marivita Cryptomonadis Gen. Nov., Sp. Nov. and Marivita Litorea Sp. Nov., of the Family Rhodobacteraceae, Isolated from Marine Habitats. Int. J. Syst. Evol. Microbiol. 2009, 59, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Kang, S.-J.; Lee, S.-Y.; Jung, Y.-T.; Lee, J.-S.; Oh, T.-K. Marivita Hallyeonensis Sp. Nov., Isolated from Seawater, Reclassification of Gaetbulicola Byunsanensis as Marivita Byunsanensis Comb. Nov. and Emended Description of the Genus Marivita. Int. J. Syst. Evol. Microbiol. 2012, 62, 839–843. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Kang, S.-J.; Lee, J.-S. Marivita Geojedonensis Sp. Nov., Isolated from Seawater. Int. J. Syst. Evol. Microbiol. 2013, 63, 423–427. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Kang, S.-J.; Jung, Y.-T.; Oh, T.-K. Gaetbulicola Byunsanensis Gen. Nov., Sp. Nov., Isolated from Tidal Flat Sediment. Int. J. Syst. Evol. Microbiol. 2010, 60, 196–199. [Google Scholar] [CrossRef]
- Budinoff, C.R.; Dunlap, J.R.; Hadden, M.; Buchan, A. Marivita Roseacus Sp. Nov., of the Family Rhodobacteraceae, Isolated from a Temperate Estuary and an Emended Description of the Genus Marivita. J. Gen. Appl. Microbiol. 2011, 57, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.-P.; Liu, Y.; Hou, T.-T.; Liu, H.-C.; Zhou, Y.-G.; Wang, F.; Liu, Z.-P. Marivita Lacus Sp. Nov., Isolated from a Saline Lake. Int. J. Syst. Evol. Microbiol. 2015, 65, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, X.-A.; Jiang, Z.-W.; Yang, X.; Zhang, X.-L.; Yang, Q. Combined Characterization of a New Member of Marivita Cryptomonadis Strain LZ-15-2 Isolated from Cultivable Phycosphere Microbiota of Highly Toxic HAB Dinoflagellate Alexandrium Catenella LZT09. Braz. J. Microbiol. 2021, 52, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Bueno de Mesquita, C.P.; Zhou, J.; Theroux, S.M.; Tringe, S.G. Methanogenesis and Salt Tolerance Genes of a Novel Halophilic Methanosarcinaceae Metagenome-Assembled Genome from a Former Solar Saltern. Genes 2021, 12, 1609. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an Efficient Tool for Accurately Reconstructing Single Genomes from Complex Microbial Communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Von Meijenfeldt, F.A.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust Taxonomic Classification of Uncharted Microbial Sequences and Bins with CAT and BAT. Genome Biol. 2019, 20, 217. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Chen, L.-X.; Anantharaman, K.; Shaiber, A.; Eren, A.M.; Banfield, J.F. Accurate and Complete Genomes from Metagenomes. Genome Res. 2020, 30, 315–333. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.-M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1000+ Type-Strain Genomes Substantially Improves Taxonomic Classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ma, J. The Global Catalogue of Microorganisms (GCM) 10K Type Strain Sequencing Project: Providing Services to Taxonomists for Standard Genome Sequencing and Annotation. Int. J. Syst. Evol. Microbiol. 2019, 69, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Feng, X.; Chen, Y.; Li, L.; Wang, X.; Hu, Z.; Wang, H.; Luo, H. Metapopulation Structure of Diatom-Associated Marine Bacteria. bioRxiv 2021. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast Selection of Best-Fit Models of Protein Evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Blankenberg, D.; Taylor, J.; Schenck, I.; He, J.; Zhang, Y.; Ghent, M.; Veeraraghavan, N.; Albert, I.; Miller, W.; Makova, K.D.; et al. A Framework for Collaborative Analysis of ENCODE Data: Making Large-Scale Analyses Biologist-Friendly. Genome Res. 2007, 17, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)Orthologs in Large-Scale Analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Krassowski, M. Krassowski/Complex-Upset. 2020. Available online: https://zenodo.org/record/5762625 (accessed on 21 December 2021).
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc Collection of Microbial Genomes and Metabolic Pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Vreeland, R.H. Mechanisms of Halotolerance in Microorganisms. Crit. Rev. Microbiol. 2009, 14, 311–356. [Google Scholar] [CrossRef]
- Vera-Gargallo, B.; Ventosa, A. Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain). Genes 2018, 9, 152. [Google Scholar] [CrossRef]
- Youssef, N.H.; Savage-Ashlock, K.N.; McCully, A.L.; Luedtke, B.; Shaw, E.I.; Hoff, W.D.; Elshahed, M.S. Trehalose/2-Sulfotrehalose Biosynthesis and Glycine-Betaine Uptake Are Widely Spread Mechanisms for Osmoadaptation in the Halobacteriales. ISME J. 2014, 8, 636–649. [Google Scholar] [CrossRef]
- Matarredona, L.; Camacho, M.; Zafrilla, B.; Bonete, M.-J.; Esclapez, J. The Role of Stress Proteins in Haloarchaea and Their Adaptive Response to Environmental Shifts. Biomolecules 2020, 10, 1390. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of Adaptation of Microorganisms of the Three Domains of Life to High Salt Concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically Driven Sequencing of Extremely Halophilic Archaea Reveals Strategies for Static and Dynamic Osmo-Response. PLOS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: http://CRAN.R-project.org/package=pheatmap (accessed on 21 December 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using Average Nucleotide Identity to Improve Taxonomic Assignments in Prokaryotic Genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the Genomic Gold Standard for the Prokaryotic Species Definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a Taxonomic Coherence between Average Nucleotide Identity and 16S rRNA Gene Sequence Similarity for Species Demarcation of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Tyson, G.W.; DeLong, E.F. Widespread Known and Novel Phosphonate Utilization Pathways in Marine Bacteria Revealed by Functional Screening and Metagenomic Analyses. Environ. Microbiol. 2010, 12, 222–238. [Google Scholar] [CrossRef]
- Elevi Bardavid, R.; Oren, A. Acid-Shifted Isoelectric Point Profiles of the Proteins in a Hypersaline Microbial Mat: An Adaptation to Life at High Salt Concentrations? Extremophiles 2012, 16, 787–792. [Google Scholar] [CrossRef]
- Buchan, A.; González, J.M.; Moran, M.A. Overview of the Marine Roseobacter Lineage. Appl. Environ. Microbiol. 2005, 71, 5665–5677. [Google Scholar] [CrossRef]
- Brinkhoff, T.; Giebel, H.-A.; Simon, M. Diversity, Ecology, and Genomics of the Roseobacter Clade: A Short Overview. Arch. Microbiol. 2008, 189, 531–539. [Google Scholar] [CrossRef]
- Ventosa, A.; Nieto, J.J.; Oren, A. Biology of Moderately Halophilic Aerobic Bacteria. Microbiol. Mol. Biol. Rev. 1998, 62, 504–544. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Bag, S.K.; Das, S.; Harvill, E.T.; Dutta, C. Molecular Signature of Hypersaline Adaptation: Insights from Genome and Proteome Composition of Halophilic Prokaryotes. Genome Biol. 2008, 9, R70. [Google Scholar] [CrossRef] [PubMed]
- Dyhrman, S.T.; Chappell, P.D.; Haley, S.T.; Moffett, J.W.; Orchard, E.D.; Waterbury, J.B.; Webb, E.A. Phosphonate Utilization by the Globally Important Marine Diazotroph Trichodesmium. Nature 2006, 439, 68–71. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueno de Mesquita, C.P.; Zhou, J.; Theroux, S.; Tringe, S.G. Methylphosphonate Degradation and Salt-Tolerance Genes of Two Novel Halophilic Marivita Metagenome-Assembled Genomes from Unrestored Solar Salterns. Genes 2022, 13, 148. https://doi.org/10.3390/genes13010148
Bueno de Mesquita CP, Zhou J, Theroux S, Tringe SG. Methylphosphonate Degradation and Salt-Tolerance Genes of Two Novel Halophilic Marivita Metagenome-Assembled Genomes from Unrestored Solar Salterns. Genes. 2022; 13(1):148. https://doi.org/10.3390/genes13010148
Chicago/Turabian StyleBueno de Mesquita, Clifton P., Jinglie Zhou, Susanna Theroux, and Susannah G. Tringe. 2022. "Methylphosphonate Degradation and Salt-Tolerance Genes of Two Novel Halophilic Marivita Metagenome-Assembled Genomes from Unrestored Solar Salterns" Genes 13, no. 1: 148. https://doi.org/10.3390/genes13010148