
Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease


1
Development, Ageing and Disease, UCL Institute of Ophthalmology, London EC1V 9EL, UK
2
Ocular Genomics and Therapeutics, The Francis Crick Institute, London NW1 1AT, UK
3
Department of Genetics, Moorfields Eye Hospital NHS Foundation Trust, London EC1V 2PD, UK
4
Department of Ophthalmology, Great Ormond Street Hospital for Children NHS Foundation Trust, London WC1N 3JH, UK
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Genes 2023, 14(7), 1325; https://doi.org/10.3390/genes14071325
Submission received: 16 May 2023
/
Revised: 11 June 2023
/
Accepted: 19 June 2023
/
Published: 23 June 2023
(This article belongs to the Special Issue Genetics and Pathogenesis of Inherited Eye Diseases)
Abstract
:NR2E3 is a nuclear hormone receptor gene required for the correct development of the retinal rod photoreceptors. Expression of NR2E3 protein in rod cell precursors suppresses cone-specific gene expression and, in concert with other transcription factors including NRL, activates the expression of rod-specific genes. Pathogenic variants involving NR2E3 cause a spectrum of retinopathies, including enhanced S-cone syndrome, Goldmann–Favre syndrome, retinitis pigmentosa, and clumped pigmentary retinal degeneration, with limited evidence of genotype–phenotype correlations. A common feature of NR2E3-related disease is an abnormally high number of cone photoreceptors that are sensitive to short wavelength light, the S-cones. This characteristic has been supported by mouse studies, which have also revealed that loss of Nr2e3 function causes photoreceptors to develop as cells that are intermediate between rods and cones. While there is currently no available cure for NR2E3-related retinopathies, there are a number of emerging therapeutic strategies under investigation, including the use of viral gene therapy and gene editing, that have shown promise for the future treatment of patients with NR2E3 variants and other inherited retinal diseases. This review provides a detailed overview of the current understanding of the role of NR2E3 in normal development and disease, and the associated clinical phenotypes, animal models, and therapeutic studies.
1. Introduction
NR2E3 (Nuclear Receptor subfamily 2 group E member 3; OMIM #604485), previously known as PNR, encodes a photoreceptor-specific orphan nuclear hormone receptor essential for the normal development of the retinal photoreceptors [1]. The gene is located on chromosome 15q23 and is comprised of 8 coding exons. NR2E3 has two isoforms: (i) a full-length transcript containing all 8 exons, producing a 410-amino acid (aa) protein, and (ii) a second transcript that retains intron 7, coding for a smaller 367-aa protein that lacks the region encoded by exon 8 [2]. Pathogenic variants in NR2E3 show significant clinical heterogeneity and have been associated with a number of retinopathies, with a lack of clear genotype–phenotype correlations [1]. A common hallmark of NR2E3-related disease is an abnormally increased number of cone photoreceptors that are sensitive to short wavelength (blue) light, the S-cones, which has been evidenced by psychophysical, electrophysiological [3,4], and histopathological [5] examination of patients, and animal studies [6]. While there is still much to be uncovered, significant progress has been made toward understanding the role of NR2E3 in retinal development and disease and, in recent years, toward the development of effective treatments through promising pre-clinical therapeutic studies.
2. NR2E3 Structure
The NR2E3 protein is a member of a large family of ligand-modulated transcription factors, the nuclear receptors. In the human genome, there are 48 nuclear receptors, which include endocrine, adopted orphan, and orphan receptors [7]. NR2E3 is an orphan receptor that shares a conserved structural organization with all nuclear receptors, consisting of several key regions: the A/B, DNA-binding, hinge, and ligand-binding domains [8,9]. In the N-terminus, the highly variable A/B domain comprises a ligand-independent activator function (AF-1). This is followed by the most conserved region, the DNA-binding domain, which consists of two Cys4 zinc fingers that contain a P-box, thought to allow the receptor to bind to unique DNA response element sites and regulate gene expression, and a D-box, proposed to be involved in protein–protein interactions. The hinge domain links the DNA-binding and ligand-binding domains and contains a nuclear localization signal that may overlap with the DNA-binding domain.
The C-terminal ligand-binding domain of nuclear receptors typically consists of 12 α-helices that fold into a conserved hydrophobic pocket where a ligand could bind to, which is unknown in the case of NR2E3. In addition to this ligand-dependent activator function (AF-2), the ligand-binding domain is also essential for homo- and heterodimerization. Tan et al. solved the crystal structure of the ligand-binding domain of NR2E3 in a ligand-free state and found that it has a dimeric arrangement, with each monomer being formed of a canonical antiparallel three-layer α-helical sandwich fold made up of 8 α-helices [7,10]. The ligand-binding pocket was found to be filled by the side chains of hydrophobic and aromatic residues and the AF-2 helix occupies the canonical cofactor binding site. It was concluded that the NR2E3 ligand-binding domain has an auto-repressed configuration.
3. NR2E3 Function
3.1. Rod and Cone Photoreceptor Differentiation
The appearance of NR2E3 in evolutionary time is thought to coincide with the emergence of rod and cone photoreceptors [1]. Prior to this, early vertebrate ancestors had only one photoreceptor cell type, which is thought to have been more structurally similar to cones than to rods [11]. Rods and cones differ in several key aspects, including their shape, photopigments, distribution within the retina, and pattern of synaptic connection [12]. Typically, the human retina contains ~5% cones and ~95% rods [13]. Cones are found at the highest density in the macula, whereas rods are more concentrated around the peripheral retina. Rods contain a single type of visual pigment, rhodopsin, for high-sensitivity low-light vision [12]. In contrast, human cones contain one of three alternative pigments (S-, M-, and L-opsins) each, which respond to short (S), medium (M), and long (L) wavelengths (i.e., blue, green, red, respectively) for color and bright-light high-resolution vision. The S-cone photoreceptor cell population is typically the least prevalent of the photoreceptor cell subtypes, accounting for 5–10% of the cone mosaic [14]. S-cones morphologically differ from M and L cones by displaying a longer and wider inner segment joining the outer segment and are most dense at ~2000 cells mm2, just outside the center fovea [15].
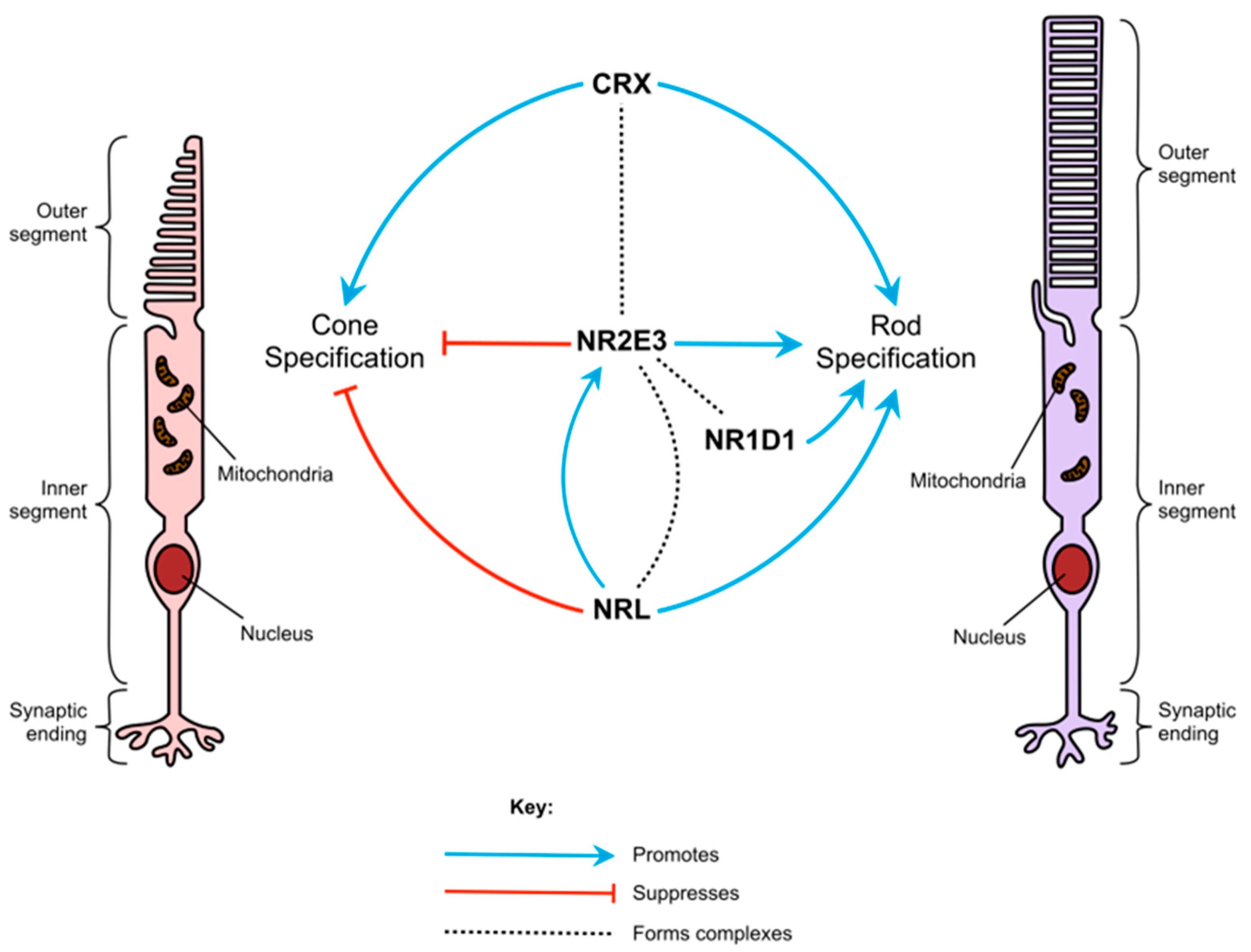
During embryonic development, rods and cones differentiate from common photoreceptor precursor cells [5]. Their differentiation is controlled by several transcription factors, including NR2E3, which ensure that rod- and cone-specific genes are confined to their corresponding photoreceptor type (Figure 1) [16]. Studies of animal models and human patients with NR2E3 variants suggest that the gene’s role in photoreceptor differentiation is two-fold, in that it suppresses the expression of cone-specific genes, such as OPNSW1 (blue opsin), GNAT2, and GNB (cone transducin subunits), and helps to activate the expression of rod-specific genes, such as the rod transducin β subunit GNB1 [17] and rhodopsin [18,19]. NR2E3 is exclusively expressed in rods and is first detected in immature rods on the foveal edge from fetal week 11.7 [20]. Without the expression of NR2E3, photoreceptor precursors differentiate to the “default” photoreceptor cell type, S-cones [18].
Other than NR2E3, the main factors involved in rod differentiation are CRX, NRL, and NR1D1 [16,21]. Evidence suggests that the transcription factors encoded by these genes, along with NR2E3, interact to form multi-protein transcriptional regulatory complexes [21]. CRX promotes the expression of both rod- and cone-specific transcripts, and pathogenic variants in CRX can cause several retinopathies, including early-onset diseases such as Leber congenital amaurosis [22] and cone-rod dystrophy [23]. Both NRL and NR2E3 are confined to rods and rod precursors and suppress cone-specific transcripts [1]. NRL also up-regulates the expression of NR2E3 and other rod-specific transcripts. Deletion of Nrl in mice also causes photoreceptors to develop as S-cones rather than rods [19].
Neither NR1D1 nor NR2E3 alone has much effect on the activity of rod-specific promoters [21]. However, when both are active together, NR1D1 and NR2E3 work synergistically to increase rhodopsin promoter activity. NR2E3 can also only bind to the promoter regions of rod-specific genes in the presence of CRX [24]. These interactions indicate that NR2E3 forms part of a complex network of signals that determine the cell fates of photoreceptor precursors. This complexity may in part account for the extensive variety of phenotypes seen in patients and animal disease models.
3.2. Role in the Adult Retina and Other Tissues
NR2E3 continues to be expressed in the adult retina, and several studies have indicated that it is involved in retinal maintenance [17,25,26]. In the mature mouse retina, NR2E3 regulates a different set of genes to those targeted in development; these include several genes responsible for the maintenance and survival of photoreceptors, such as phototransduction-related genes Opnsw1 and Gnb1 [14]. Furthermore, Olivares et al. found that Nr2e3 is involved in several gene networks in the adult retina and regulates genes associated with age-related macular degeneration, including Flt1, Abca1, and Alcam [25].
Nr2e3 protein expression has been found in murine liver cells, suggesting its role is more widespread than was previously considered [27]. However, the role of NR2E3 in the liver and other tissues is not well understood. Higher levels of NR2E3 were associated with good clinical outcomes in liver cancer patients [28], while loss of NR2E3 was correlated with the development of liver disease and cancer [27]. Expression of NR2E3 showed a similar association in breast cancer patients, where it appears to regulate the estrogen receptor α [29].
4. Clinical Phenotype
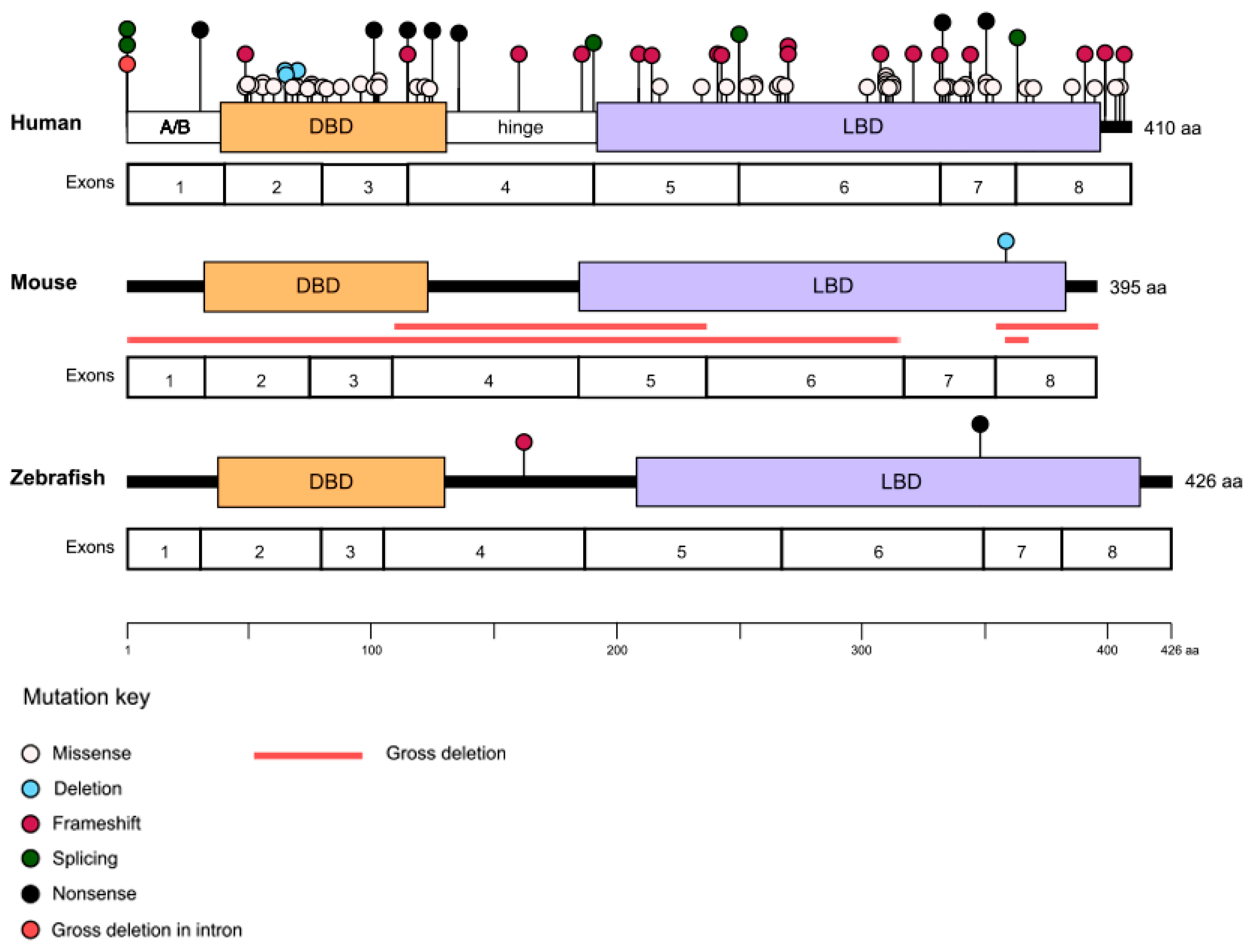
There are currently more than 80 identified disease-causing variants of NR2E3, which cause a variety of retinopathies, most of which show autosomal recessive inheritance (Figure 2 and Table 1). Among the recessively inherited disorders are enhanced S-cone syndrome (ESCS; MIM #268100), Goldmann–Favre syndrome (GFS; MIM #268100), retinitis pigmentosa (RP; MIM #611131), and clumped pigmentary retinal degeneration (CPRD). In addition, NR2E3 is associated with an autosomal dominant form of RP.
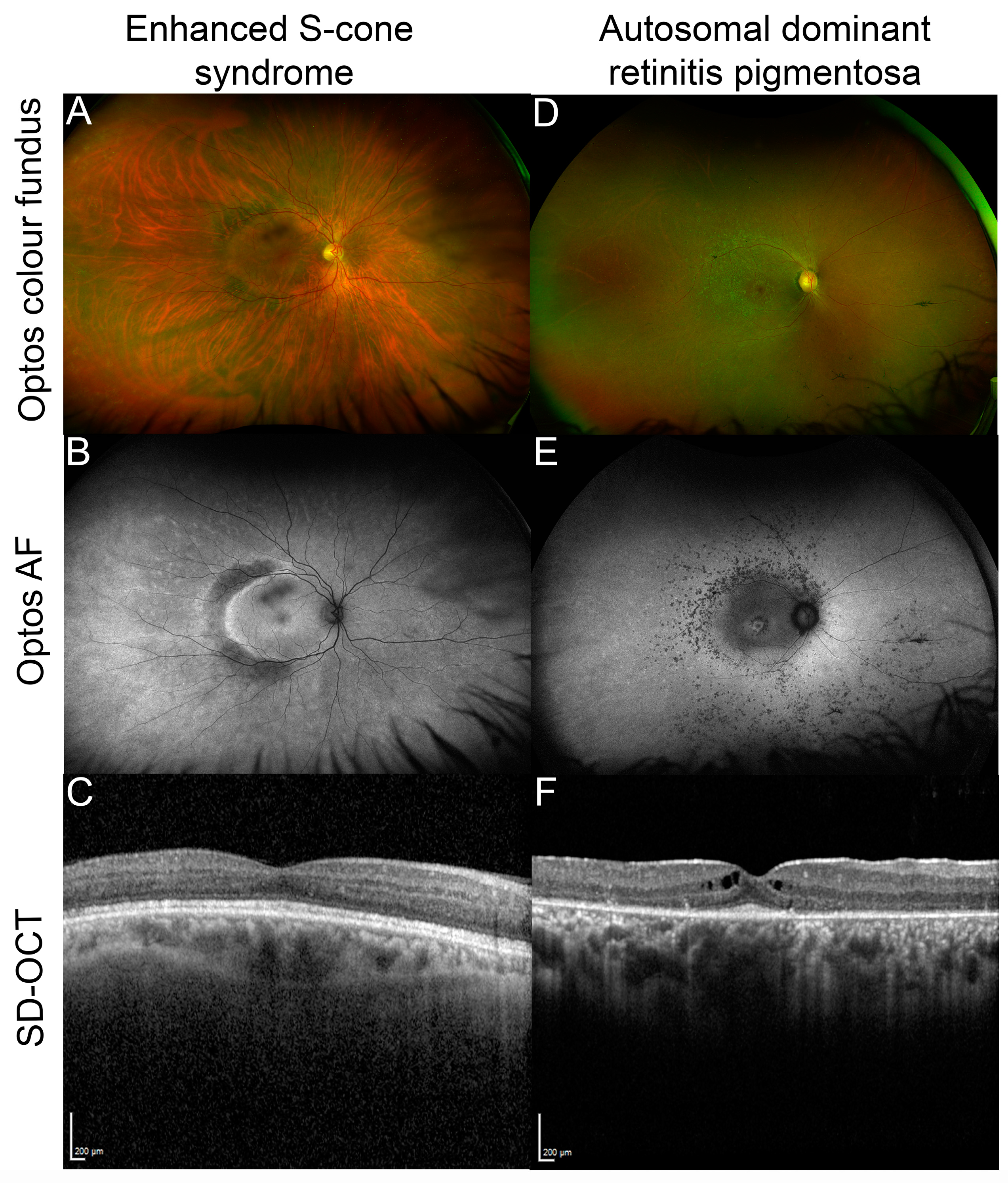
Pathogenic variants in NR2E3 were initially described in patients with ESCS, a developmental condition that causes enhanced sensitivity to blue light, early onset night blindness (nyctalopia), and abnormal ERG responses due to an overabundance of S-cones and lack of functional rods [30]. Patients also have varying degrees of sensitivity to green and red light (due to varying abundance of M-cones and L-cones). Visual function for patients with these variants is highly variable, even within families, and can range from normal to severely reduced [31]. Additional clinical findings include hypermetropia, astigmatism, macular holes, vessel attenuation, and degenerative changes including subretinal white dots or yellow flecks and characteristic clumped or nummular pigment deposition observed in the mid-peripheral fundus [31,32,33]. Retinal images taken from a patient with ESCS are shown in Figure 3A–C. GFS is similar to ESCS, and the disorders are now considered to share the same clinical spectrum [33,34], with GFS representing a more severe form. In addition to enhanced S-cone function, early onset nyctalopia, and clumped fundus pigmentation, GFS is typically characterized by degenerative changes in the vitreous humor, macular and peripheral retinoschisis (splitting of retinal layers), and posterior subcapsular cataracts [35]. CPRD is a further disorder discovered to be on the NR2E3 phenotypic spectrum, sharing some clinical features with ESCS and GFS (i.e., clumped pigmentation throughout the mid-peripheral fundus and nyctalopia early in life), with ERG responses more similar to that of RP patients [33,36]. However, an assessment of 11 confirmed NR2E3 patients with CPRD, ESCS, or GFS revealed functional defects of little or absent rod function, regardless of diagnosis [33]. The same study found that pathogenic NR2E3 variants accounted for approximately half of CPRD cases. In all NR2E3-associated disorders, there is degeneration of photoreceptors over time and patients often suffer from a progressive decline in vision [37,38]. However, follow-up assessments of some 50 patients have found that the best-corrected visual acuity remains stable over time for many patients [31]. In this study, the follow-up time ranged from 0 to 34 years, with a mean of 6.1 years.
ESCS, GFS, and CPRD are all linked to shared recessive biallelic NR2E3 variants, and patients show considerable clinical heterogeneity even when carrying identical mutations [39]. The majority of the NR2E3 pathogenic variants are found in either the DNA- or ligand-binding domains of the protein (Figure 2) [8,40]. For instance, the most common variant of NR2E3 found in patients is a missense mutation in exon 6, c.932G>A p.(Arg311Gln), which occurs in the ligand-binding domain [30,33]. However, there are exceptions, including one of the most common NR2E3 variants reported in the U.S., c.119-2A>C, which falls within the canonical splice acceptor site of intron 1 and has been shown to induce skipping on exon 2 [41]. It has not been possible to establish clear genotype–phenotype correlations among the recessively inherited NRE23 diseases [8,33,39,41,42]. A number of factors may contribute to the high phenotypic variability, including the complex interactions between NR2E3 and other molecules involved in photoreceptor cell fate determination, the presence of modifier genes, and environmental influences.
NR2E3 variants have also been identified as causing both autosomal recessive [43] and autosomal dominant RP [44]. RP is a common form of inherited retinal disease characterized by progressive loss of rod photoreceptors (presenting with nyctalopia and peripheral field loss) with subsequent cone degeneration, causing loss of central vision. In NR2E3-RP patients, night blindness is usually the first reported symptom starting in childhood or adolescence [43,44]. In typical RP, bone spicule-like pigment deposits are seen in the mid-peripheral retina; however, in some patients, clumped pigmentation has also been observed [37,43,45]. Retinal images taken from a patient with NR2E3-related autosomal dominant RP are shown in Figure 3D–F.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Published patient NR2E3 variants and associated phenotypes.
| Region | Variant | Mutation Type | Amino Acid Change | Protein Domain | Reported Phenotypes | References |
|---|---|---|---|---|---|---|
| Intron 1 | c.119-2A>C | Splicing | ESCS, GFS, CPRD, RP | [30,33,41] | ||
| c.119-3C>G | Splicing | ESCS | [46] | |||
| c.119-57_166 | Gross deletion | RP | [47] | |||
| Exon 1 | c.95G>A | Nonsense | p.Trp32 * | A/B | RP | [48] |
| Exon 2 | c.142C>T | Missense | p.Arg48Cys | DBD | ESCS | [40] |
| c.143_144delGCins25 | Indel/ Frameshift | p.Arg48Glufs*66 | DBD | RP | [49] | |
| c.145G>A | Missense | p.Val49Met | DBD | ESCS | [46] | |
| c.151G>A | Missense | p.Gly51Arg | DBD | ESCS | [40] | |
| c.166G>A | Missense | p.Gly56Arg | DBD | RP | [44] | |
| c.166G>C | Missense | p.Gly56Arg | DBD | RP | [50] | |
| c.188C>A | Missense | p.Ala63Asp | DBD | Cone–rod dystrophy | [51] | |
| c.194_202del | Deletion | p.Asn65_Cys67del | DBD | RP, ESCS | [30,52] | |
| c.196_201delGGCTGC | Deletion | p.Gly66_Cys67del | DBD | ESCS | [53] | |
| c.202A>G | Missense | p.Ser68Gly | DBD | ESCS | [54] | |
| c.211T>C | Missense | p.Phe71Leu | DBD | ESCS | [31] | |
| c.211_213delTTC | Deletion | p.Phe71del | DBD | ESCS | [42] | |
| c.223G>A | Missense | p.Val75Ile | DBD | RP | [55] | |
| c.226C>T | Missense | p.Arg76Trp | DBD | ESCS | [30] | |
| c.227G>A | Missense | p.Arg76Gln | DBD | ESCS | [30] | |
| c.242A>G | Missense | p.Tyr81Cys | DBD | ESCS | [46] | |
| Exon 3 | c.248G>A | Missense | p.Cys83Tyr | DBD | ESCS | [56] |
| c.263G>T | Missense | p.Gly88Val | DBD | ESCS | [57] | |
| c.290G>A | Missense | p.Arg97His | DBD | ESCS, RD | [30,58] | |
| c.305C>A | Missense | p.Ala102Asp | DBD | ESCS, RP, RD | [59,60,61] | |
| c.309C>A | Nonsense | p.Cys103 * | DBD | RP | [62] | |
| c.310C>T | Missense | p.Arg104Trp | DBD | ESCS | [30] | |
| c.311G>A | Missense | p.Arg104Gln | DBD | ESCS, RP, RD | [63,64,65] | |
| c.328C>T | Nonsense | p.Gln110 * | DBD | RD, RP | [16,66] | |
| c.328dupC | Insertion/ Frameshift | p.Gln110Profs*31 | DBD | GFS | [67] | |
| Exon 4 | c.352G>A | Missense | p.Val118Met | DBD | RP | [68] |
| c.364C>T | Missense | p.Arg122Cys | DBD | RP, RD | [16,69] | |
| c.371C>T | Missense | p.Pro124Leu | DBD | RP | [70] | |
| c.373C>T | Nonsense | p.Arg125 * | DBD | ESCS, RP | [71,72] | |
| c.406G>T | Nonsense | p.Glu136 * | DBD | RP | [66] | |
| c.481delA | Deletion/ Frameshift | p.Thr161Hisfs*18 | Hinge | RP, ESCS | [57,73] | |
| c.554delA | Deletion/ Frameshift | p.Lys185Serfs*66 | Hinge | RP | [74] | |
| Intron 4 | c.571+2T>C | Splicing | RP | [75] | ||
| Exon 5 | c.626dupA | Insertion/ Frameshift | p.Tyr209 * | LBD | RP | [74] |
| c.639_640insT | Insertion/ Frameshift | p.Pro214Serfs*9 | LBD | RD | [76] | |
| c.646G>A | Missense | p.Gly216Ser | LBD | GFS, RP | [72,77] | |
| c.701G>C | Missense | p.Trp234Ser | LBD | ESCS | [30] | |
| c.724_725delTC | Deletion/ Frameshift | p.Ser242Glnfs*17 | LBD | ESCS, RP, cone–rod dystrophy | [78,79,80] | |
| c.731delT | Deletion/ Frameshift | p.Leu244Argfs*7 | LBD | RP | [81] | |
| c.739C>T | Missense | p.Arg247Trp | LBD | ESCS | [31] | |
| Intron 5 | c.747+1G>C | Splicing | ESCS | [39] | ||
| Exon 6 | c.755T>C | Missense | p.Leu252Pro | LBD | ESCS | [82] |
| c.767C>A | Missense | p.Ala256Glu | LBD | ESCS, RD | [33,65] | |
| c.767C>T | Missense | p.Ala256Val | LBD | ESCS | [83] | |
| c.788T>C | Missense | p.Leu263Pro | LBD | ESCS | [57] | |
| c.790G>A | Missense | p.Gly264Arg | LBD | ESCS | [84] | |
| c.797T>A | Missense | p.Ile266Asn | LBD | RD | [61] | |
| c.808_809delCT | Deletion/ Frameshift | p.Leu270Alafs*70 | LBD | ESCS | [85] | |
| c.827_843del17 | Deletion/ Frameshift | p.Leu270Alafs*70 | LBD | ESCS, GFS, CPRD | [33] | |
| c.908T>C | Missense | p.Leu303Pro | LBD | ESCS | [31] | |
| c.919_920delAT | Deletion/ Frameshift | p.Ile307Leufs*33 | LBD | ESCS | [86] | |
| c.925C>G | Missense | p.Arg309Gly | LBD | ESCS | [30] | |
| c.925C>T | Missense | p.Arg309Trp | LBD | GFS | [87] | |
| c.926G>T | Missense | p.Arg309Leu | LBD | GFS | [72] | |
| c.926G>A | Missense | p.Arg309Gln | LBD | ESCS | [31] | |
| c.930C>G | Missense | p.Phe310Leu | LBD | ESCS | [88] | |
| c.931C>T | Missense | p.Arg311Trp | LBD | RD, RP | [66,89] | |
| c.932G>A | Missense | p.Arg311Gln | LBD | RP, ESCS, GFS, CPRD | [30,33,41,43] | |
| c.951delC | Deletion/ Frameshift | p.Thr318Argfs*6 | LBD | RP | [90] | |
| c.967dupA | Insertion/ Frameshift | p.Met323Asnfs*18 | LBD | RP | [52] | |
| Exon 7 | c.994G>A | Missense | p.Glu332Lys | LBD | ESCS | [59] |
| c.994G>T | Nonsense | p.Glu332 * | LBD | RD | [91] | |
| c.1000C>G | Missense | p.Arg334Gly | LBD | ESCS | [63] | |
| c.1007T>C | Missense | p.Leu336Pro | LBD | ESCS | [57] | |
| c.1018G>A | Missense | p.Glu340Lys | LBD | ESCS | [92] | |
| c.1025T>C | Missense | p.Val342Ala | LBD | ESCS | [59] | |
| c.1025T>G | Missense | p.Val342Gly | LBD | RP | [93] | |
| c.1034_1038delTGCAG | Deletion/ Frameshift | p.Leu345 * | LBD | RP | [41] | |
| c.1048C>G | Missense | p.Gln350Glu | LBD | RD | [76] | |
| c.1048C>T | Nonsense | p.Gln350 * | LBD | ESCS | [94] | |
| c.1049A>G | Missense | p.Gln350Arg | LBD | ESCS, RP | [42,95] | |
| c.1057C>G | Missense | p.Leu353Val | LBD | ESCS | [57] | |
| Intron 7 | c.1101-1G>A | Splicing | ESCS | [46] | ||
| Exon 8 | c.1112T>G | Missense | p.Leu371Trp | LBD | ESCS | [92] |
| c.1118T>C | Missense | p.Leu373Pro | LBD | ESCS, RD | [16,96] | |
| c.1154G>C | Missense | p.Arg385Pro | LBD | ESCS | [30] | |
| c.1171_1172delTT | Deletion/ Frameshift | p.Phe391Profs*15 | LBD | RP | [97] | |
| c.1184T>C | Missense | p.Ile395Thr | LBD | RP | [98] | |
| c.1194delT | Deletion/ Frameshift | p.Pro399Glnfs*79 | ESCS | [59] | ||
| c.1217A>G | Missense | p.Asp406Gly | GFS | [99] | ||
| c.1220T>A | Missense | p.Met407Lys | ESCS | [30] | ||
| c.1223delT | Deletion/ Frameshift | p.Phe408Serfs*7 | RD | [16] |
DBD, DNA-binding domain; LBD, ligand-binding domain; ESCS, enhanced S-cone syndrome; CPRD, clumped pigmentary retinal degeneration; GFS, Goldmann–Favre syndrome; RP, retinitis pigmentosa; RD, unspecified retinal dystrophy; * premature termination codon (PTC). The pathogenic variants are listed in the Human Gene Mutation Database (HGMD) Professional version, accessed on 16 January 2023.
There is a genotype–phenotype association for the autosomal dominant RP, with all cases linked to an NR2E3 missense variant, c.166G>A p.(Gly56Arg), which occurs in the first zinc finger of the DNA-binding domain [44]. This variant has been found to cause 1–2% of autosomal dominant RP in North America [51] and to have a frequency of 3.5% in a large Spanish cohort [37]. Functional analysis showed that the absence of DNA-binding but competition for dimer formation may explain the dominant negative activity exhibited by the p.(Gly56Arg) mutant protein [45]. Furthermore, the p.(Gly56Arg) mutant NR2E3 protein was found to show a distinct in vivo protein–protein interaction with CRX, comparable to that of wild-type NR2E3, and unlike the protein with biallelic recessive variants located in the DNA-binding domain [100]. Escher et al. investigated a family with autosomal dominant RP caused by the heterozygous p.(Gly56Arg) variant, in which two family members that carried compound heterozygous variants (p.Gly56Arg/p.Arg311Gln) had an ESCS-like phenotype [45]; it was suggested that the p.(Arg311Gln) variant may have a beneficial modifying effect on p.(Gly56Arg) due to increased photoreceptor-specific gene expression caused by impaired corepressor binding.
5. Animal Models
One commonly used NR2E3 animal model is the rd7 mouse, which was initially considered to have a naturally occurring recessively inherited 380 bp deletion in the coding region of Nr2e3, resulting in a frameshift premature stop codon [101]. However, Chen et al. later reported in 2006 a 10-fold increase in a 9 kb photoreceptor-specific Nr2e3 transcript, which was found to arise from the antisense insertion of a long interspersed nuclear element (LINE-1) (or L1) into exon 5. This L1 insertion subsequently blocks splicing, leading to incompletely spliced transcripts and their accumulation of mutant Nr2e3 in photoreceptor nuclei [102]. These mice suffer from progressive photoreceptor degeneration starting at 12 months and have a 1.5 to 2-fold increase in S-cone numbers [1]. At the age of 1 month, the outer nuclear layer of rd7 mouse retinas shows patterns of waves, whorls, and rosettes, which gradually disappear between 5 and 16 months [101]. ERGs are normal until 5 months, after which there is a progressive reduction in signals for both rods and cones. A mottled pigment can be seen in the retina at the age of 16 months, along with a reduction in outer nuclear layer thickness. The retinas of these mice also included some cells that are intermediates between rods and cones [1]. Cheng et al. found that 50% of cells expressing S-opsin in the rd7 mouse also express Nrl, which is not seen in wild-type mice. In the rd7 retina, cells that should develop into rods show downregulated expression of rod-specific genes (such as Rho, Gnb1, and Pde6b) and upregulated expression of cone-specific genes (such as Opnsw, Gnb3, and Pde6c) compared with those of wild-type [18].
A recent longitudinal study using spectral domain optical coherence tomography (SD-OCT) to compare the retinas of rd7 mice and ESCS patients found that the disease progression correlates well between the two species, and identified characteristics on the patient scans that may be equivalent to the whorls and rosettes seen in mice [103].
An additional Nr2e3 knockout mouse model (Nr2e3−/−) mouse was previously generated through the ablation of exons 1–6, showing a phenotype and gene expression profile similar to the rd7 mouse [104]. In recent years, two new mouse models have been generated by Aísa-Marín et al. using the CRISPR/Cas9 D10A nickase system [2]. Allele Δ27 was an in-frame deletion of 27 bp in exon 8 that ablates the dimerization domain, whereas allele ΔE8 (full deletion of exon 8) produced only the short isoform, which lacks the C-terminal part of the LBD involved in repressor activity.
Both models showed retinal invaginations similar to the rosettes found in the rd7 mouse; however, the ΔE8 model displayed an RP-like phenotype with progressive retinal degeneration, while Δ27 had a more ESCS-like disease with developmental defects.
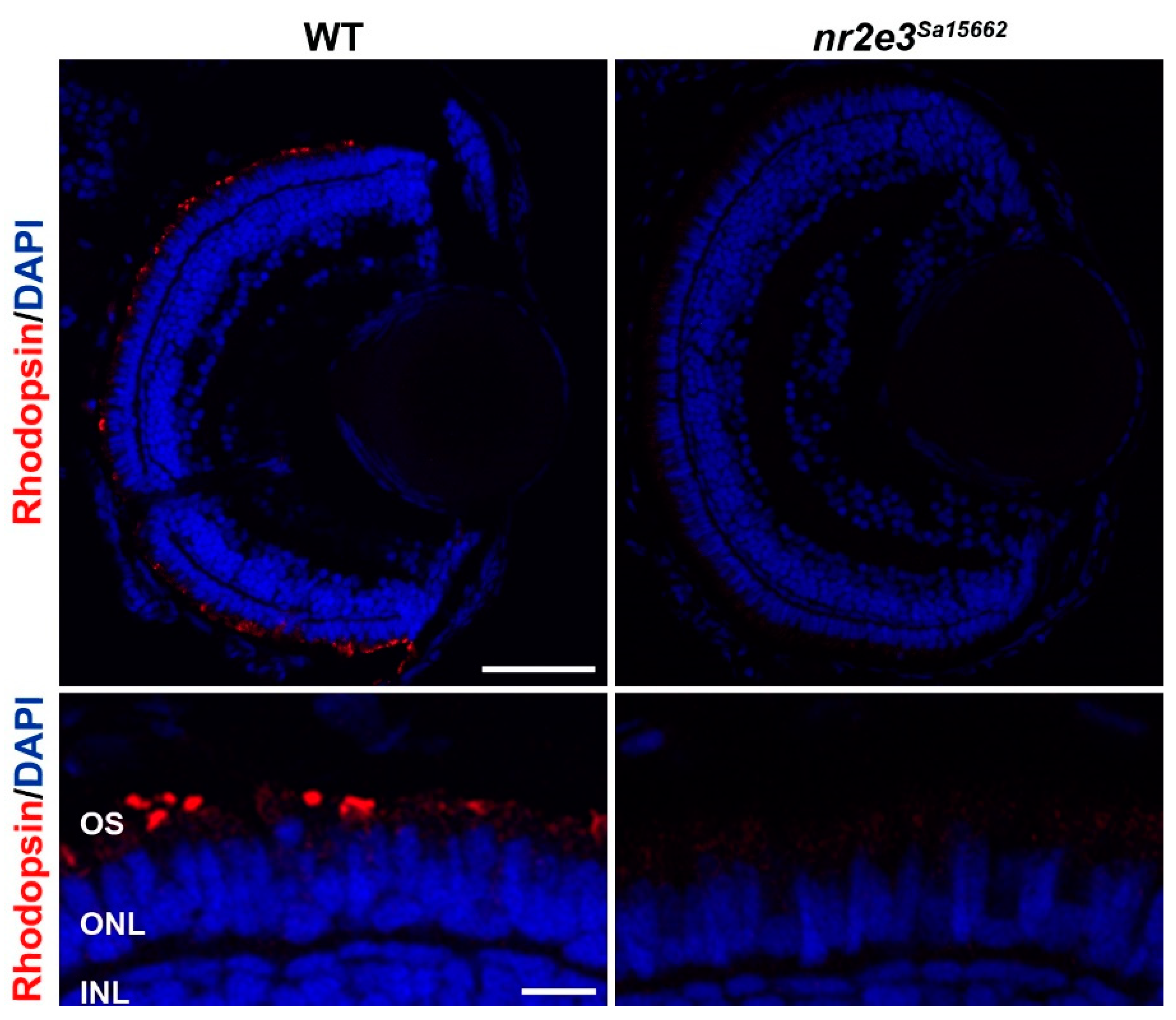
Zebrafish have been used to study the role of NR2E3. Xie et al. used CRISPR/Cas9 to create an nr2e3 knockout line with a 37 bp deletion c.485_521del, causing a frameshift premature stop codon (p.Leu162Glnfs*30) [105]. These fish showed no rhodopsin expression and a lack of rod photoreceptors at 10 days post fertilization, which were still absent at ages 6 and 10 months. Other rod-specific genes, such as gnat1 and pde6b, were not expressed at the mRNA or protein level. These fish were found to suffer from selective degeneration of L- and M-cones, with their outer segments beginning to shorten around the age of 1 month. However, the variant appeared to have no effect on the number of UV- or S-cones. The nr2e3 Sanger zebrafish line Sa15662 harboring a nonsense mutation in exon 6 (c.1036A>T, p.[Lys346*]) also shows an absence of rod photoreceptors (Figure 4) (unpublished data).
As in humans, the expression of nr2e3/Nr2e3 in zebrafish and mice is largely confined to the photoreceptors, although in zebrafish the expression of nr2e3 is transiently expressed in both rod and cone precursors during early photoreceptor development [105]. Other than the initially normal ERGs, the mouse rd7 model appears to phenocopy human patients with NR2E3 pathogenic variants in a fairly faithful manner [38]. The zebrafish model does not mimic the human disease as closely, as it does not show any increase in S- or UV-cone development. However, the lack of rod cell development and the progressive degeneration of L- and M-cones are reminiscent of human patients with NR2E3 variants. The differences between human patients, rd7 mice, and zebrafish nr2e3 models are most likely due to the evolutionary history of rods and cones. Mammalian S-cones are thought to be most closely related to teleost UV-cones, whereas teleost M-cones are evolutionarily closer to the mammalian rods than cones [11].
6. Treatments
Although there are currently no approved therapies for the primary genetic defects associated with NR2E3, several strategies for treating NR2E3-related retinal disease and other inherited retinopathies are currently under development and have shown promise. The treatment strategy will likely depend on the clinical phenotype, with developmental effects on the ESCS spectrum, such as the low number or absence of rod photoreceptors, posing a greater challenge as the ideal window for therapeutic intervention may be prenatal. In contrast, the late onset of RP provides a longer and more accessible time period for potential intervention.
One of the leading treatment avenues being pursued for many inherited retinal diseases is the use of viral gene therapy for the replacement of the defective gene. A phase 1/2 clinical trial (NCT05203939) has commenced testing AAV-NR2E3 gene therapy (OCU400) in adults with autosomal recessive and dominant NR2E3 retinopathy. This is based on the work of Li et al. [26], who investigated the use of AAV vectors to over-express Nr2e3 in 5 different mouse models of RP, via subretinal injection in neonatal mice or adult mice. This method was found to reduce retinal degeneration caused by mutations in several genes, as well as NR2E3, highlighting this as a potential broad-spectrum therapy for multiple retinopathies. In addition, subretinal delivery of Nr1d1 using expression constructs was able to ameliorate retinal degeneration in the rd7 mouse, further demonstrating the beneficial effects of modifier genes in mediating disease progression [106]. Interestingly, intravitreal injection of Nr2e3 antagonist photoregulin-1 has been shown to prevent photoreceptor death in rod degeneration mouse models Pde6brd1 and RhoP23H [107], with similar results being found in knockout experiments of Nrl in the adult retina [108]. This demonstrates the marked difference in roles of Nr2e3 in the developing versus adult retinas.
A potential agnostic gene therapy approach for patients suffering from advanced stages of RP is the use of optogenetics, which involves the transduction of light-sensitive ion channels called channel rhodopsin or related photosensitive molecules, which can open existing ion channels on the remaining retinal cells, including ganglion and bipolar cells [109,110]. Clinical trials are underway (NCT02556736; NCT03326336; NCT04945772). However, while these methods may restore some measure of light sensitivity to patients, they would be unlikely to result in high-resolution vision.
Preclinically, CRISPR/Cas9 gene editing was used to correct NR2E3 pathogenic variants in induced pluripotent stem cells (iPSCs) generated from two patients with ESCS, using a homology-directed repair (HDR)-based strategy [111]. Patient 1 carried a homozygous c.119-2A>C splice site variant, and patient 2 had compound heterozygous p.(Arg73Ser) and p.(Arg311Gln) variants. They achieved relatively high efficiency (~72–83% of clones initially screened showed incorporation of HDR cassette) with minimal off-target mutagenesis. As most of the NR2E3 pathogenic variants are point mutations or small deletions rather than large indels or complex rearrangements, CRISPR/Cas9 may be applicable to NR2E3-related diseases in the future.
The frequent autosomal dominant NR2E3 RP-associated variant, p.(Gly56Arg), was targeted using antisense oligonucleotides (AONs), which were designed to bind and silence expression of the mutant mRNA transcripts by inducing RNAse-H1 cleavage [112]. Wild-type or mutant NR2E3 was over-expressed in RPE-1 cells before treatment with the AONs, all of which showed a general knock-down in both the mutant and wild-type NR2E3 at the mRNA and protein level, although a preferential mutant protein-specific knock-down was observed for most of the AONs. While this investigation showed the accessibility of the region for AON-induced knockdown, further modifications are needed to increase allele-specificity to ensure this is an effective therapeutic approach in the future.
Although it is unlikely that the gene-based therapies described would reverse developmental photoreceptor defects in adults with ESCS-type disease, NR2E3 continues to be expressed in adult life, and boosting its function or that of its modifier genes with such treatments might help to maintain normal photoreceptor function and prolong survival. Alternatively, in patients with ESCS or late-stage RP, stem cell therapies to replace photoreceptor loss may be the most suitable future option [113,114]. However, it may not be possible for implanted cells to form proper synaptic connections in the retinas of patients with developmental defects.
While there is currently no approved treatment for genetic defects underlying NR2E3 disorders, the associated disease complications, such as hypermetropia and retinoschisis, should be monitored and management offered where appropriate to reduce further sight loss. For instance, macular retinoschisis and cystoid macular edema can be effectively treated using the oral carbonic anhydrase inhibitor, acetazolamide [115,116].
7. Future Perspectives
In spite of the variety of studies conducted on NR2E3 and its role in the developing and mature retina, there are still significant gaps in our understanding. The high levels of disease heterogeneity, particularly among patients with recessive mutations, demonstrate the complex interactions of this gene that have yet to be understood. A greater understanding of the underlying disease mechanisms and genotype–phenotype correlations is needed to better inform genetic counseling and the most effective treatment approaches. Further investigation into the function of NR2E3 will be aided by the use of iPSCs, which have already been generated from patients with NR2E3 pathogenic variants. iPSC-derived retinal organoids have recapitulated the enhanced S-cone phenotype and have been used for in vitro therapeutic studies [111,117,118,119]. For in vivo disease modeling, the CRISPR/Cas9 system could be used to create higher-order animal models harboring specific mutations, which can further aid in pre-clinical therapeutic work. In recent years, NR2E3 has been highlighted as an important modifier of retinal disease and, in addition to the treatment of patients with NR2E3-related conditions, NR2E3 gene supplementation could be pursued as a broad-spectrum therapy for various other retinopathies, which is particularly promiseng for the significant proportion of patients that remain without a molecular diagnosis.
Author Contributions
Data curation, N.W and M.T..; writing—original draft preparation, N.W and M.T.; writing—review & editing M.T., N.W. and M.M.; visualization, N.W. and M.M.; funding acquisition, M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Wellcome Trust 205174/Z/16/Z and Retina UK (GR598).
Institutional Review Board Statement
The contents of this review was conducted in accordance with the Declaration of Helsinki, and approved by the National Ethics Committee (London—Camden & Kings Cross Research Ethics Committee) (protocol code 12/LO/0141 and date of approval: 30 September 2022). The animal procedures and protocols for this review was approved by the UCL Animal Welfare and Ethical Review Body, in addition to the UK Home Office (License no. PPL PC916FDE7 on 10 January 2023). All approved standard protocols followed the guidelines of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research Ethics.
Informed Consent Statement
All images are anonymous and written informed consent was provided by participants under the above ethics.
Data Availability Statement
This is a review so previous data is referenced.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, J.; Rattner, A.; Nathans, J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J. Neurosci. 2005, 25, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aísa-Marín, I.; López-Iniesta, M.J.; Milla, S.; Lillo, J.; Navarro, G.; de la Villa, P.; Marfany, G. Nr2e3 functional domain ablation by CRISPR-Cas9D10A identifies a new isoform and generates retinitis pigmentosa and enhanced S-cone syndrome models. Neurobiol. Dis. 2020, 146, 105122. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, V.C.; Zaidi, Q.; Hood, D.C.; Spehar, B.; Cideciyan, A.V.; Jacobson, S.G. The Enhanced S Cone Syndrome: An Analysis of Receptoral and Post-receptoral Changes. Vis. Res. 1996, 36, 3711–3722. [Google Scholar] [CrossRef] [Green Version]
- Hood, D.C.; Cideciyan, A.V.; Roman, A.J.; Jacobson, S.G. Enhanced S cone syndrome: Evidence for an abnormally large number of S cones. Vis. Res. 1995, 35, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.-X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, N.B.; Naggert, J.K.; Nishina, P.M. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum. Mol. Genet. 2001, 10, 1619–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.E.; Zhou, X.E.; Soon, F.F.; Li, X.; Li, J.; Yong, E.L.; Melcher, K.; Xu, H.E. The crystal structure of the orphan nuclear receptor NR2E3/PNR ligand binding domain reveals a dimeric auto-repressed conformation. PLoS ONE 2013, 8, e74359. [Google Scholar] [CrossRef] [Green Version]
- Schorderet, D.F.; Escher, P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum. Mutat. 2009, 30, 1475–1485. [Google Scholar] [CrossRef]
- Mollema, N.; Haider, N.B. Focus on Molecules: Nuclear hormone receptor Nr2e3: Impact on retinal development and disease. Exp. Eye Res. 2010, 91, 116–117. [Google Scholar] [CrossRef]
- Von Alpen, D.; Tran, H.V.; Guex, N.; Venturini, G.; Munier, F.L.; Schorderet, D.F.; Haider, N.B.; Escher, P. Differential Dimerization of Variants Linked to Enhanced S-Cone Sensitivity Syndrome (ESCS) Located in the NR2E3 Ligand-Binding Domain. Hum. Mutat. 2015, 36, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Bowmaker, J.K. Evolution of colour vision in vertebrates. Eye 1998, 12, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Mustafi, D.; Engel, A.H.; Palczewski, K. Structure of cone photoreceptors. Prog. Retin. Eye Res. 2009, 28, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Lamb, T.D. Why rods and cones? Eye 2016, 30, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Calkins, D.J. Seeing with S cones. Prog. Retin. Eye Res. 2001, 20, 255–287. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Allen, K.A.; Sloan, K.R.; Lerea, C.L.; Hurley, J.B.; Klock, I.B.; Milam, A.H. Distribution and morphology of human cone photoreceptors stained with anti-blue opsin. J. Comp. Neurol. 1991, 312, 610–624. [Google Scholar] [CrossRef]
- Murro, V.; Mucciolo, D.P.; Sodi, A.; Passerini, I.; Giorgio, D.; Virgili, G.; Rizzo, S. Novel clinical findings in autosomal recessive NR2E3-related retinal dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Mollema, N.; Gaule, M.; Yuan, Y.; Sachs, A.J.; Nystuen, A.M.; Naggert, J.K.; Nishina, P.M. Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction. Exp. Eye Res. 2009, 89, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Khan, N.W.; Roger, J.E.; Swaroop, A. Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum. Mol. Genet. 2011, 20, 4102–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, L.L.; Lillo, C.; Lyubarsky, A.L.; Nikonov, S.S.; Philp, N.; Mears, A.J.; Swaroop, A.; Williams, D.S.; Pugh, E.N. Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2156–2167. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.M.B.; Schulte, D.; Hendrickson, A.E. Expression of photoreceptor-associated molecules during human fetal eye development. Mol. Vis. 2003, 9, 401–409. [Google Scholar]
- Cheng, H.; Khanna, H.; Oh, E.C.; Hicks, D.; Mitton, K.; Swaroop, A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 2004, 13, 1563–1575. [Google Scholar] [CrossRef]
- Freund, C.L.; Wang, Q.-L.; Chen, S.; Muskat, B.L.; Wiles, C.D.; Sheffield, V.C.; Jacobson, S.G.; Mclnnes, R.R.; Zack, D.J.; Stone, E.M. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat. Genet. 1998, 18, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.S.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.-H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef] [Green Version]
- Olivares, A.M.; Jelcick, A.S.; Reinecke, J.; Leehy, B.; Haider, A.; Morrison, M.A.; Cheng, L.; Chen, D.F.; DeAngelis, M.M.; Haider, N.B. Multimodal Regulation Orchestrates Normal and Complex Disease States in the Retina. Sci. Rep. 2017, 7, 690. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Datta, S.; Brabbit, E.; Love, Z.; Woytowicz, V.; Flattery, K.; Capri, J.; Yao, K.; Wu, S.; Imboden, M.; et al. Nr2e3 is a genetic modifier that rescues retinal degeneration and promotes homeostasis in multiple models of retinitis pigmentosa. Gene Ther. 2021, 28, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanal, T.; Leung, Y.-K.; Jiang, W.; Timchenko, N.; Ho, S.-M.; Kim, K. NR2E3 is a key component in p53 activation by regulating a long noncoding RNA DINO in acute liver injuries. FASEB J. 2019, 33, 8335–8348. [Google Scholar] [CrossRef] [PubMed]
- Khanal, T.; Choi, K.; Leung, Y.-K.; Wang, J.; Kim, D.; Janakiram, V.; Cho, S.-G.; Puga, A.; Ho, S.-M.; Kim, K. Loss of NR2E3 represses AHR by LSD1 reprogramming, is associated with poor prognosis in liver cancer. Sci. Rep. 2017, 7, 10662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Kim, K.; Kim, S.; Hennessy, B.T.; Kim, S.M.; Park, E.S.; Lim, J.Y.; Li, J.; Lu, Y.; Gonzalez-Angulo, A.M.; et al. Reconstruction of nuclear receptor network reveals that NR2E3 is a novel upstream regulator of ESR1 in breast cancer. EMBO Mol. Med. 2012, 4, 52–67. [Google Scholar] [CrossRef]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef]
- de Carvalho, E.R.; Robson, A.G.; Arno, G.; Boon, C.J.; Webster, A.A.; Michaelides, M. Enhanced S-Cone Syndrome: Spectrum of Clinical, Imaging, Electrophysiologic, and Genetic Findings in a Retrospective Case Series of 56 Patients. Ophthalmol. Retina 2021, 5, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Jacobson, S.G.; Foerster, M.H.; Kellner, U.; Weleber, R.G. Diagnostic Clinical Findings of a New Syndrome with Night Blindness, Maculopathy, and Enhanced S Cone Sensitivity. Am. J. Ophthalmol. 1990, 110, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Sandberg, M.A.; Caruso, R.C.; Berson, E.L.; Dryja, T.P. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch. Ophthalmol. 2003, 121, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, S.G.; Román, A.J.; Román, M.I.; Gass, J.D.M.; Parker, J.A. Relatively Enhanced S Cone Function in the Goldmann-Favre Syndrome. Am. J. Ophthalmol. 1991, 111, 446–453. [Google Scholar] [CrossRef]
- Fishman, G.A.; Jampol, L.M.; Goldberg, M.F. Diagnostic features of the Favre-Goldmann syndrome. Br. J. Ophthalmol. 1976, 60, 345–353. [Google Scholar] [CrossRef] [Green Version]
- To, K.W.; Adamian, M.; Jakobiec, F.A.; Berson, E.L. Clinical and Histopathologic Findings in Clumped Pigmentary Retinal Degeneration. Arch. Ophthalmol. 1996, 114, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Kelly, F.; Hoyos, M.G.; Martinez, M.A.L.; Lopez-Molina, M.I.; Riveiro-Alvarez, R.; Jose, P.F.-S.; Avila-Fernandez, A.; Corton, M.; Millan, J.M.; Sandoval, B.G.; et al. Dominant Retinitis Pigmentosa, p.Gly56Arg Mutation in NR2E3: Phenotype in a Large Cohort of 24 Cases. PLoS ONE 2016, 11, e0149473. [Google Scholar] [CrossRef] [Green Version]
- Garafalo, A.V.; Calzetti, G.; Cideciyan, A.V.; Roman, A.J.; Saxena, S.; Sumaroka, A.; Choi, W.; Wright, A.F.; Jacobson, S.G. Cone Vision Changes in the Enhanced S-Cone Syndrome Caused by NR2E3Gene Mutations. Investig. Opthalmol. Vis. Sci. 2018, 59, 3209–3219. [Google Scholar] [CrossRef] [Green Version]
- Bandah, D.; Merin, S.; Ashhab, M.; Banin, E.; Sharon, D. The Spectrum of Retinal Diseases Caused by NR2E3 Mutations in Israeli and Palestinian Patients. Arch. Ophthalmol. 2009, 127, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Kuniyoshi, K.; Hayashi, T.; Sakuramoto, H.; Nakao, A.; Sato, T.; Utsumi, T.; Tsuneoka, H.; Shimomura, Y. Novel Mutations in Enhanced S-cone Syndrome. Ophthalmology 2013, 120, 431.e1-6. [Google Scholar] [CrossRef]
- Bernal, S.; Solans, T.; Gamundi, M.; Hernan, I.; De Jorge, L.; Carballo, M.; Navarro, R.; Tizzano, E.; Ayuso, C.; Baiget, M. Analysis of the involvement of the NR2E3 gene in autosomal recessive retinal dystrophies. Clin. Genet. 2008, 73, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Pachydaki, S.I.; Klaver, C.; Barbazetto, I.A.; Roy, M.S.; Gouras, P.; Allikmets, R.; Yannuzzi, L.A. Phenotypic features of patients with NR2E3 mutations. Arch Ophthalmol. 2009, 127, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, S.; Rozet, J.M.; Takezawa, S.I.; Coutinho dos Santos, L.; Lopes, L.; Gribouval, O.; Penet, C.; Perrault, I.; Ducroq, D.; Souied, E.; et al. The photoreceptor cell-specific nuclear receptor gene (PNR) accounts for retinitis pigmentosa in the Crypto-Jews from Portugal (Marranos), survivors from the Spanish Inquisition. Hum. Genet. 2000, 107, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. Recurrent Mutation in the First Zinc Finger of the Orphan Nuclear Receptor NR2E3 Causes Autosomal Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Escher, P.; Gouras, P.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.-C.; Hayashi, M.; Zernant, J.; Merriam, J.E.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Audo, I.; Michaelides, M.; Robson, A.G.; Hawlina, M.; Vaclavik, V.; Sandbach, J.M.; Neveu, M.M.; Hogg, C.R.; Hunt, D.M.; Moore, A.T.; et al. Phenotypic Variation in Enhanced S-cone Syndrome. Investig. Opthalmol. Vis. Sci. 2008, 49, 2082–2093. [Google Scholar] [CrossRef]
- van Huet, R.A.; Pierrache, L.H.; Meester-Smoor, M.A.; Klaver, C.C.; van den Born, L.I.; Hoyng, C.B.; de Wijs, I.J.; Collin, R.W.; Hoefsloot, L.H.; Klevering, B.J. The efficacy of microarray screening for autosomal recessive retinitis pigmentosa in routine clinical practice. Mol. Vis. 2015, 21, 461–476. [Google Scholar]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Kannabiran, C.; Singh, H.; Sahini, N.; Jalali, S.; Mohan, G. Mutations in TULP1, NR2E3, and MFRP genes in Indian families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2012, 18, 1165–1174. [Google Scholar]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef]
- I Gire, A.; Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2007, 13, 1970–1975. [Google Scholar] [PubMed]
- Bravo-Gil, N.; Méndez-Vidal, C.; Romero-Pérez, L.; Pozo, M.G.-D.; la Rúa, E.R.-D.; Dopazo, J.; Borrego, S.; Antiñolo, G. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci. Rep. 2016, 6, 23910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udar, N.; Small, K.; Chalukya, M.; Silva-Garcia, R.; Marmor, M. Developmental or degenerative—NR2E3 gene mutations in two patients with enhanced S cone syndrome. Mol. Vis. 2011, 17, 519–525. [Google Scholar]
- Park, S.P.; Hong, I.H.; Tsang, S.H.; Lee, W.; Horowitz, J.; Yzer, S.; Allikmets, R.; Chang, S. Disruption of the human cone photoreceptor mosaic from a defect in NR2E3 transcription factor function in young adults. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 2299–2309. [Google Scholar] [CrossRef]
- Maeda, A.; Yoshida, A.; Kawai, K.; Arai, Y.; Akiba, R.; Inaba, A.; Takagi, S.; Fujiki, R.; Hirami, Y.; Kurimoto, Y.; et al. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018, 62, 451–457. [Google Scholar] [CrossRef]
- Rocha-Sousa, A.; Hayashi, T.; Gomes, N.L.; Penas, S.; Brandão, E.; Rocha, P.; Urashima, M.; Yamada, H.; Tsuneoka, H.; Falcão-Reis, F. A novel mutation (Cys83Tyr) in the second zinc finger of NR2E3 in enhanced S-cone syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Reddick, A.C.; Schwartz, S.B.; Ferguson, J.S.; Aleman, T.S.; Kellner, U.; Jurklies, B.; Schuster, A.; Zrenner, E.; Wissinger, B.; et al. Mutation analysis ofNR2E3 andNRL genes in Enhanced S Cone Syndrome. Hum. Mutat. 2004, 24, 439. [Google Scholar] [CrossRef]
- Huang, X.-F.; Huang, F.; Wu, K.-C.; Wu, J.; Chen, J.; Pang, C.-P.; Lu, F.; Qu, J.; Jin, Z.-B. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Hull, S.; Arno, G.; Sergouniotis, P.I.; Tiffin, P.; Borman, A.D.; Chandra, A.; Robson, A.; Holder, G.E.; Webster, A.R.; Moore, A.T. Clinical and Molecular Characterization of Enhanced S-Cone Syndrome in Children. JAMA Ophthalmol. 2014, 132, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Lingao, M.D.; Ganesh, A.; Karthikeyan, A.S.; Al Zuhaibi, S.; Al-Hosni, A.; Al Khayat, A.; Capasso, J.; Trumler, A.A.; Stroh, E.; Al Shekaili, H.; et al. Macular cystoid spaces in patients with retinal dystrophy. Ophthalmic Genet. 2016, 37, 377–383. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Gekka, T.; Goto-Omoto, S.; Takeuchi, T.; Kubo, A.; Kitahara, K. Novel NR2E3 mutations (R104Q, R334G) associated with a mild form of enhanced S-cone syndrome demonstrate compound heterozygosity. Ophthalmology 2005, 112, 2115. [Google Scholar] [CrossRef]
- Kimchi, A.; Khateb, S.; Wen, R.; Guan, Z.; Obolensky, A.; Beryozkin, A.; Kurtzman, S.; Blumenfeld, A.; Pras, E.; Jacobson, S.G.; et al. Nonsyndromic Retinitis Pigmentosa in the Ashkenazi Jewish Population: Genetic and Clinical Aspects. Ophthalmology 2018, 125, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, M.; Duignan, E.; Malone, C.P.G.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-Based Population Next-Generation Sequencing for Inherited Retinal Degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, X.; Chen, L.J.; Chiang, S.W.; Tam, P.O.; Lai, T.Y.; Chan, C.K.; Wang, N.; Lam, D.S.; Pang, C.P. Association of NR2E3 but not NRL mutations with retinitis pigmentosa in the Chinese population. Investigative Ophthalmol. Vis. Sci. 2010, 51, 2229–2235. [Google Scholar] [CrossRef] [Green Version]
- Bocquet, B.; Marzouka, N.A.D.; Hebrard, M.; Manes, G.; Sénéchal, A.; Meunier, I.; Hamel, C.P. Homozygosity mapping in autosomal recessive retinitis pigmentosa families detects novel mutations. Mol. Vis. 2013, 19, 2487–2500. [Google Scholar]
- Takahashi, V.K.L.; Xu, C.L.; Takiuti, J.T.; Apatoff, M.B.L.; Duong, J.K.; Mahajan, V.B.; Tsang, S.H. Comparison of structural progression between ciliopathy and non-ciliopathy associated with autosomal recessive retinitis pigmentosa. Orphanet J. Rare Dis. 2019, 14, 187. [Google Scholar] [CrossRef] [Green Version]
- Cassiman, C.; Spileers, W.; De Baere, E.; De Ravel, T.; Casteels, I. Peculiar fundus abnormalities and pathognomonic electrophysiological findings in a 14-month-old boy with NR2E3 mutations. Ophthalmic Genet. 2013, 34, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.-C.C.; Yu, H.-C.; Martin, R.; Cirulli, E.T.; Schenker-Ahmed, N.M.; Hicks, M.; Cohen, I.V.; Jönsson, T.J.; Heister, R.; Napier, L.; et al. Precision medicine integrating whole-genome sequencing, comprehensive metabolomics, and advanced imaging. Proc. Natl. Acad. Sci. USA 2020, 117, 3053–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, D.; Votruba, M. A novel NR2E3 gene mutation in autosomal recessive retinitis pigmentosa with cystic maculopathy. Acta Ophthalmol. 2018, 96, e535–e536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khuzaei, S.; Broadgate, S.; Halford, S.; Jolly, J.K.; Shanks, M.; Clouston, P.; Downes, S.M. Novel Pathogenic Sequence Variants in NR2E3 and Clinical Findings in Three Patients. Genes 2020, 11, 1288. [Google Scholar] [CrossRef]
- Seo, G.H.; Kim, T.; Choi, I.H.; Park, J.Y.; Lee, J.; Kim, S.; Won, D.G.; Oh, A.; Lee, Y.; Choi, J.; et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020, 98, 562–570. [Google Scholar] [CrossRef]
- Collin, R.W.J.; Born, L.I.V.D.; Klevering, B.J.; de Castro-Miró, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-Resolution Homozygosity Mapping Is a Powerful Tool to Detect Novel Mutations Causative of Autosomal Recessive RP in the Dutch Population. Investig. Opthalmol. Vis. Sci. 2011, 52, 2227–2239. [Google Scholar] [CrossRef] [Green Version]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Haer-Wigman, L.; Van Zelst-Stams, W.A.G.; Pfundt, R.; Born, L.I.V.D.; Klaver, C.; Verheij, J.B.G.M.; Hoyng, C.B.; Breuning, M.H.; Boon, C.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Cottriall, C.L.; Jolly, J.K.; Shanks, M.; Clouston, P.; Issa, P.C.; MacLaren, R.E. Electrophysiological verification of enhanced S-cone syndrome caused by a novel c.755T>C NR2E3 missense variant. Ophthalmic Genet. 2019, 40, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Goldberg, J.L.; Hartley, K.L.; Stone, E.M.; Liu, M. Atypical Mild Enhanced S-Cone Syndrome with Novel Compound Heterozygosity of the NR2E3 Gene. Am. J. Ophthalmol. 2007, 144, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Termühlen, J.; Alex, A.F.; Glöckle, N.; Kellner, U.; Fiedler, B.; Eter, N.; Uhlig, C.E. A new mutation in enhanced S-cone syndrome. Acta Ophthalmol. 2018, 96, e539–e540. [Google Scholar] [CrossRef] [PubMed]
- Collison, F.T.; Park, J.C.; Fishman, G.A.; Stone, E.M.; McAnany, J.J. Two-color pupillometry in enhanced S-cone syndrome caused by NR2E3 mutations. Doc. Ophthalmol. 2016, 132, 157–166. [Google Scholar] [CrossRef]
- Kuniyoshi, K.; Hayashi, T.; Sakuramoto, H.; Mishima, H.; Tsuneoka, H.; Tsunoda, K.; Iwata, T.; Shimomura, Y. New truncation mutation of the NR2E3 gene in a Japanese patient with enhanced S-cone syndrome. Jpn. J. Ophthalmol. 2016, 60, 476–485. [Google Scholar] [CrossRef]
- Bai, Z.; Xie, Y.; Liu, L.; Shao, J.; Liu, Y.; Kong, X. Genetic investigation of 211 Chinese families expands the mutational and phenotypical spectra of hereditary retinopathy genes through targeted sequencing technology. BMC Med. Genom. 2021, 14, 92. [Google Scholar] [CrossRef]
- Bechet, L.; Atia, R.; Zeitz, C.; Mohand-Saïd, S.; Sahel, J.-A.; Barale, P.-O.; Audo, I. Management of a case of Enhanced S-cone syndrome with massive foveoschisis treated with pars plana vitrectomy with silicone oil tamponade. Ophthalmic Genet. 2021, 42, 615–618. [Google Scholar] [CrossRef]
- Patel, N.; Alkuraya, H.; Alzahrani, S.; Nowailaty, S.; Seidahmed, M.; Alhemidan, A.; Ben-Omran, T.; Ghazi, N.; Al-Aqeel, A.; Al-Owain, M.; et al. Mutations in known disease genes account for the majority of autosomal recessive retinal dystrophies. Clin. Genet. 2018, 94, 554–563. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Ripamonti, C.; Aboshiha, J.; Henning, G.B.; Sergouniotis, P.I.; Michaelides, M.; Moore, A.T.; Webster, A.R.; Stockman, A. Vision in Observers with Enhanced S-Cone Syndrome: An Excess of S-Cones but Connected Mainly to Conventional S-Cone Pathways. Investig. Opthalmol. Vis. Sci. 2014, 55, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemiatkowska, A.M.; Arimadyo, K.; Moruz, L.M.; Astuti, G.D.; De Castro-Miro, M.; Zonneveld, M.N.; Strom, T.M.; De Wijs, I.J.; Hoefsloot, L.H.; Faradz, S.M.; et al. Molecular genetic analysis of retinitis pigmentosa in Indonesia using genome-wide homozygosity mapping. Mol. Vis. 2011, 17, 3013–3024. [Google Scholar] [PubMed]
- Nakamura, Y.; Hayashi, T.; Kozaki, K.; Kubo, A.; Omoto, S.; Watanabe, A.; Toda, K.; Takeuchi, T.; Gekka, T.; Kitahara, K. Enhanced S-cone syndrome in a Japanese family with a nonsense NR2E3 mutation (Q350X). Acta Ophthalmol. Scand. 2004, 82, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Maltese, P.E.; Colombo, L.; Martella, S.; Rossetti, L.; El Shamieh, S.; Sinibaldi, L.; Passarelli, C.; Coppè, A.M.; Buzzonetti, L.; Falsini, B.; et al. Genetics of Inherited Retinal Diseases in Understudied Ethnic Groups in Italian Hospitals. Front. Genet. 2022, 13, 914345. [Google Scholar] [CrossRef]
- Minnella, A.M.; Pagliei, V.; Savastano, M.C.; Federici, M.; Bertelli, M.; Maltese, P.E.; Placidi, G.; Corbo, G.; Falsini, B.; Caporossi, A. Swept source optical coherence tomography and optical coherence tomography angiography in pediatric enhanced S-cone syndrome: A case report. J. Med. Case Rep. 2018, 12, 287. [Google Scholar] [CrossRef]
- Song, F.; Owczarek-Lipska, M.; Ahmels, T.; Book, M.; Aisenbrey, S.; Menghini, M.; Barthelmes, D.; Schrader, S.; Spital, G.; Neidhardt, J. High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies. Genes 2021, 12, 1269. [Google Scholar] [CrossRef]
- Gao, F.-J.; Li, J.-K.; Chen, H.; Hu, F.-Y.; Zhang, S.-H.; Qi, Y.-H.; Xu, P.; Wang, D.-D.; Wang, L.-S.; Chang, Q.; et al. Genetic and Clinical Findings in a Large Cohort of Chinese Patients with Suspected Retinitis Pigmentosa. Ophthalmology 2019, 126, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Manayath, G.J.; Namburi, P.; Periasamy, S.; Kale, J.A.; Narendran, V.; Ganesh, A. A novel mutation in the NR2E3 gene associated with Goldmann-Favre syndrome and vasoproliferative tumor of the retina. Mol. Vis. 2014, 20, 724–731. [Google Scholar]
- Roduit, R.; Escher, P.; Schorderet, D.F. Mutations in the DNA-binding domain of NR2E3 affect in vivo dimerization and interaction with CRX. PLoS ONE 2009, 4, e7379. [Google Scholar] [CrossRef]
- Akhmedov, N.B.; Piriev, N.I.; Chang, B.; Rapoport, A.L.; Hawes, N.L.; Nishina, P.M.; Nusinowitz, S.; Heckenlively, J.R.; Roderick, T.H.; Kozak, C.A.; et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 5551–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Rattner, A.; Nathans, J. Effects of L1 retrotransposon insertion on transcript processing, localization and accumulation: Lessons from the retinal degeneration 7 mouse and implications for the genomic ecology of L1 elements. Hum. Mol. Genet. 2006, 15, 2146–2156. [Google Scholar] [CrossRef] [Green Version]
- Iannaccone, A.; Brabbit, E.; Lopez-Miro, C.; Love, Z.; Griffiths, V.; Kedrov, M.; Haider, N.B. Interspecies Correlations between Human and Mouse NR2E3-Associated Recessive Disease. J. Clin. Med. 2021, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Webber, A.L.; Hodor, P.; Thut, C.J.; Vogt, T.F.; Zhang, T.; Holder, D.J.; Petrukhin, K. Dual role of Nr2e3 in photoreceptor development and maintenance. Exp. Eye Res. 2008, 87, 35–48. [Google Scholar] [CrossRef]
- Xie, S.; Han, S.; Qu, Z.; Liu, F.; Li, J.; Yu, S.; Reilly, J.; Tu, J.; Liu, X.; Lu, Z.; et al. Knockout of Nr2e3 prevents rod photoreceptor differentiation and leads to selective L-/M-cone photoreceptor degeneration in zebrafish. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, N.M.; Yuan, Y.; Leehy, B.D.; Baid, R.; Kompella, U.; DeAngelis, M.M.; Escher, P.; Haider, N.B. Modifier genes as therapeutics: The nuclear hormone receptor Rev Erb Alpha (Nr1d1) rescues Nr2e3 associated retinal disease. PLoS ONE 2014, 9, e87942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, P.A.; Tang, S.; Shimchuk, A.A.; Ding, S.; Reh, T.A. Potential of Small Molecule–Mediated Reprogramming of Rod Photoreceptors to Treat Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2016, 57, 6407–6415. [Google Scholar] [CrossRef] [Green Version]
- Montana, C.L.; Kolesnikov, A.V.; Shen, S.Q.; Myers, C.A.; Kefalov, V.J.; Corbo, J.C. Reprogramming of adult rod photoreceptors prevents retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 1732–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simunovic, M.; Shen, W.; Lin, J.; Protti, D.; Lisowski, L.; Gillies, M. Optogenetic approaches to vision restoration. Exp. Eye Res. 2019, 178, 15–26. [Google Scholar] [CrossRef]
- Tochitsky, I.; Kienzler, M.A.; Isacoff, E.; Kramer, R.H. Restoring Vision to the Blind with Chemical Photoswitches. Chem. Rev. 2018, 118, 10748–10773. [Google Scholar] [CrossRef]
- Bohrer, L.R.; Wiley, L.A.; Burnight, E.R.; Cooke, J.A.; Giacalone, J.C.; Anfinson, K.R.; Andorf, J.L.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes 2019, 10, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naessens, S.; Ruysschaert, L.; Lefever, S.; Coppieters, F.; De Baere, E. Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrell, D.; Comander, J. Current Stem-Cell Approaches for the Treatment of Inherited Retinal Degenerations. Semin. Ophthalmol. 2019, 34, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, S.J.; Llonch, S.; Borsch, O.; Ader, M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog. Retin. Eye Res. 2019, 69, 1–37. [Google Scholar] [CrossRef]
- Iannaccone, A.; Fung, K.H.; Eyestone, M.E.; Stone, E.M. Treatment of Adult-Onset Acute Macular Retinoschisis in Enhanced S-cone Syndrome with Oral Acetazolamide. Am. J. Ophthalmol. 2009, 147, 307–312.e2. [Google Scholar] [CrossRef] [Green Version]
- Chatzistergiou, V.; Papasavvas, I.; Escher, P.; Durig, J.; Vaudaux, J.; Pournaras, J.-A.; Ambresin, A. Optical Coherence Tomography Analysis of Cystoid Macular Edema in Retinal Dystrophy Treated with Oral Acetazolamide: Two Cases. Klin. Mon. Für Augenheilkd. 2020, 237, 484–486. [Google Scholar] [CrossRef]
- Wiley, L.A.; Burnight, E.R.; DeLuca, A.P.; Anfinson, K.R.; Cranston, C.M.; Kaalberg, E.E.; Penticoff, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci. Rep. 2016, 6, 30742. [Google Scholar] [CrossRef] [Green Version]
- Terray, A.; Slembrouck, A.; Nanteau, C.; Chondroyer, C.; Zeitz, C.; Sahel, J.-A.; Audo, I.; Reichman, S.; Goureau, O. Generation of an induced pluripotent stem cell (iPSC) line from a patient with autosomal dominant retinitis pigmentosa due to a mutation in the NR2E3 gene. Stem Cell Res. 2017, 24, 1–4. [Google Scholar] [CrossRef]
- Diakatou, M.; Dubois, G.; Erkilic, N.; Sanjurjo-Soriano, C.; Meunier, I.; Kalatzis, V. Allele-Specific Knockout by CRISPR/Cas to Treat Autosomal Dominant Retinitis Pigmentosa Caused by the G56R Mutation in NR2E3. Int. J. Mol. Sci. 2021, 22, 2607. [Google Scholar] [CrossRef]
Figure 1.
The role of NR2E3 in photoreceptor cell specification. A schematic representation of how NR2E3 and other transcription factors interact to regulate rod and cone photoreceptor specification.
Figure 1.
The role of NR2E3 in photoreceptor cell specification. A schematic representation of how NR2E3 and other transcription factors interact to regulate rod and cone photoreceptor specification.

Figure 2.
Pathogenic variants in NR2E3. The types and locations of the variants identified to date are marked on the human, mouse, and zebrafish NR2E3 proteins with the corresponding exons displayed underneath. Approximate locations of the A/B domain, DNA binding domain (DBD), hinge domain, and ligand binding domain (LBD) are marked on the human protein.
Figure 2.
Pathogenic variants in NR2E3. The types and locations of the variants identified to date are marked on the human, mouse, and zebrafish NR2E3 proteins with the corresponding exons displayed underneath. Approximate locations of the A/B domain, DNA binding domain (DBD), hinge domain, and ligand binding domain (LBD) are marked on the human protein.

Figure 3.
Examples of NR2E3 patient retinal phenotypes. (A) Optos color fundus image of enhanced S-cone syndrome with (B) corresponding optos fundus autofluorescence (FAF) image showing diffuse peripheral hypoautofluorescence with a half-ring of pronounced hyper-AF along the temporal macular rim and (C) spectral domain optical coherence tomography (SD-OCT) through the macula of the same patient. (D) Optos color fundus image of autosomal dominant NR2E3-related retinitis pigmentosa with (E) corresponding optos FAF image that shows nummular hypoautofluorescent areas around the arcades, a hyperAF ring at the macula and another more diffuse ring along the arcades, and (F) SD-OCT through the macula of the patient showing cystoid macular edema and a restricted ellipsoid zone.
Figure 3.
Examples of NR2E3 patient retinal phenotypes. (A) Optos color fundus image of enhanced S-cone syndrome with (B) corresponding optos fundus autofluorescence (FAF) image showing diffuse peripheral hypoautofluorescence with a half-ring of pronounced hyper-AF along the temporal macular rim and (C) spectral domain optical coherence tomography (SD-OCT) through the macula of the same patient. (D) Optos color fundus image of autosomal dominant NR2E3-related retinitis pigmentosa with (E) corresponding optos FAF image that shows nummular hypoautofluorescent areas around the arcades, a hyperAF ring at the macula and another more diffuse ring along the arcades, and (F) SD-OCT through the macula of the patient showing cystoid macular edema and a restricted ellipsoid zone.

Figure 4.
Lack of rod differentiation in nr2e3 mutant zebrafish. Immunohistochemical staining for rhodopsin shows a lack of rod photoreceptors (red) in the nr2e3Sa15662 mutant retina at 5 days post fertilization. Nuclei are counterstained with DAPI (blue). Scale bars are 50 µm (top) and 10 µm (bottom). Retinal layers are indicated: photoreceptor outer segments (OS), outer nuclear layer (ONL), and inner nuclear layer (INL). Image kindly prepared by Dr. Manuela Lahne from Prof. Mariya Moosajee’s group.
Figure 4.
Lack of rod differentiation in nr2e3 mutant zebrafish. Immunohistochemical staining for rhodopsin shows a lack of rod photoreceptors (red) in the nr2e3Sa15662 mutant retina at 5 days post fertilization. Nuclei are counterstained with DAPI (blue). Scale bars are 50 µm (top) and 10 µm (bottom). Retinal layers are indicated: photoreceptor outer segments (OS), outer nuclear layer (ONL), and inner nuclear layer (INL). Image kindly prepared by Dr. Manuela Lahne from Prof. Mariya Moosajee’s group.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Toms, M.; Ward, N.; Moosajee, M. Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease. Genes 2023, 14, 1325. https://doi.org/10.3390/genes14071325
AMA Style
Toms M, Ward N, Moosajee M. Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease. Genes. 2023; 14(7):1325. https://doi.org/10.3390/genes14071325
Chicago/Turabian StyleToms, Maria, Natasha Ward, and Mariya Moosajee. 2023. "Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease" Genes 14, no. 7: 1325. https://doi.org/10.3390/genes14071325
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.