
Competitive Metabolism of Polycyclic Aromatic Hydrocarbons (PAHs): An Assessment Using In Vitro Metabolism and Physiologically Based Pharmacokinetic (PBPK) Modeling


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. In Vitro Metabolism Studies
2.3. Competitive Inhibition Metabolism Assays
2.4. Analyte Quantitation
2.5. Data Analysis
2.6. PBPK Interaction Model
3. Results
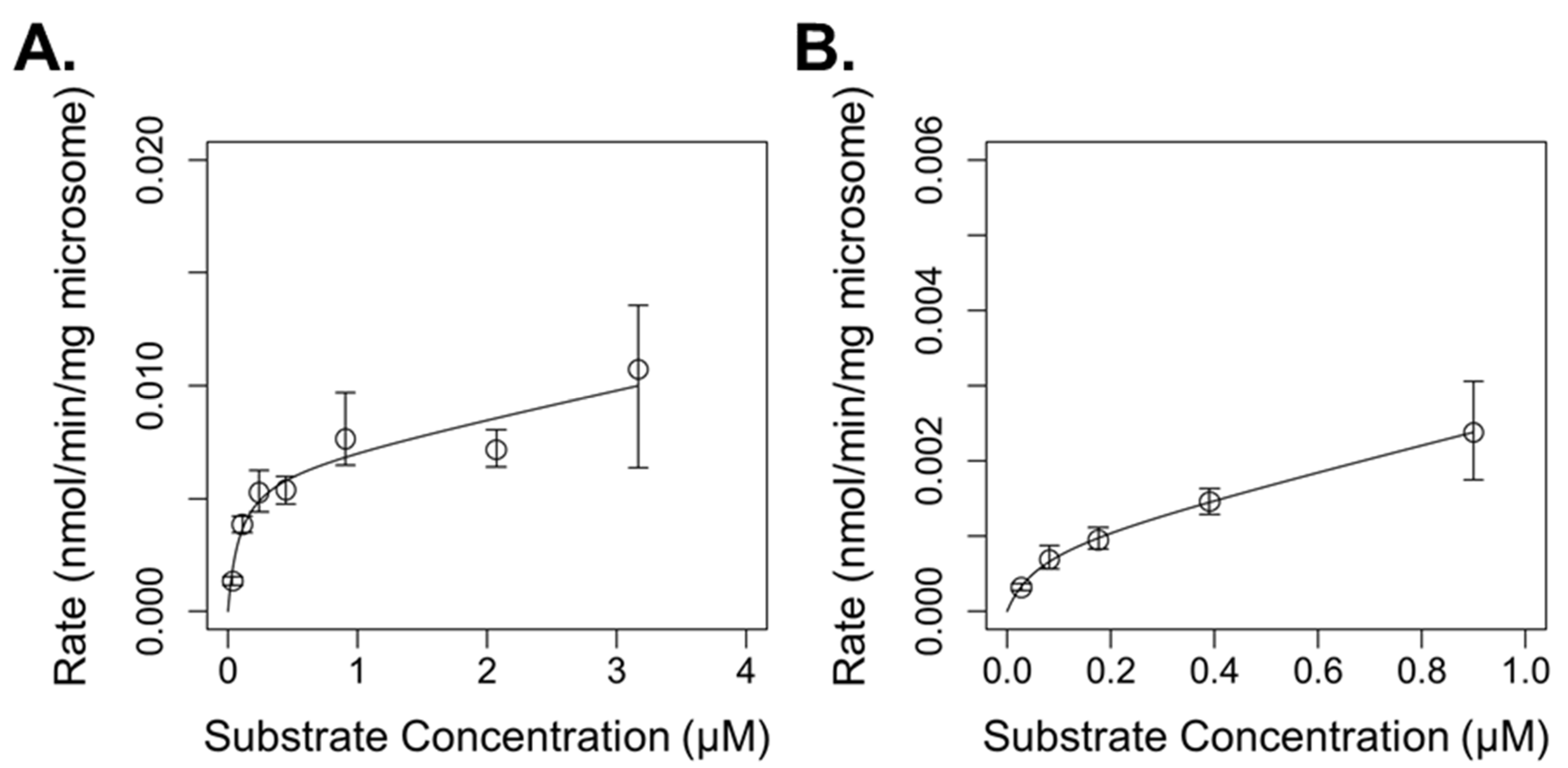
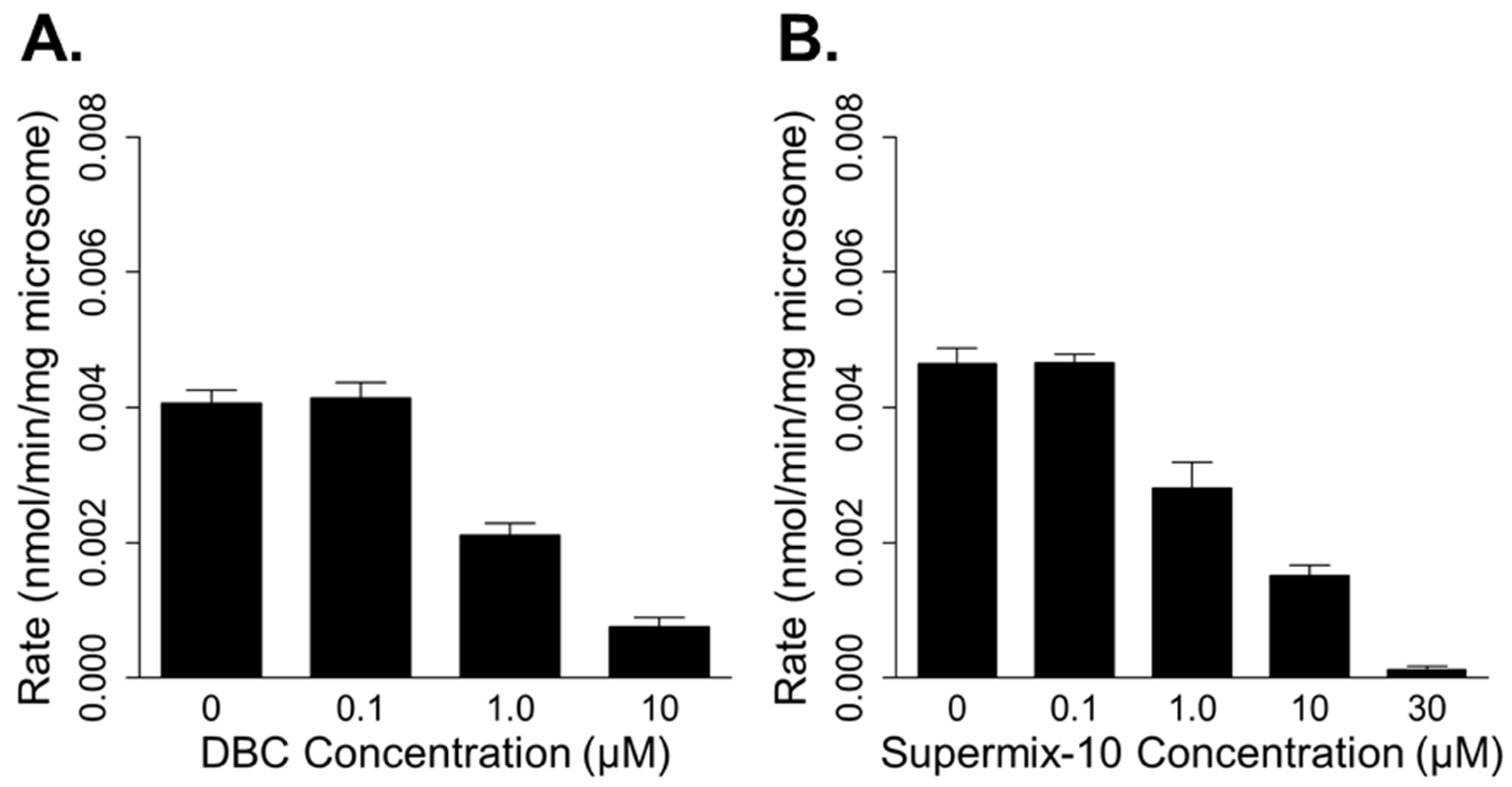
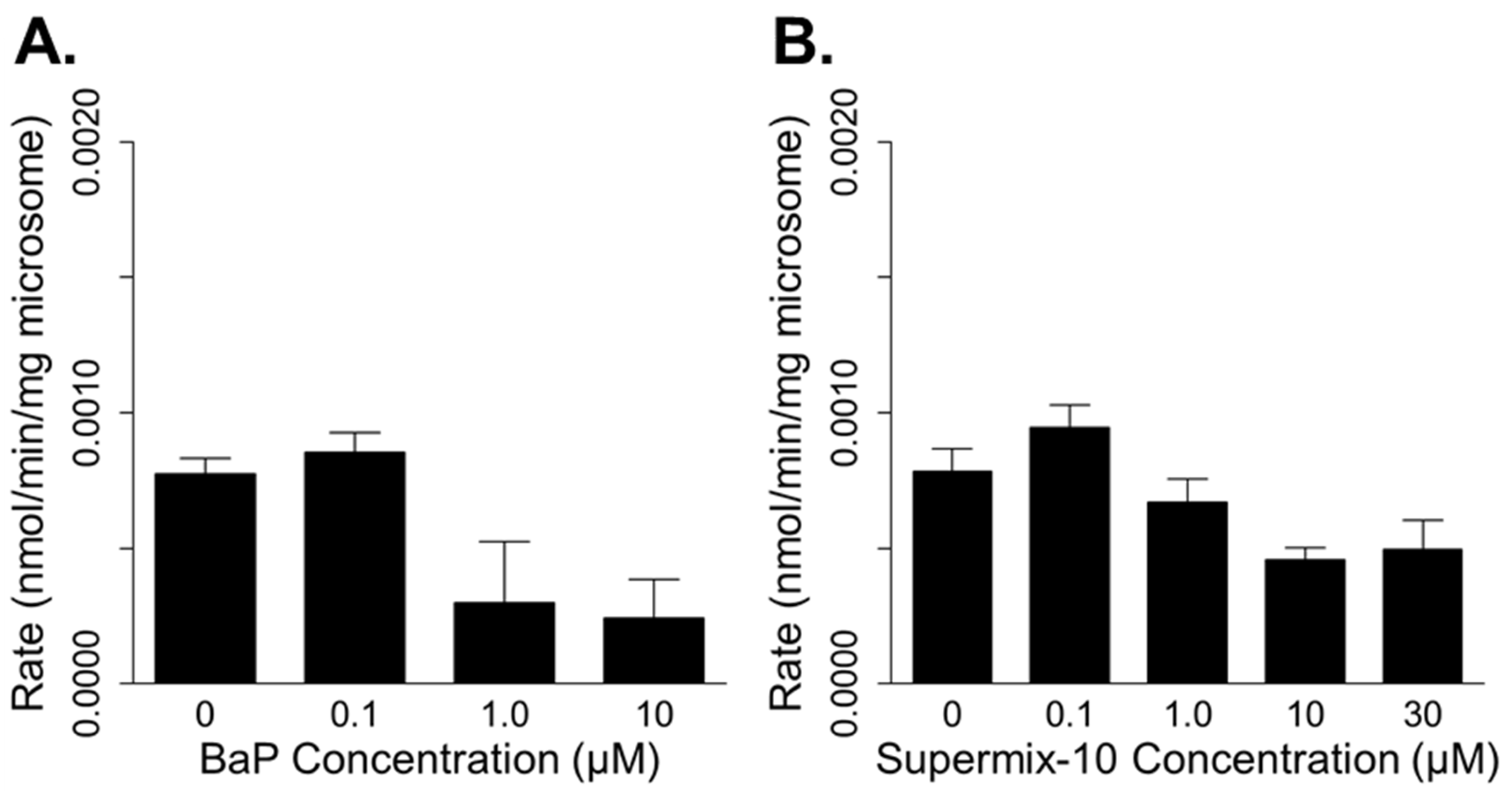
3.1. In Vitro Metabolism
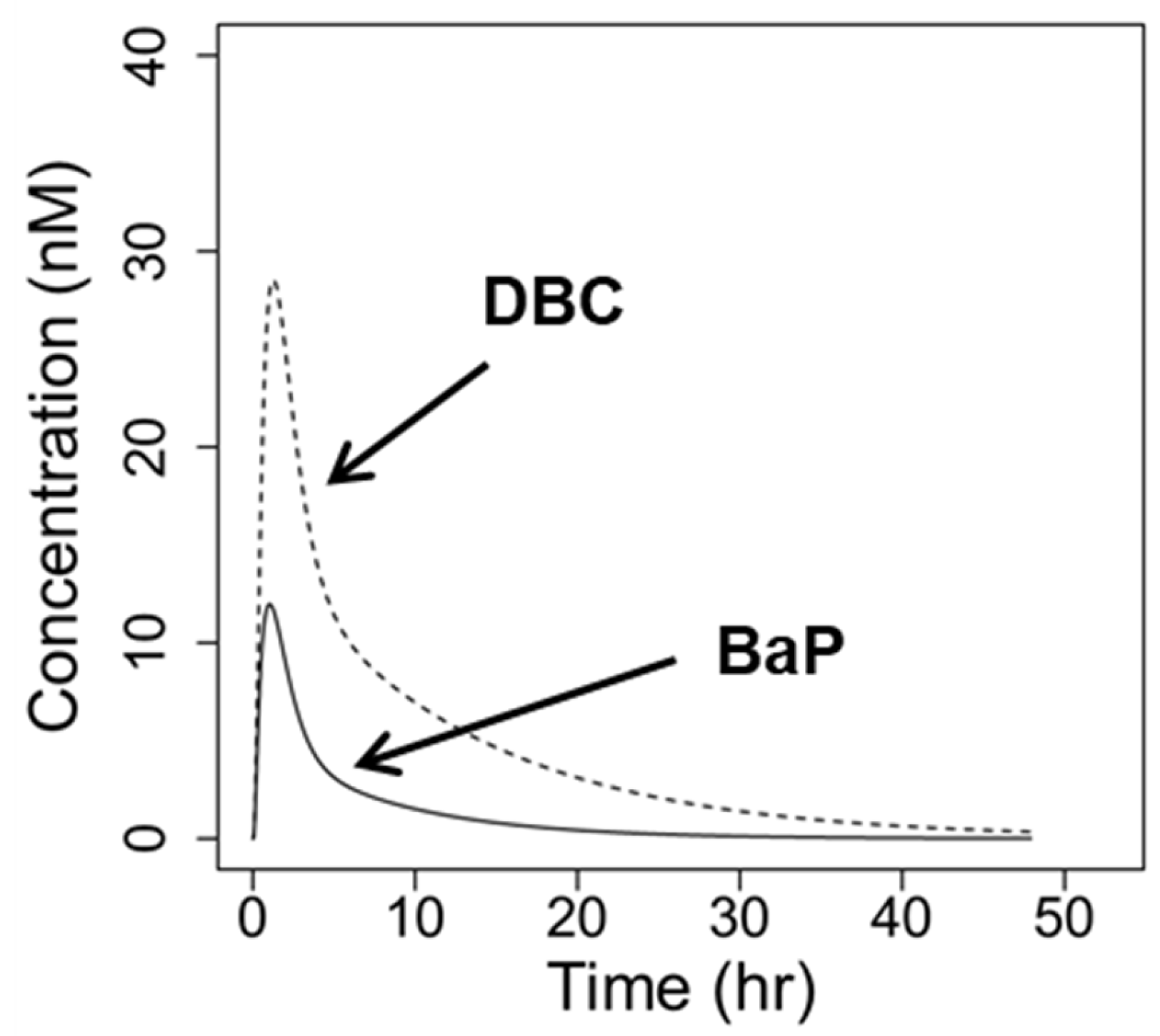
3.2. Interaction PBPK Modeling
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kim, K.-H.; Jahan, S.A.; Kabir, E.; Brown, R.J.C. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef] [PubMed]
- de Vos, R.H.; van Dokkum, W.; Schouten, A.; de Jong-Berkhout, P. Polycyclic aromatic hydrocarbons in Dutch total diet samples (1984–1986). Food Chem. Toxicol. 1990, 28, 263–268. [Google Scholar] [CrossRef]
- Dennis, M.J.; Massey, R.C.; McWeeny, D.J.; Knowles, M.E.; Watson, D. Analysis of polycyclic aromatic hydrocarbons in UK total diets. Food Chem. Toxicol. 1983, 21, 569–574. [Google Scholar] [CrossRef]
- Lodovici, M.; Dolara, P.; Casalini, C.; Ciappellano, S.; Testolin, G. Polycyclic aromatic hydrocarbon contamination in the Italian diet. Food Addit. Contam. 1995, 12, 703–713. [Google Scholar] [CrossRef]
- Menzie, C.A.; Potocki, B.B.; Santodonato, J. Exposure to carcinogenic PAHs in the environment. Environ. Sci. Technol. 1992, 26, 1278–1284. [Google Scholar] [CrossRef]
- Dennis, M.J.; Massey, R.C.; Cripps, G.; Venn, I.; Howarth, N.; Lee, G. Factors affecting the polycyclic aromatic hydrocarbon content of cereals, fats and other food products. Food Addit. Contam. 1991, 8, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Jakszyn, P.; Agudo, A.; Ibáñez, R.; García-Closas, R.; Pera, G.; Amiano, P.; González, C.A. Development of a food database of nitrosamines, heterocyclic amines, and polycyclic aromatic hydrocarbons. J. Nutr. 2004, 134, 2011–2014. [Google Scholar] [CrossRef]
- Skupińska, K.; Misiewicz, I.; Kasprzycka-Guttman, T. Polycyclic aromatic hydrocarbons: Physicochemical properties, environmental appearance and impact on living organisms. Acta Pol. Pharm. 2004, 61, 233–240. [Google Scholar]
- EPA. Development of a Relative Potency Factor (RPF) Approach for Polycyclic Aromatic Hydrocarbon (PAH) Mixtures; U.S. Environmental Protection Agency: Washington, DC, USA, 2010.
- Shimada, T. Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab. Pharmacokinet. 2006, 21, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef]
- Baird, W.M.; Hooven, L.A.; Mahadevan, B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ. Mol. Mutagenesis 2005, 45, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Stansbury, K.H.; Walker, N.J.; Trush, M.A.; Strickland, P.T.; Sutter, T.R. Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis 1998, 19, 1847–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulc, M.; Indra, R.; Moserova, M.; Schmeiser, H.H.; Frei, E.; Arlt, V.M.; Stiborova, M. The impact of individual cytochrome P450 enzymes on oxidative metabolism of benzo[a]pyrene in human livers. Environ. Mol. Mutagenesis 2016, 57, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.W.; Ladd, M.A.; Hoffman, A.D.; Fitzsimmons, P.N. Biotransformation of Polycyclic Aromatic Hydrocarbons by Trout Liver S9 Fractions: Evaluation of Competitive Inhibition Using a Substrate Depletion Approach. Environ. Toxicol. Chem. 2019, 38, 2729–2739. [Google Scholar] [CrossRef]
- Stringfellow, W.T.; Aitken, M.D. Competitive metabolism of naphthalene, methylnaphthalenes, and fluorene by phenanthrene-degrading pseudomonads. Appl. Environ. Microbiol. 1995, 61, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.E.; Hosted, T.J., Jr.; Lavoie, E.J. Fluoranthene and pyrene enhance benzo[a]pyrene—DNA adduct formation in vivo in mouse skin. Cancer Lett. 1984, 24, 327–333. [Google Scholar] [CrossRef]
- Crowell, S.R.; Amin, S.G.; Anderson, K.A.; Krishnegowda, G.; Sharma, A.K.; Soelberg, J.J.; Williams, D.E.; Corley, R.A. Preliminary physiologically based pharmacokinetic models for benzo[a]pyrene and dibenzo[def,p]chrysene in rodents. Toxicol. Appl. Pharmacol. 2011, 257, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pande, P.; Madeen, E.P.; Williams, D.E.; Crowell, S.R.; Ognibene, T.J.; Turteltaub, K.W.; Corley, R.A.; Smith, J.N. Translating dosimetry of Dibenzo[def,p]chrysene (DBC) and metabolites across dose and species using physiologically based pharmacokinetic (PBPK) modeling. Toxicol. Appl. Pharmacol. 2022, 438, 115830. [Google Scholar] [CrossRef]
- Geier, M.C.; James Minick, D.; Truong, L.; Tilton, S.; Pande, P.; Anderson, K.A.; Teeguardan, J.; Tanguay, R.L. Systematic developmental neurotoxicity assessment of a representative PAH Superfund mixture using zebrafish. Toxicol. Appl. Pharmacol. 2018, 354, 115–125. [Google Scholar] [CrossRef]
- Krzeminski, J.; Lin, J.M.; Amin, S.; Hecht, S.S. Synthesis of Fjord region diol epoxides as potential ultimate carcinogens of dibenzo[a,l]pyrene. Chem. Res. Toxicol. 1994, 7, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Luch, A.; Coffing, S.L.; Tang, Y.M.; Schneider, A.; Soballa, V.; Greim, H.; Jefcoate, C.R.; Seidel, A.; Greenlee, W.F.; Baird, W.M.; et al. Stable expression of human cytochrome P450 1B1 in V79 Chinese hamster cells and metabolically catalyzed DNA adduct formation of dibenzo[a,l]pyrene. Chem. Res. Toxicol. 1998, 11, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Kumar, S.; Amin, S. A Highly Abbreviated Synthesis of Dibenzo[def,p]chrysene and Its 12-Methoxy Derivative, a Key Precursor for the Synthesis of the Proximate and Ultimate Carcinogens of Dibenzo[def,p]chrysene. J. Org. Chem. 2004, 69, 3979–3982. [Google Scholar] [CrossRef]
- Hummel, J.M.; Madeen, E.P.; Siddens, L.K.; Uesugi, S.L.; McQuistan, T.; Anderson, K.A.; Turteltaub, K.W.; Ognibene, T.J.; Bench, G.; Krueger, S.K.; et al. Pharmacokinetics of [(14)C]-Benzo[a]pyrene (BaP) in humans: Impact of Co-Administration of smoked salmon and BaP dietary restriction. Food Chem. Toxicol. 2018, 115, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Madeen, E.; Corley, R.A.; Crowell, S.; Turteltaub, K.; Ognibene, T.; Malfatti, M.; McQuistan, T.J.; Garrard, M.; Sudakin, D.; Williams, D.E. Human in Vivo Pharmacokinetics of [(14)C]Dibenzo[def,p]chrysene by Accelerator Mass Spectrometry Following Oral Microdosing. Chem. Res. Toxicol. 2015, 28, 126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeen, E.; Siddens, L.K.; Uesugi, S.; McQuistan, T.; Corley, R.A.; Smith, J.; Waters, K.M.; Tilton, S.C.; Anderson, K.A.; Ognibene, T.; et al. Toxicokinetics of benzo[a]pyrene in humans: Extensive metabolism as determined by UPLC-accelerator mass spectrometry following oral micro-dosing. Toxicol. Appl. Pharmacol. 2019, 364, 97–105. [Google Scholar] [CrossRef]
- Madeen, E.P.; Ognibene, T.J.; Corley, R.A.; McQuistan, T.J.; Henderson, M.C.; Baird, W.M.; Bench, G.; Turteltaub, K.W.; Williams, D.E. Human Microdosing with Carcinogenic Polycyclic Aromatic Hydrocarbons: In Vivo Pharmacokinetics of Dibenzo[def,p]chrysene and Metabolites by UPLC Accelerator Mass Spectrometry. Chem. Res. Toxicol. 2016, 29, 1641–1650. [Google Scholar] [CrossRef] [Green Version]
- Vermillion Maier, M.L.; Siddens, L.K.; Pennington, J.M.; Uesugi, S.L.; Anderson, K.A.; Tidwell, L.G.; Tilton, S.C.; Ognibene, T.J.; Turteltaub, K.W.; Smith, J.N.; et al. Benzo[a]pyrene (BaP) metabolites predominant in human plasma following escalating oral micro-dosing with [14C]-BaP. Environ. Int. 2022, 159, 107045. [Google Scholar] [CrossRef]
- Crowell, S.R.; Hanson-Drury, S.; Williams, D.E.; Corley, R.A. In vitro metabolism of benzo[a]pyrene and dibenzo[def,p]chrysene in rodent and human hepatic microsomes. Toxicol. Lett. 2014, 228, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.N.; Mehinagic, D.; Nag, S.; Crowell, S.R.; Corley, R.A. In vitro metabolism of benzo[a]pyrene-7,8-dihydrodiol and dibenzo[def,p]chrysene-11,12 diol in rodent and human hepatic microsomes. Toxicol. Lett. 2017, 269, 23–32. [Google Scholar] [CrossRef]
- Stoddard, E.G.; Nag, S.; Martin, J.; Tyrrell, K.J.; Gibbins, T.; Anderson, K.A.; Shukla, A.K.; Corley, R.; Wright, A.T.; Smith, J.N. Exposure to an Environmental Mixture of Polycyclic Aromatic Hydrocarbons Induces Hepatic Cytochrome P450 Enzymes in Mice. Chem. Res. Toxicol. 2021, 34, 2145–2156. [Google Scholar] [CrossRef]
- Allan, S.E.; Sower, G.J.; Anderson, K.A. Estimating risk at a Superfund site using passive sampling devices as biological surrogates in human health risk models. Chemosphere 2011, 85, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, S.; Béliveau, M.; Tardif, R.; Krishnan, K. A PBPK modeling-based approach to account for interactions in the health risk assessment of chemical mixtures. Toxicol. Sci. 2001, 63, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennison, J.E.; Andersen, M.E.; Yang, R.S. Characterization of the pharmacokinetics of gasoline using PBPK modeling with a complex mixtures chemical lumping approach. Inhal. Toxicol. 2003, 15, 961–986. [Google Scholar] [CrossRef] [PubMed]
- Tardif, R.; Charest-Tardif, G.; Brodeur, J.; Krishnan, K. Physiologically Based Pharmacokinetic Modeling of a Ternary Mixture of Alkyl Benzenes in Rats and Humans. Toxicol. Appl. Pharmacol. 1997, 144, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Quinney, S.K.; Zhang, X.; Lucksiri, A.; Gorski, J.C.; Li, L.; Hall, S.D. Physiologically Based Pharmacokinetic Model of Mechanism-Based Inhibition of CYP3A by Clarithromycin. Drug Metab. Dispos. 2010, 38, 241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Zhang, L.; Grillo, J.; Liu, Q.; Bullock, J.; Moon, Y.; Song, P.; Brar, S.; Madabushi, R.; Wu, T.; et al. Applications of Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation During Regulatory Review. Clin. Pharmacol. Ther. 2011, 89, 259–267. [Google Scholar] [CrossRef]
- Yamazaki, S.; Evers, R.; De Zwart, L. Physiologically-based pharmacokinetic modeling to evaluate in vitro-to-in vivo extrapolation for intestinal P-glycoprotein inhibition. CPT Pharmacomet. Syst. Pharmacol. 2022, 11, 55–67. [Google Scholar] [CrossRef]
- FDA. Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry; EPA/600/R-05/043F.; US Food and Drug Administration: Washington, DC, USA, 2020. [Google Scholar]
- Jia, L.; Liu, X. The conduct of drug metabolism studies considered good practice (II): In vitro experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EPA. U.S. IRIS Toxicological Review of Benzo[a]pyrene (Final Report); EPA/635/R-17/003F; U.S. Environmental Protection Agency: Washington, DC, USA, 2017.
- ATSDR. Public Health Assessment: Portland Harbor, Multnomah County, Oregon; Oregon Department of Human Services Superfund Health Investigation and Education Program; US Department of Health and Human Services, Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2006.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CAS | Molecular Weight | Molar Ratio | Molar Fraction | Carcinogenic A |
|---|---|---|---|---|---|
| (g/mol) | |||||
| benzo(a)anthracene | 56-55-3 | 228.29 | 2.47 | 0.05 | EPA, IARC, ACGIH |
| retene | 483-65-8 | 234.34 | 7.43 | 0.15 | |
| pyrene | 129-00-0 | 202.25 | 14.50 | 0.28 | |
| phenanthrene | 85-01-8 | 178.23 | 1.72 | 0.03 | |
| naphthalene | 91-20-3 | 128.17 | 3.48 | 0.07 | EPA, IARC, ACGIH |
| fluorene | 86-73-7 | 166.22 | 1.84 | 0.04 | |
| fluoranthene | 206-44-0 | 202.25 | 14.24 | 0.28 | |
| chrysene | 218-01-9 | 228.29 | 2.59 | 0.05 | EPA, ACGIH |
| acenaphthylene | 208-96-8 | 154.21 | 2.00 | 0.04 | |
| 2-methylnaphthalene | 91-57-6 | 142.20 | 1.00 | 0.02 |
| Model | BIC | |
|---|---|---|
| BaP | DBC | |
| Michaelis-Menten | –71.1 | –71.6 |
| Michaelis-Menten-clearance | –73.5 | –88.3 |
| Double Michaelis-Menten | –71.5 | –86.8 |
| Compound | Parameter | |||||||
|---|---|---|---|---|---|---|---|---|
| Vmax1 | Km1 | Clint1 | Clint2 | |||||
| nmol/min/mg Micro | 95% CI | µM | 95% CI | mL/min | 95% CI | mL/min | 95% CI | |
| BaP | 0.0063 | 0.0044–0.0083 | 0.088 | 0.044–0.15 | 0.072 | 0.054–0.10 | 0.0012 | 1.7 × 10−7–0.0028 |
| DBC | 0.00090 | 0.00044–0.0023 | 0.060 | 0.014–0.22 | 0.015 | 0.0097–0.032 | 0.0017 | 6.5 × 10−7–0.0028 |
| Substrate | Inhibitor | |||||
|---|---|---|---|---|---|---|
| KiBaP | KiDBC | KiSM10 | ||||
| µM | 95% CI | µM | 95% CI | µM | 95% CI | |
| BaP | 0.44 | 0.36–0.54 | 0.75 | 0.49–1.1 | ||
| DBC | 0.061 | 0.041–0.12 | 0.63 | 0.36–1.2 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, J.N.; Gaither, K.A.; Pande, P. Competitive Metabolism of Polycyclic Aromatic Hydrocarbons (PAHs): An Assessment Using In Vitro Metabolism and Physiologically Based Pharmacokinetic (PBPK) Modeling. Int. J. Environ. Res. Public Health 2022, 19, 8266. https://doi.org/10.3390/ijerph19148266
Smith JN, Gaither KA, Pande P. Competitive Metabolism of Polycyclic Aromatic Hydrocarbons (PAHs): An Assessment Using In Vitro Metabolism and Physiologically Based Pharmacokinetic (PBPK) Modeling. International Journal of Environmental Research and Public Health. 2022; 19(14):8266. https://doi.org/10.3390/ijerph19148266
Chicago/Turabian StyleSmith, Jordan N., Kari A. Gaither, and Paritosh Pande. 2022. "Competitive Metabolism of Polycyclic Aromatic Hydrocarbons (PAHs): An Assessment Using In Vitro Metabolism and Physiologically Based Pharmacokinetic (PBPK) Modeling" International Journal of Environmental Research and Public Health 19, no. 14: 8266. https://doi.org/10.3390/ijerph19148266