
Evaluating the Oxidative Stress in Inflammation: Role of Melatonin


Abstract
:
1. Introduction
2. Oxidative Stress, Inflammation and Chronic Diseases
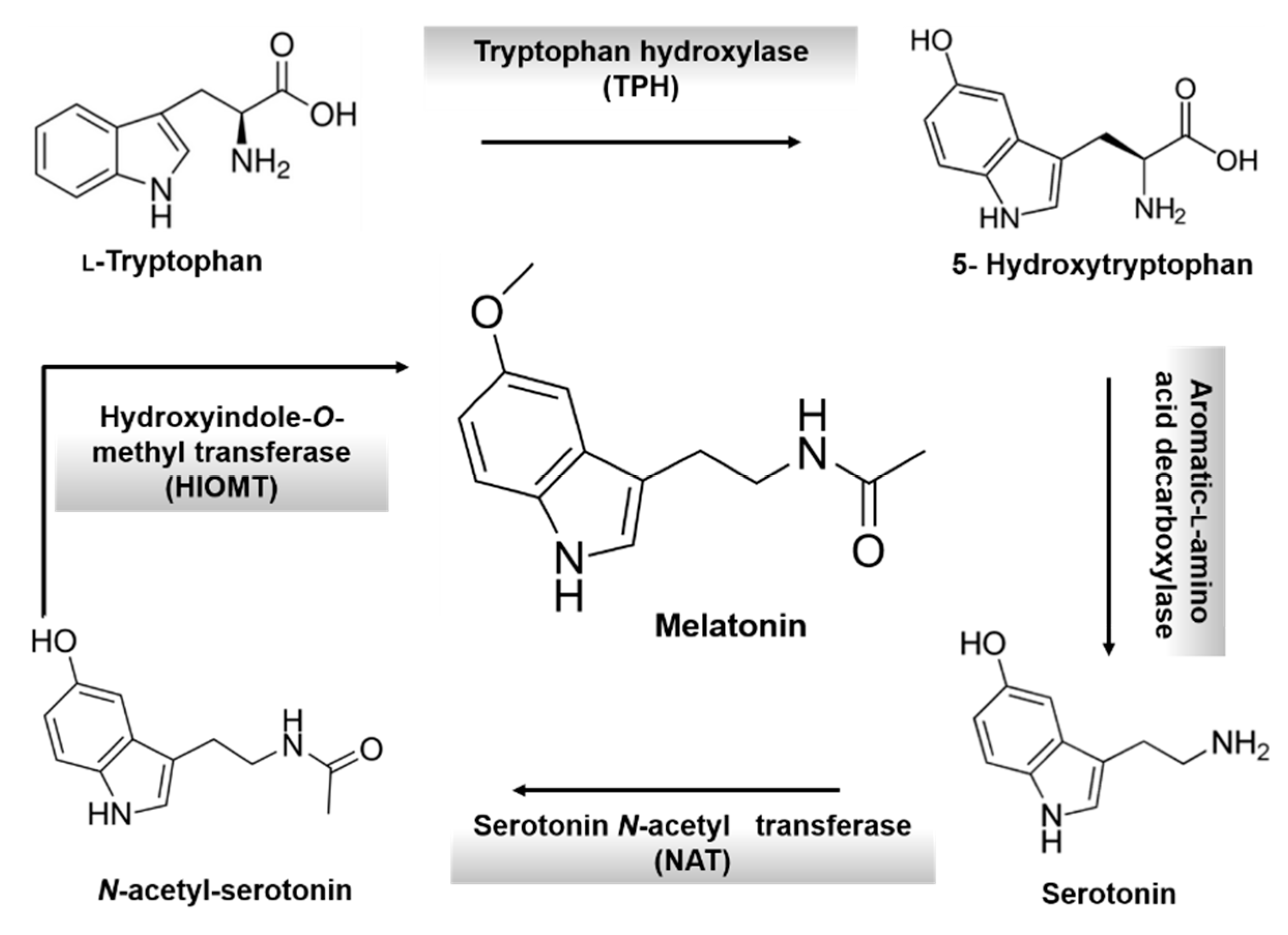
3. Melatonin: An Overview

4. Role of Melatonin in Cancer

{kind=link}
{kind=link}
{kind=link}
| Tumor | Treatment | Results and Conclusions | References |
|---|---|---|---|
| Metastatic non-small cell lung cancer | MLT + CisP + etoposide | Better tolerance to chemotherapy. Improve the efficacy of chemotherapy in terms of both survival and quality of life. | [68] |
| Metastatic cancer | MLT | Decline in VEGF secretion and control of the neoplasic growth. | [69] |
| Chronic lymphocytic leukemia | Cyclophosphamide + somastostatin + bromocriptine + retinoids + MLT + ACTH | Partial remission after 2 months and continued treatment. Patients hadn’t got disease recurrence. No toxicity. | [70] |
| Metastatic melanoma | MLT + IL-2 + Cisp | No Cisplatin-related neurotoxicity was observed. Effective and well tolerated treatment. Clinical efficacy at least comparable to that obtained with a first-line therapy of dacarbazine plus interferon-α. | [71] |
| Non-Hodgkin’s lymphomas (NHL) | Cyclophosphamide + somatostatin + bromocriptin + retinoids + MLT + ACTH | 70% of participants had a partial response. 20% of participants had stable disease. 10% progressed on therapy. The combination was effective in treatment of low-grade NHL at advanced stage. | [72] |
| Advances solid neoplasms: non-small cell lung cancer (NSCLC) or gastrointestinal tumors | NSCLC: MLT + CisP + etoposide or gemcitabine; Colorectal cancer: MLT + OxiP + 5-FU or MLT + Etoposide or MLT + 5-FU + FA; Gastric cancer: MLT + CisP + Epirubicin + 5-FU + FA or MLT + 5-FU + FA | Regression rate achieved in MLT patients treated significantly higher than in those treated with chemotherapy alone and 2-year survival rate significantly higher in patients concomitantly treated with MLT. | [73] |
| Metastatic solid tumour: lung cancer, breast cancer, gastrointestinal tract neoplasms, head and neck cancers | Lung cancer: MLT + CisP + etoposide or MLT + gemcitabine; Breast cancer: MLT + Doxorubicin or MLT + Mitoxantrone or MLT + Paclitaxel; Gastrointestinal tumors: MLT + 5-FU + FA; Head and neck cancers: MLT + 5-FU + CisP | 1-year survival rate and the objective tumour regression rate significantly higher in patients concomitantly treated with MLT than in those who received chemotherapy alone. MLT significantly reduced the frequency of thrombocytopenia, neurotoxicity, cardiotoxicity, stomatitis and asthenia. | [74] |
| Lymph node relapses due to malignant melanoma. | MLT | Disease-free survival in melanoma patients surgically treated for regional node recurrence was significantly higher in MLT-treated individuals than in controls. | [75] |
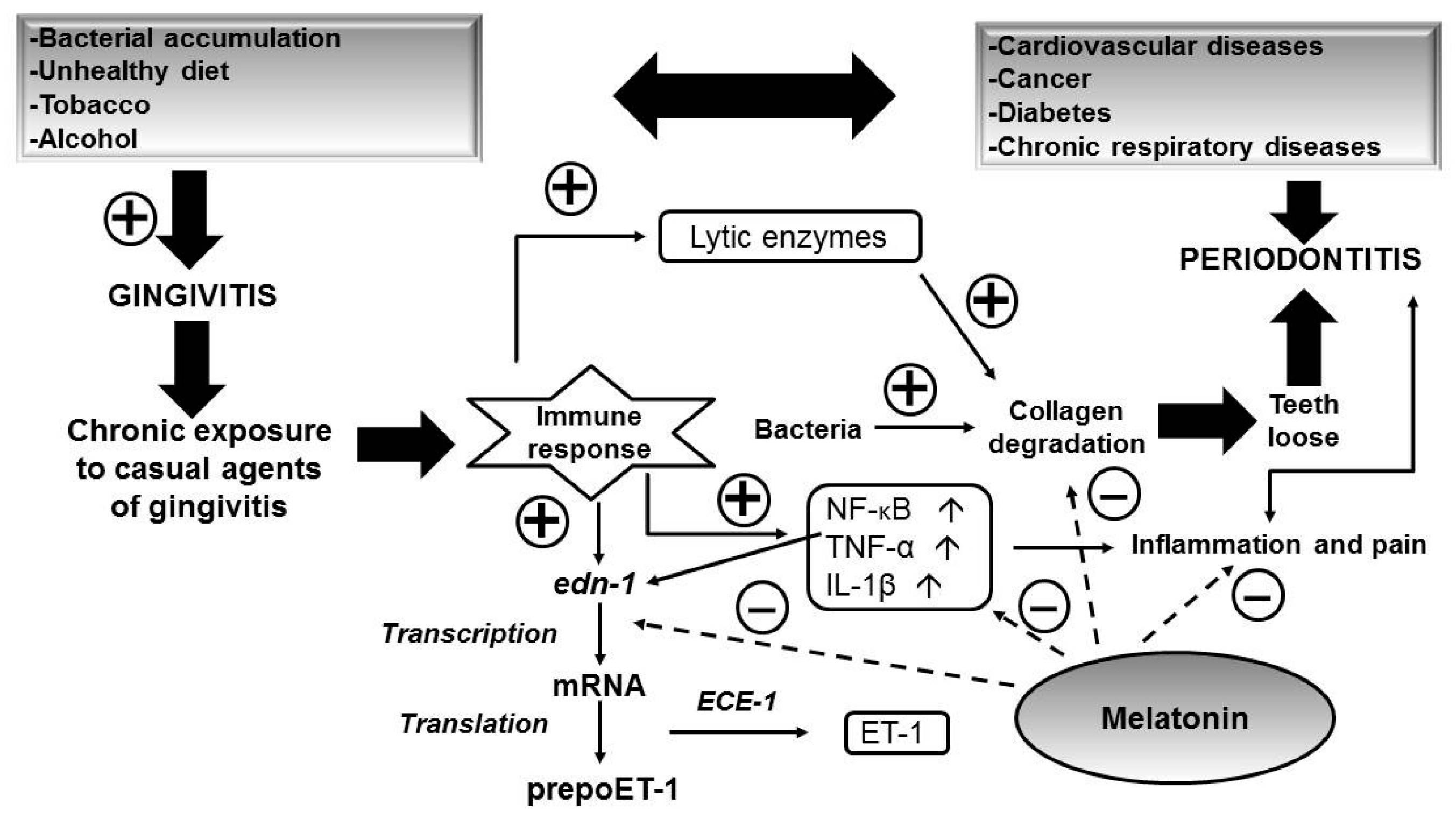
5. Role of Melatonin in Oral Health

6. Role of Melatonin in Kidney Chronic Disease
7. Role of Melatonin in Fibromyalgia
8. Role of Melatonin in Inflammatory Bowel Disease
| Study | Colitis Model | Results | Effects |
|---|---|---|---|
| Trivedi and Jena [143] | Dextran sulphate sodium (DSS) induced colitis | ↓ Colon length observed in mice with UC | Increase intestinal surface |
| ↓ Levels of inflammatory markers: MPO, IL-17, IL-6, TNF-α, NF-κB, COX-2, STAT3 | Anti-inflammatory at the systemic site | ||
| ↑ Nrf2, NQO-1 and GSH | ↓ Oxidative stress involved in UC | ||
| Antifibrotic effect: ↓ MMP-9 and CTGF | Decrease in the loss of function of intestinal tissue | ||
| ↓ 8-Oxo-de expression | ↓ Oxidative DNA damage | ||
| ↓ Occluding expression | ↓ Elevated gut permeability | ||
| ↓ LPS plasma levels | ↓ Gut bacteria in the systemic circulation | ||
| Li et al. [144] | 2,4,6-Trinitrobenzene sulfonic acid induced colitis | ↓ mRNA levels for TNF-α and ICAM-1 colon tissues | ↓ Colitis symptoms: rectal bleeding and occult blood and ↓ frequency and severity of mucosa damage dramatically |
| ↓ NF-κB-DNA translocation and mRNA expression by ↑ IκBα | ↓ Expression of inflammatory cytokines | ||
| ↓ Inflammatory infiltrate of neutrophils, lymphocytes, and macrophages | ↓ Severity of mucosa injury and alleviate colitis symptoms | ||
| Trivedi et al. [145] | 1,2-Dimethylhydrazine dihydrochloride (DMH) and DSS induced colitis-associated colon carcinogenesis (CACC) | ↓ Tumor multiplicity, significantly ↓ in number of aberrant and abnormal crypts in the colon | Ameliorative effect on the progression of colon carcinogenesis |
| Significantly ↓ levels of inflammatory markers: MPO, IL-17, IL-6, TNF-α | Anti-inflammatory effect | ||
| Significantly ↓ in NF-κB, COX-2, and STAT3 levels in the colon of mice with CACC | |||
| Significant ↓ TBARS and ↑ GSH levels in the colon | Antioxidant effect | ||
| Significantly ↓ autophagy as revealed from the expression pattern of Beclin1, LC3-II/LC3-I ratio, and p62 | ↓ Autophagy in the colon of mice with CACC | ||
| Significant ↑ Nrf2, NQO-1, and HO-1 | ↓ Oxidative stress | ||
| ↓ CACC-associated DNA damage as well as oxidative DNA damage in the colon of mice | Protective role in CACC | ||
| Tahan et al. [146] | Acetic acid-induced colitis | Significant ↓ TNF-α, IL-1β , IL-6, myeloperoxidase (MPO), and malondialdehyde (MDA) levels | Anti-inflammatory effect |
| Significant ↑ GSH and SOD levels | Antioxidant effect | ||
| Significant ↓ macro and microscopic lesion scores of the UC group | Protective role in UC | ||
| Sayyed et al. [147] | Acetic acid-induced colitis | ↓ NF-κB inmuno histochemical expression | Anti-inflammatory effect |
| ↓ LP Levels | ↓ Intestinal permeability | ||
| ↓ PTX3 Levels | ↓ Neutrophil infiltration and proinflammatories Cytokines | ||
| Dong et al. [148] | Acetic acid or 2,4,6-trinitrobenzene sulfonic acid (TNBS) induced colitis | ↓ Severity of gut injury and significantly ↓ of colon mucosal damage index (CMDI) | Protective role in UC |
| Significantly ↓ of NO content and iNOS expression in colonic tissue | Antioxidant effect | ||
| ↓ PGE2 and expression of COX-2 | Anti-inflammatory effect |
9. Role of Melatonin in Rheumatoid Arthritis
10. Conclusions
Author Contributions
Conflicts of Interest
References
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Moniczewski, A.; Gawlik, M.; Smaga, I.; Niedzielska, E.; Krzek, J.; Przegaliński, E.; Pera, J.; Filip, M. Oxidative stress as an etiological factor and a potential treatment target of psychiatric disorders. Part 1. Chemical aspects and biological sources of oxidative stress in the brain. Pharmacol. Rep. 2015, 67, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Specificity of a third kind: Reactive oxygen and nitrogen intermediates in cell signalling. J. Clin. Investig. 2003, 111, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive Oxygen species in living systems: Source, biochemistry and role in human disease. Am. J. Med. 1991, 91, 14–22. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.H.H.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Mc Cord, J. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef]
- Hartman, K.G.; Bortner, J.D.; Falk, G.W.; Yu, J.; Martín, M.G.; Rustgi, A.K.; Lynch, J.P. Modeling inflammation and oxidative stress in gastrointestinal disease development usign novel organotypic culture systems. Stem Cell Res. Ther. 2013, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Buelna-Chontal, M.; Zazueta, C. Redox activation of Nrf2 & NF-κB: A double end sword? Cell. Signal. 2013, 25, 2548–2557. [Google Scholar] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive pro-inflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Kang, B.S.; Lee, H.L.; Son, S.J.; Hwang, S.H.; Kim, D.S.; Park, J.S.; Cho, H.J. Spinal NF-κB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur. J. Neurosci. 2004, 19, 3375–3381. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.; Ramana, K.V. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid. Med. Cell. Longev. 2013, 690545, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, S.; Hammam, O.; Murata, M.; Kawanishi, S. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxid. Med. Cell. Longev. 2013, 387014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Babatunji, E.O.; Abiola, F.A.; Abidemi, P.K. Reactive oxygen species, apoptosis, antimicrobial peptides and human inflammatory diseases. Pharmaceuticals 2015, 8, 151–175. [Google Scholar]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Smaga, I.; Niedzielska, E.; Gawlik, M.; Moniczewski, A.; Krzek, J.; Przegalin, E.; Pera, J.; Filip, M. Oxidative stress as an etiological factor and a potential treatment target of psychiatric disorders. Part 2. Depression, anxiety, schizophrenia and autism. Pharmacol. Rep. 2015, 67, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, C.; Orsucci, D.; Caldarazzo, E.; Siciliano, G.; Bonuccelli, U.; Mancuso, M. Alzheimer’s pathogenesis and its link to the mitochondrion. Oxid. Med. Cell. Longev. 2015, 803942, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3 β (GSK-3β) signaling: Implications for Parkinson’s disease. Pharmacol. Res. 2008, 97, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Hawa, N.S.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015. [Google Scholar] [CrossRef]
- Pickup, J.C. Inflammation and activate innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 2004, 27, 813–823. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, F.; Nibali, L.; Parkar, M.; Patel, K.; Suvan, J.; Donos, N. Oxidative stress, systemic inflammation, and severe periodontitis. J. Dent. Res. 2010, 89, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Tucker, P.; Scanlan, A.; Dalbo, V.J. Chronic kidney disease influences multiple systems: Describing the relationship between oxidative stress, inflammation, kidney damage, and concomitant disease. Oxid. Med. Cell. Longev. 2015, 806358, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Piechota-Polanczyk, A.; Fichna, J. Review article: The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Park, Y.J.; Yoo, S.A.; Kim, W.U. Role of endoplasmic reticulum stress in rheumatoid arthritis pathogenesis. J. Korean Med. Sci. 2014, 29, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyan, V. Endothelial dysfunction in cirrhosis: Role of inflammation and oxidative stress. World J. Hepatol. 2015, 27, 443–459. [Google Scholar]
- Sánchez-Domínguez, B.; Bullón, P.; Román-Malo, L.; Marín-Aguilar, F.; Alcocer-Gómez, E.; Carrión, A.M.; Sánchez-Alcazar, J.A.; Cordero, M.D. Oxidative stress, mitochondrial dysfunction and, inflammation common events in skin of patients with Fibromyalgia. Mitochondrion 2015, 21, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Linker-Israeli, M.; Hallegua, D.; Silverman, S.; Silver, D.; Weisman, M.H. Cytokines play an aetiopathogenetic role in fibromyalgia: A hypothesis and pilot study. Rheumatology 2001, 40, 473–479. [Google Scholar] [CrossRef]
- Macchi, M.M.; Bruce, J.N. Human pineal physiology and functional significance of melatonin. Front. Neuroendocrinol. 2004, 25, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.B.; Case, J.D.; Takahashi, Y. Isolation of melatonin and 5- methoxyindole-3-acetic acid from bovine pineal glands. J. Biol. Chem. 1960, 235, 1992–1997. [Google Scholar] [PubMed]
- Klein, D.C.; Moore, R.Y. Pineal N-acetyltransferase and hydroxyindole-O-methyltransferase: Control by the retinohy-pothalamic tract and the suprachiasmatic nucleus. Brain Res. 1979, 174, 245–262. [Google Scholar] [CrossRef]
- Axelrod, J.; Weissbach, H. Enzymatic O-methylation of N- acetylserotonin to melatonin. Science 1960, 131, 1312–1312. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Paredes, S.D.; Manchester, L.C.; Tan, D.X. Reducing oxidative/nitrosative stress: A newly-discovered genre for melatonin. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species. J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Copes, N.; O’Neal-Moffitt, G.; Jin, J.; Buzzeo, R.; Mamcarz, M.; Tan, J.; Cao, C.; Olcese, J.M.; Arendash, G.W.; et al. Melatonin treatment restores mitochondrial function in Alzheimer’s mice: A mitochondrial protective role of melatonin membrane receptor signaling. J. Pineal Res. 2011, 51, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Sawhney, G.; Pandhi, P. The therapeutic potential of melatonin: A review of the science. Med. Gen. Med. 2004, 6, 46. [Google Scholar]
- Arendt, J. Safety of melatonin in long-term use. J. Biol. Rhythm. 1997, 12, 673–681. [Google Scholar] [CrossRef]
- Schernhammer, E.S.; Giobbie-Hurder, A.; Gantman, K.; Savoie, J.; Scheib, R.; Parker, L.M.; Chen, W.Y. A randomized controlled trial of oral melatonin supplementation and breast cancer biomarkers. Cancer Causes Control 2012, 23, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Luboshitzky, R.; Shen-Orr, Z.; Nave, R.; Lavi, S.; Lavie, P. Melatonin administration alters semen quality in healthy men. J. Androl. 2002, 23, 572–578. [Google Scholar] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R. The NF-κB regulatory network. Cardiovacsc. Toxicol. 2006, 6, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Taniguichi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Papandreou, C.N. The Bmi-1/NF-κB/VEGF story: Another hint for protea-some involvement in glioma angiogenesis? J. Cell Commun. Signal. 2013, 7, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Steagall, R.J.; Daniels, C.R.; Dalal, S.; Joyner, W.L.; Singh, M.; Singh, K. Extracellular ubiquitin increases expression of angiogenic molecules and stimulates angiogenesis in cardiac microvascular endothelial cells. Microcirculation 2013, 21, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, R.; Yanagisawa, M. Endothelin system: The double-edged sword in health and disease. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 851–876. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.; Loizidon, M.; Taylor, I. Endothelin-1: A multi-functional molecule in cancer. Br. J. Cancer 2003, 88, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Stow, L.R.; Jacobs, M.E.; Wingo, C.S.; Cain, B.D. Endothelin-1 gene regulation. FASEB J. 2011, 25, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Casado, J.; Sergio, M.; Jimenez Ruiz, S.M.; Zurita, M.S.; Gonzalez-Puga, C.; Rejon, J.D.; Gila, A.; Muñoz de Rueda, P.; Pavón, E.J.; et al. Melatonin reduces endothelin-1 expression and secretion in colon cancer cells through the inactivation of FoxO-1 and NF-κB. J. Pineal Res. 2014, 56, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Dai1, M.; Cui, P.; Yu, M.; Han, J.; Li, H.; Xiu, R. Melatonin modulates the expression of VEGF and HIF-1α induced by CoCl2 in cultured cancer cells. J. Pineal Res. 2008, 44, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; Mcknight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, G.H.; Fardid, R. Oral administration of melatonin modulates the expression of tumor necrosis factor-α (TNF-α) gene in irradiated rat cervical spinal cord. Rep. Pract. Oncol. Radiother. 2015, 20, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Farriol, M.; Venereo, Y.; Orta, X.; Castellanos, J.M.; Segovia-Silvestre, T. In vitro effects of melatonin on cell proliferation in a colon adenocarcinoma line. J. Appl. Toxicol. 2000, 20, 21–24. [Google Scholar] [CrossRef]
- Leon-Blanco, M.M.; Guerrero, J.M.; Reiter, R.J.; Calvo, J.R.; Pozo, D. Melatonin inhibits telomerase activity in the MCF-7 tumor cell line both in vivo and in vitro. J. Pineal Res. 2003, 35, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Blask, D.E.; Dauchy, R.T.; Sauer, L.A.; Krause, J.A. Melatonin uptake and growth prevention in rat hepatoma 7288 CTC in response to dietary melatonin: Melatonin receptor-mediated inhibition of tumor linoleic acid metabolism to the growth signaling molecule 13-hydroxydecadienoic acid and the potential role of phytomelatonin. Carcinogenesis 2004, 25, 951–960. [Google Scholar] [PubMed]
- Padillo, F.J.; Ruiz-Rabelo, J.F.; Cruz, A.; Perea, M.D.; Tasset, I.; Montilla, P. Melatonin and celecoxib improve the outcomes in hamsterrs with experimental pancreatic cancer. J. Pineal Res. 2010, 49, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rabelo, J.; Vázquez, R.; Arjona, A.; Perea, D.; Montilla, P.; Túnez, I. Improvement of capecitabine antitumoral activity by melatonin in pancreatic cancer. Pancreas 2011, 40, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rabelo, J.F.; Vázquez, R.; Perea, M.D.; Cruz, A.; González, R.; Romero, A. Beneficial propierties of melatonin in an experimental model of pancreatic cancer. J. Pineal Res. 2007, 43, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Chilelli, M.; Villa, S.; Cerizza, L.; Tancini, G. Five years survival in metastatic non-small cell lung cancer patients treated with chemotherapy alone or chemotherapy and melatonin: A randomized trial. J. Pineal Res. 2003, 35, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Rovelli, F.; Malugani, F.; Bucovec, R.; Conti, A.; Maestroni, G.J. Anti-angiogenic activity of melatonin in advanced cancer patients. Neuroendocrinol. Lett. 2001, 22, 45–47. [Google Scholar] [PubMed]
- Todisco, M. Chronic lymphocytic leukemia: Long-lasting remission with combination of cyclophosphamide, somatostatin, bromocriptine, retinoids, melatonin, and ACTH. Cancer Biother. Radiopharm. 2009, 24, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Vaghi, M.; Ardizzoia, A.; Malugani, F.; Fumagalli, E.; Bordin, V.; Fumagalli, L.; Bordoni, A.; Mengo, S.; Gardani, G.S.; et al. A phase II study of chemoneuroimmunotherapy with platinum, subcutaneous low-dose interleukin-2 and the pineal neurohormone melatonin (P.I.M.) as a second-line therapy in metastatic melanoma patients progressing on dacarbazine plus interferon-α. In Vivo 2002, 16, 93–96. [Google Scholar] [PubMed]
- Todisco, M.; Casaccia, P.; Rossi, N. Cyclophosphamide plus somatostatin, bromocriptin, retinoids, melatonin and ACTH in the treatment of low-grade non-Hodgkin’s lymphomas at advanced stage: Results of a phase II trial. Cancer Biother. Radiopharm. 2001, 16, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P. Biochemotherapy with standard chemotherapies plus the pineal hormone melatonin in the treatment of advanced solid neoplasms. Pathol. Biol. 2007, 51, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Barni, S.; Mandalà, M.; Ardizzoia, A.; Paolorossi, F.; Vaghi, M.; Longarini, R.; Malugani, F.; Tancini, G. Decreased toxicity and increased efficacy of cancer chemotherapy using the pineal hormone melatonin in metastatic solid tumour patients with poor clinical status. Eur. J. Cancer 1999, 35, 1688–1692. [Google Scholar] [CrossRef]
- Lissoni, P.; Brivio, O.; Brivio, F.; Barni, S.; Tancini, G.; Crippa, D.; Meregalli, S. Adjuvant therapy with the pineal hormone melatonin in patients with lymph node relapse due to malignant melanoma. J. Pineal Res. 1996, 21, 239–342. [Google Scholar] [CrossRef]
- Flo, A.; Flo, V.; Calpena, A.C.; Clares, B. The role of antioxidant in sunscreens: The case of melatonin. In Sunscreens. Properties, Role in Skin Cancer Prevention and Health Effects; Sharp, S.H., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2015; pp. 39–76. [Google Scholar]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, C.; Pommergaard, H.C.; Rosenberg, J.; Gögenur, I. Melatonin’s protective effect against UV radiation: A systematic review of clinical and experimental studies. Photodermatol. Photoimmunol. Photomed. 2014, 30, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.F.; Ramírez, M.L.; Campmany, A.C.; Martínez, A.R.; Naveros, B.C. In vivo and in vitro evaluation of the use of a newly developed melatonin loaded emulsion combined with UV filters as a protective agent against skin irradiation. J. Dermatol. Sci. 2013, 69, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, S. Periodontal diseases: Pathogenesis. Ann. Periodontol. 1996, 1, 821–878. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.W. Pathogenic mechanisms in periodontal disease. Adv. Dent. Res. 1994, 8, 320–328. [Google Scholar] [PubMed]
- World Health Organization (WHO). Oral Health. Fact Sheet N° 318. Available online: http://www.who.int/mediacentre/factsheets/fs318/en/ (accessed on 11 July 2015).
- Scardina, G.A.; Messina, P. Good oral health and diet. J. Biomed. Biotechnol. 2012, 720692, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.H.; Dikshit, M.; Bhaduri, D.; Aghamolaei, T.; Moosavy, S.H.; Azarpaykan, A. Interaction of alcohol use and specific types of smoking on the development of oral cancer. Int. J. High Risk Behav. Addict. 2014, 1, 2120–2120. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.J.; Zheng, T.; Marshall, J.R.; Boffetta, P.; Niu, S.; Brasure, J.; Merletti, F.; Boyle, P. Alcohol, tobacco, diet and the risk of oral cancer: A pooled analysis of three case-control studies. Eur. J. Cancer B 1995, 31, 181–187. [Google Scholar] [CrossRef]
- Moreno-López, L.A.; Esparza-Gómez, G.C.; González-Navarro, A.; Cerero-Lapiedra, R.; González-Hernández, M.J.; Domı́nguez-Rojas, V. Risk of oral cancer associated with tobacco smoking, alcohol consumption and oral hygiene: A case-control study in Madrid, Spain. Oral Oncol. 2000, 36, 170–174. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Bissett, S.M. Periodontitis: Oral complication of diabetes. Endocrinol. Metab. Clin. N. Am. 2013, 42, 849–867. [Google Scholar] [CrossRef] [PubMed]
- Irani, F.C.; Wassall, R.R.; Preshaw, P.M. Impact of periodontal status on oral health-related quality of life in patients with and without type 2 diabetes. J. Dent. 2015, 43, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.; Asman, B. Increased release of free oxygen radicals from peripheral neutrophils in adult periodontitis after Feδ-receptor stimulation. J. Clin. Periodontol. 1996, 23, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Waddington, R.J.; Moseley, R.; Embery, G. Reactive oxygen species: A potential role in the pathogenesis of periodontal diseases. Oral Dis. 2000, 6, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Avezov, K.; Reznick, A.; Aizenbud, D. Oxidative stress in the oral cavity: Sources and pathological outcomes. Respir. Physiol. Neurobiol. 2015, 209, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Firatli, E.; Unal, T.; Onan, U. Antioxidant activities of some chemotherapeutics. A possible mechanism in reducing gingival inflammation. J. Clin. Periodontol. 1994, 21, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Kasznicki, J.; Drzewoski, J.; Pawlowska, E.; Szczepanska, J.; Reiter, R.J. Perspectives on the use of melatonin to reduce cytotoxic and genotoxic effects of methacrylate-based dental materials. J. Pineal Res. 2011, 51, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Moreno, G.; Guardia, J.; Ferrera, M.J.; Cutando, A.; Reiter, R.J. Melatonin in diseases of the oral cavity. Oral Dis. 2010, 16, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Cutando, A.; Gomez-Moreno, G.; Arana, C.; Acuna-Castroviejo, D.; Reiter, R.J. Melatonin: Potential functions in the oral cavity. J. Periodontol. 2007, 78, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Almughrabi, O.M.; Marzouk, K.M.; Hasanato, R.M.; Shafik, S.S. Melatonin levels in periodontal health and disease. J. Periodontal Res. 2013, 48, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Srinath, R.; Acharya, A.B.; Thakur, S.L. Salivary and gingival crevicular fluid melatonin in periodontal health and disease. J. Periodontol. 2010, 81, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Moreno, G.; Cutando-Soriano, A.; Arana, C.; Galindo, P.; Bolaños, J.; Acuña-Castroviejo, D. Melatonin expression in periodontal disease. J. Periodontal Res. 2007, 42, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, N.; Tani-Ishii, N.; Tsukinoki, K.; Chieda, K.; Watanabe, K. Expression of toll-like receptor 2 and 4 in dental pulp. J. Endod. 2007, 33, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gao, Y.J.; Ji, R.R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012, 28, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Ji-Guo, L.; Jia-Ji, L.; Zhao-Ling, W.; Wen-Ke, C.; Pei-Na, W.; Qian, J.; An-Sheng, Z.; Gao-Yi, W.; Guo-Xiong, Z.; Long-Xing, N. Melatonin attenuates inflammation of acute pulpitis subjected to dental pulp injury. Am. J. Transl. Res. 2015, 7, 66–78. [Google Scholar]
- Adem, K.; Sumeyra, A.; Seckin, O.; Ummuhan, T.; Yıldıray, K.; Cenk, F.C.; Sinan, T. Immune modulatory and antioxidant effects of melatonin in experimental periodontitis in rats. Free Radic. Biol. Med. 2013, 55, 21–26. [Google Scholar]
- Gomez-Florit, M.; Ramis, J.M.; Monjo, M. Anti-fibrotic and anti-inflammatory properties of melatonin on human gingival fibroblasts in vitro. Biochem. Pharmacol. 2013, 86, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Nakade, O.; Koyama, H.; Ariji, H.; Yajima, A.; Kaku, T. Melatonin stimulates proliferation and type I collagen synthesis in human bone cells in vitro. J. Pineal Res. 1999, 27, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Avery, S.V. Molecular targets of oxidative stress. Biochem. J. 2011, 434, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 33, 20313–20316. [Google Scholar] [CrossRef]
- Halliwell, B.; Chirico, S.; Crawford, M.A.; Bjerve, K.S.; Gey, K.F. Lipid peroxidation: Its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S. [Google Scholar] [PubMed]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997, 17, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kurcer, Z.E.; Oguz, H.; Ozbilge, F.; Baba, N.; Aksoy, H.; Celik, H.; Cakir, H.; Gezen, M.R. Melatonin protects from ischemia/reperfusion-induced renal injury in rats: This effect is not mediated by proinflammatory cytokines. J. Pineal Res. 2007, 36, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Abraham, P.; Kolli, V.K.; Rabi, S. Melatonin attenuates methotrexate-induced oxidative stress and renal damage in rats. Cell Biochem. Funct. 2010, 28, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Yoshida, M.; Nishijima, H.; Yokosuka, M.; Ligo, M.; Ohtani-Kaneko, R.; Shimada, A.; Hasegawa, T.; Akama, Y.; Hirata, K. Melatonin, a pineal secretory product with antioxidant properties, protects against cisplatin-induced nephrotoxicity in rats. J. Pineal Res. 2001, 30, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Dei-Anane, G.; Liang, R. Donor preconditioning with taurine protects kidney grafts from injury after experimental transplantation. J. Surg. Res. 2008, 146, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Thurman, R.G. Reperfusion injury after liver preservation for transplantation. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nickkholgh, A.; Yi, X.; Bruns, H.; Gross, M.L.; Hoffmann, K.; Mohr, E.; Zorn, M.; Büchler, M.W.; Schemmer, P. Melatonin protects kidney grafts from ischemia/reperfusion injury through inhibition of NF-κB and apoptosis after experimental kidney transplantation. J. Pineal Res. 2009, 46, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.M.; Jones, J.; Turk, D.C.; Russell, I.J.; Matallana, L. An internet survey of 2596 people with fibromyalgia. BMC Musculoskelet. Dis. 2007, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Kalyan-Raman, U.P.; Kalyan-Raman, K.; Yunus, M.B. Muscle pathology in primary fibromyalgia syndrome: A light microscopic, histochemical and ultrastructural study. J. Rheumatol. 1984, 11, 808–813. [Google Scholar] [PubMed]
- Lucas, H.J.; Brauch, C.M.; Settas, L. Fibromyalgia—New concepts of pathogenesis and treatment. Int. J. Immunopathol. Pharmacol. 2006, 19, 5–10. [Google Scholar] [PubMed]
- De Zanette, S.A.; Vercelino, R.; Laste, G.; Rozisky, J.R.; Schwertner, A.; Machado, C.B.; Xavier, F.; Custódio de Souza, I.C.; Deitos, A.; Torres, I.L.S.; et al. Melatonin analgesia is associated with improvement of the descending endogenous pain-modulating system in fibromyalgia: A phase II, randomized, double-dummy, controlled trial. BMC Pharmacol. Toxicol. 2014, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Al-Khalifa, I.I.; Jasim, N.A.; Gorial, F.I. Adjuvant use of melatonin for treatment of fibromyalgia. J. Pineal Res. 2011, 50, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B. Inflammatory bowel disease: Epidemiology, pathogenesis, and therapeutic opportunities. Inflamm. Bowel Dis. 2006, 12, 3–9. [Google Scholar] [CrossRef]
- Geboes, K. Crohn’s disease, ulcerative colitis or indeterminate colitis—How important is it to differentiate? Acta Gastroenterol. Belg. 2001, 64, 197–200. [Google Scholar] [PubMed]
- Sartor, R.B. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Ulcerative colitis. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Coller, J.A.; Corman, M.L.; Nugent, F.W.; Veidenheimer, M.C. Anal complications in Crohn’s disease. Dis. Colon Rectum 1981, 24, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Plauth, M.; Jenss, H.; Meyle, J. Oral manifestations of Crohn’s disease: An analysis of 79 cases. J. Clin. Gastroenterol. 1991, 13, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Seril, D.N.; Liao, J.; Yang, G.Y.; Yang, C.S. Oxidative stress and ulcerative colitis-associated carcinogenesis: Studies in humans and animal models. Carcinogenesis 2003, 24, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Boldeanu, M.V.; Siloşi, I.; Ghiluşi, M.; Cojocaru, M.; Biciuşcă, B.; Avrămescu, C.S.; Cojocaru, I.M.; Ciurea, T.; Albu, D.F.; Siloşi, C.A. Investigation of inflammatory activity in ulcerative colitis. Rom. J. Morphol. Embryol. 2014, 55, 1345–1351. [Google Scholar] [PubMed]
- Radema, S.A.; van Deventer, S.J.; Cerami, A. Interleukin-1-β is expressed predominantly by enterocytes in experimental colitis. Gastroenterology 1991, 100, 1180–1186. [Google Scholar] [PubMed]
- Sakamoto, C. Roles of COX-1 and COX-2 in gastrointestinal pathophysiology. J. Gastroenterol. 1998, 33, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, A.S.; Inoue, A.; Ohashi, R. Long pentraxin 3 (PTX3) expression and release by neutrophils in vitro and in ulcerative colitis. Pathol. Int. 2011, 61, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Amaral, F.A.; Fagundes, C.T.; Coelho, F.M.; Arantes, R.M.; Sousa, L.P. The long pentraxin PTX3 is crucial for tissue inflammation after intestinal ischemia and reperfusion in mice. Am. J. Pathol. 2009, 174, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Bottazzi, B.; Garlanda, C.; Salvatori, G.; Jeannin, P.; Manfredi, A.; Mantovani, A. Pentraxins as a key component of innate immunity. Curr. Opin. Immunol. 2006, 18, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Ouyang, Q.; Jia, D.; Xia, Q. Activation of nuclear factor-κB and its relationship with cytokine gene expression in colonic mucosa of ulcerative colitis patients. Chin. J. Int. Med. 2002, 41, 252–255. [Google Scholar]
- Jobin, C.; Hellerbrand, C.; Licato, L.L.; Brenner, D.A.; Sartor, R.B. Mediation by NF-κB of cytokine induced expression of intercellular adhesion molecule 1(ICAM-1) in an intestinal epithelial cell line, a process blocked by proteasome inhibitors. Gut 1998, 42, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, S.; Araki, T.; Tanaka, K.; Hashimoto, K.; Okita, Y.; Fujikawa, H.; Okugawa, Y.; Toiyama, Y.; Inoue, Y.; Uchida, K.; et al. Identification of patients with developing ulcerative colitis–associated neoplasia by nitrative DNA damage marker 8-nitroguanin expression in rectal mucosa. J. Clin. Gastroenterol. 2013, 47, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Schulzke, J.D. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; Francés, R.; Such, J. Pathological bacterial translocation in cirrhosis: Pathophysiology, diagnosis and clinical implications. Liver Int. 2013, 33, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.P.; Jena, G.B. Melatonin reduces ulcerative colitis-associated local and systemic damage in mice: Investigation on possible mechanisms. Dig. Dis. Sci. 2013, 58, 3460–3474. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Yu, J.P.; Yu, H.G.; Xu, X.M.; Yu, L.L.; Liu, J.; Luo, H.S. Melatonin reduces inflammatory injury through inhibiting NF-κB activation in rats with colitis. Mediat. Inflamm. 2005, 4, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.P.; Jena, G.B.; Kumar, V. Melatonin modulated autophagy and Nrf2 signaling pathways in mice with colitis-associated colon carcinogenesis. Mol. Carcinog. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Tahan, G.; Gramignoli, R.; Marongiu, F.; Aktolga, S.; Cetinkaya, A.; Tahan, V.; Dorko, K. Melatonin expresses powerful anti-inflammatory and antioxidant activities resulting in complete improvement of acetic-acid-induced colitis in rats. Dig. Dis. Sci. 2011, 56, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, H.G.; Jaumdally, R.J.; Idriss, N.K.; Dalia, A.; Blann, A. The effect of melatonin on plasma markers of inflammation and on expression of nuclear factor-κ β in acetic acid-induced colitis in the rat. Dig. Dis. Sci. 2013, 58, 3156–3164. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.G.; Mei, Q.; Yu, J.P.; Xu, J.M.; Xiang, L.; Xu, Y. Effects of melatonin on the expression of iNOS and COX-2 in rat models of colitis. World J. Gastroenterol. 2003, 9, 1307–1311. [Google Scholar] [PubMed]
- Chojnacki, C.; Wisniewska-jarosinska, M.; Walecka-kapica, E.; Klupinska, G.; Jaworek, J.; Chojnacki, J. Evaluation of melatonin effectiveness in the adjuvant treatment of ulcerative colitis. J. Physiol. Pharmacol. 2011, 62, 327–334. [Google Scholar] [PubMed]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- Aho, K.; von Essen, R.; Kurki, P.; Palosuo, T.; Heliovaara, M. Antikeratin antibody and antiperinuclear factor as markers for subclinical rheumatoid disease process. J. Rheumatol. 1993, 20, 1278–1281. [Google Scholar] [PubMed]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J; Johnson, D.S.; Lahey, L.J.; Wagner, C.A.; Cheng, D.; Thiele, G.M.; Michaud, K.; Sayles, H.; Reimold, A.M.; Caplan, L.; et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheum. 2014, 66, 813–821. [Google Scholar]
- Isomaki, H. Long-term outcome of rheumatoid arthritis. Scand. J. Rheumatol. 1992, 95, 3–8. [Google Scholar] [CrossRef]
- Pincus, T.; Callahan, L.F.; Sale, W.G.; Brooks, A.L.; Payne, L.E.; Vaughn, W.K. Severe functional declines, work disability, and increased mortality in seventy five rheumatoid arthritis patients studied over nine years. Arthritis Rheum. 1984, 27, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Lo Gullo, A.; Mandraffino, G.; Imbalzano, E.; Mamone, F.; Aragona, C.O.; D’Ascola, A.; Loddo, S.; Cinquegrani, A.; Alibrandi, A.; Mormina, E.; et al. Toll-like receptor 3 and interleukin 1β expression in CD34+ cells from patients with rheumatoid arthritis: association with inflammation and vascular involvement. Clin. Exp. Rheumatol. 2014, 32, 922–929. [Google Scholar] [PubMed]
- Lo Gullo, A.; Mandraffino, G.; Sardo, M.A.; D’Ascola, A.; Mamone, F.; Loddo, S.; Alibrandi, A.; Imbalzano, E.; Mandraffino, R.; Mormina, E.; et al. Circulating progenitor cells in rheumatoid arthritis: Association with inflammation and oxidative stress. Scand. J. Rheumatol. 2014, 43, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Hollan, I.; Nebuloni, M.; Bottazzi, B.; Mikkelsen, K.; Førre, O.T.; Almdahl, S.M.; Mantovani, A.; Fagerland, M.W.; Aukrust, P.; Meroni, P.L.; Feiring Heart Biopsy Study Group. Pentraxin 3, a novel cardiovascular biomarker, is expressed in aortic specimens of patients with coronary artery disease with and without rheumatoid arthritis. Cardiovasc. Pathol. 2013, 22, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Van de Stadt, L.A.; de Vrieze, H.; Derksen, N.I.; Brouwer, M.; Wouters, D.; van Schaardenburg, D.; Wolbink, G.; Rispens, T. Antibodies to IgG4 hinge can be found in rheumatoid arthritis patients during all stages of disease and may exacerbate chronic antibody-mediated inflammation. Arthritis Rheum. 2014, 66, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- García-González, A.; Gaxiola-Robles, R.; Zenteno-Savín, T. Oxidative stress in patients with rheumatoid arthritis. Rev. Investig. Clin. 2015, 67, 46–53. [Google Scholar]
- Ali, A.M.; Habeeb, R.A.; El-Azizi, N.O.; Khattab, D.A.; Abo-Shady, R.A.; Elkabarity, R.H. Higher nitric oxide levels are associated with disease activity in Egyptian rheumatoid arthritis patients. Rev. Bras. Reumatol. 2014, 54, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, G.J.; Sulli, A.; Pizzorni, C.; Villaggio, B.; Cutolo, M. Melatonin in rheumatoid arthritis: Synovial macrophages show melatonin receptors. Ann. N. Y. Acad. Sci. 2002, 966, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Villaggio, B.; Candido, F.; Valenti, S.; Giusti, M.; Felli, L.; Sulli, A.; Accardo, S. Melatonin influences interleukin-12 and nitric oxide production by primary cultures of rheumatoid synovial macrophages and THP-1 cells. Ann. N. Y. Acad. Sci. 1999, 2, 246–254. [Google Scholar] [CrossRef]
- Bang, J.; Chang, H.W.; Jung, H.R.; Cho, C.H.; Hur, J.A.; Lee, S.I.; Choi, T.H.; Kim, S.H. Melatonin attenuates clock gene Cryptochrome1, which may aggravates mouse anti-type II collagen antibody-induced arthritis. Reumathol. Int. 2012, 32, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Kalpakcioglu, B.; Senel, K. The role of melatonin in rheumatic diseases. Infect. Disord. Drug Targets 2009, 9, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Forrest, C.M.; Mackay, G.M.; Stoy, N.; Stone, T.W.; Darlington, L.G. Inflammatory status and kynurenine metabolism in rheumatoid arthritis treated with melatonin. Br. J. Clin. Pharmacol. 2008, 64, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, G.J.; Otsa, K.; Cutolo, M. Melatonin treatment does not improve rheumatoid arthritis. Br. J. Clin. Pharmacol. 2008, 65, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; García, A.P.; Cano, P.; Esquifino, A.I. Melatonin role in experimental arthritis. Curr. Drug Targets Immune Endocr. Metab. Disord. 2004, 4, 1–10. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, A.; Calpena, A.C.; Clares, B. Evaluating the Oxidative Stress in Inflammation: Role of Melatonin. Int. J. Mol. Sci. 2015, 16, 16981-17004. https://doi.org/10.3390/ijms160816981
Sánchez A, Calpena AC, Clares B. Evaluating the Oxidative Stress in Inflammation: Role of Melatonin. International Journal of Molecular Sciences. 2015; 16(8):16981-17004. https://doi.org/10.3390/ijms160816981
Chicago/Turabian StyleSánchez, Aroha, Ana Cristina Calpena, and Beatriz Clares. 2015. "Evaluating the Oxidative Stress in Inflammation: Role of Melatonin" International Journal of Molecular Sciences 16, no. 8: 16981-17004. https://doi.org/10.3390/ijms160816981