Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels

Department of Medicine and Clinical Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(1), 315; https://doi.org/10.3390/ijms19010315
Submission received: 25 December 2017
/
Revised: 18 January 2018
/
Accepted: 19 January 2018
/
Published: 21 January 2018
(This article belongs to the Special Issue Ion Transporters and Channels in Physiology and Pathophysiology)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Upon stimulation with agonists and shear stress, the vascular endothelium of different vessels selectively releases several vasodilator factors such as nitric oxide and prostacyclin. In addition, vascular endothelial cells of many vessels regulate the contractility of the vascular smooth muscle cells through the generation of endothelium-dependent hyperpolarization (EDH). There is a general consensus that the opening of small- and intermediate-conductance Ca2+-activated K+ channels (SKCa and IKCa) is the initial mechanistic step for the generation of EDH. In animal models and humans, EDH and EDH-mediated relaxations are impaired during hypertension, and anti-hypertensive treatments restore such impairments. However, the underlying mechanisms of reduced EDH and its improvement by lowering blood pressure are poorly understood. Emerging evidence suggests that alterations of endothelial ion channels such as SKCa channels, inward rectifier K+ channels, Ca2+-activated Cl− channels, and transient receptor potential vanilloid type 4 channels contribute to the impaired EDH during hypertension. In this review, we attempt to summarize the accumulating evidence regarding the pathophysiological role of endothelial ion channels, focusing on their relationship with EDH during hypertension.
1. Introduction
Endothelial cells play a critical role in the regulation of vascular tone through the release of several vasorelaxing and vasoconstricting factors [1]. In addition to the release of relaxing factors such as nitric oxide (NO) and prostaglandins, endothelial cells relax the vascular smooth muscle cells through the generation of smooth muscle hyperpolarization in an endothelium-dependent manner [2,3,4]. Although the mechanisms by which endothelial cells produce smooth muscle hyperpolarization may vary depending on the vascular beds and species, both diffusible factors and contact-mediated pathways contribute to the endothelium-dependent smooth muscle hyperpolarization [5,6,7].
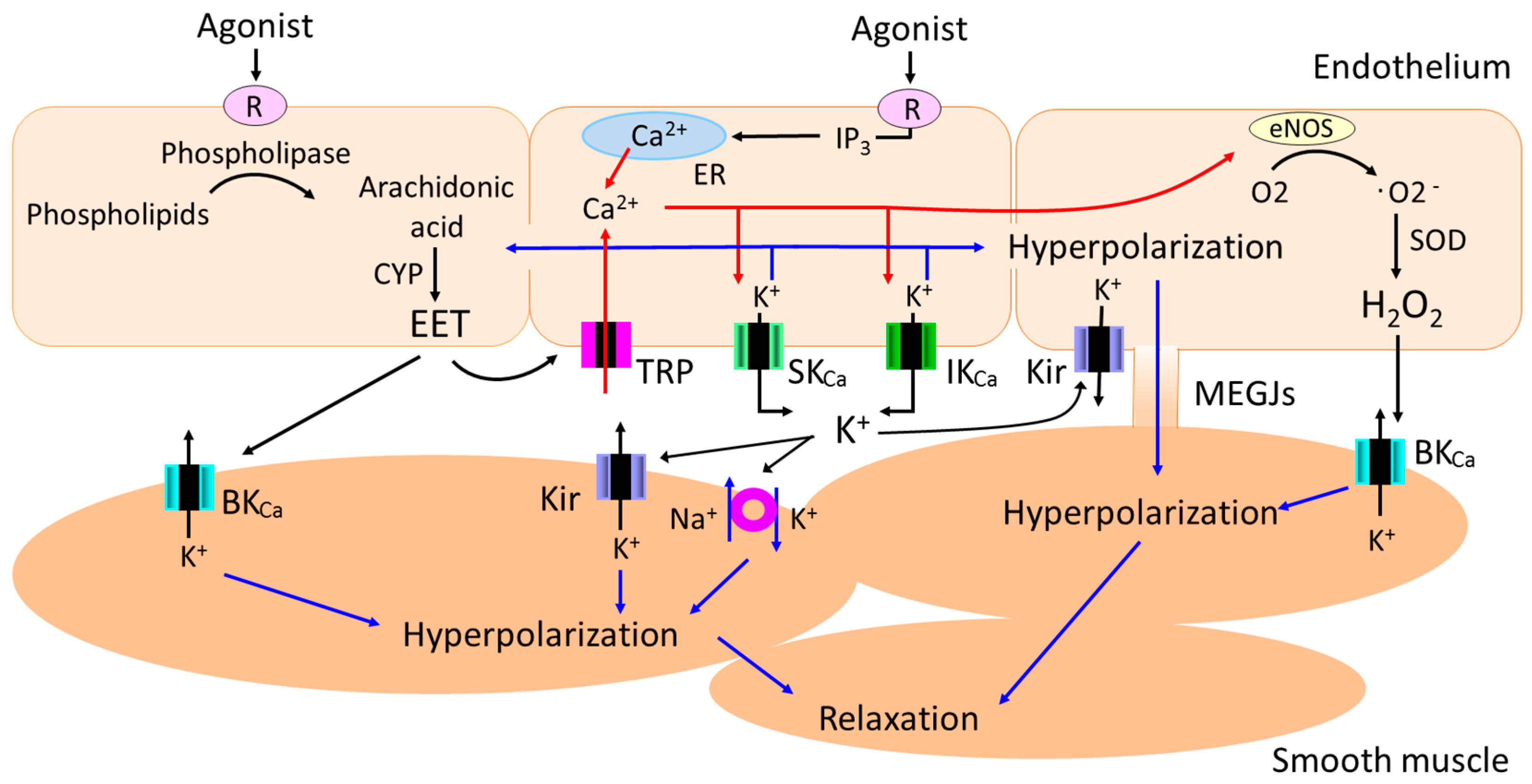
In certain vascular beds and specific conditions, diffusible factors such as epoxyeicosatrienoic acids (EETs) [8,9], K+ ions [10], C-type natriuretic peptide [11], hydrogen peroxide (H2O2) [12] and hydrogen sulfide (H2S) [13] function as endothelium-derived hyperpolarizing factors (EDHFs). For instance, EETs (which play an important role in the regulation of vascular tone, hemostasis, and inflammation [14,15,16]) released from the endothelial cells transfer to the adjacent smooth muscle cells and produce smooth muscle hyperpolarization through the opening of large conductance Ca2+-activated K+ (BKCa) channels (Figure 1).
Endothelial cells also produce smooth muscle hyperpolarization in a contact-dependent manner [5,6,7]. Specifically, endothelium-dependent hyperpolarization (EDH) initiated in endothelial cells with a rise in intracellular calcium and the subsequent activation of small (SKCa) and intermediate conductance (IKCa) Ca2+-activated K+ channels spreads to adjacent smooth muscle cells via myoendothelial gap junctions (MEGJs) in a number of vascular beds [17,18,19,20,21,22,23,24,25]. Although there is a consensus that the intracellular release of Ca2+ from the endoplasmic reticulum (ER) and subsequent activation of the SKCa and IKCa channels in the endothelium is a prerequisite for the generation of EDH [5,6,7], several studies suggest that Ca2+ influx through endothelial non-selective cation channels of the transient receptor potential (TRP) family also plays an important role in EDH via the downstream activation of SKCa and IKCa channels in some vascular beds [26,27,28,29,30]. In certain vascular beds in specific conditions, diffusible factors such as EETs and K+ ions generate EDH through the activation of endothelial TRP channels [14] and endothelial inward rectifier K+ (Kir) channels [31], respectively (Figure 1). As EDH plays a dominant role in endothelium-dependent relaxation in resistance arteries [32,33,34], and pressure in resistance arteries contribute substantially to total peripheral resistance, alterations in the EDH pathway would contribute not only to endothelial function but also to the regulation of arterial blood pressure.
Hypertension is the most important risk factor for cardiovascular disease across the world [35]. Prolonged hypertension causes vascular endothelial dysfunction, which in turn facilitates the progress of atherosclerosis, finally leading to cardiovascular disease [36,37,38]. It is therefore of great importance to uncover the responsible mechanisms and find effective treatments for endothelial dysfunction during hypertension. Although a reduction in NO bioavailability and an enhanced production of endothelium-derived contracting factors contribute to the endothelial dysfunction during hypertension [36,37,39], we have shown that the impaired EDH-mediated hyperpolarization and relaxation contribute to the endothelial dysfunction in mesenteric arteries of spontaneously hypertensive rats (SHR) [40,41]—the most commonly used animal model for human essential hypertension [42]. Reduced EDH during hypertension has also been reported in other models of hypertension [41].
However, the precise mechanisms by which prolonged hypertension impairs EDH-mediated responses are not fully understood. The change in the number and/or function of MEGJs does not seem to be a major contributing factor to impaired EDH-mediated responses during hypertension: first, because previous studies did not observe a positive correlation between EDH-mediated responses and the number of MEGJs in arteries of SHR [43,44]; second, smooth muscle hyperpolarization to 1-EBIO, an endothelial IKCa channel activator [45], was not altered in mesenteric arteries of SHR, indicating that electrical propagation from endothelial cells to smooth muscle cells via MEGJs is preserved during hypertension [44,46].
Vascular endothelial cells express various types of functional ion channels, including SKCa channels, IKCa channels, inward rectifier K+ (Kir) channels, ATP-sensitive K+ (KATP) channels, voltage-gated K+ (Kv) channels, Ca2+-activated Cl− channels (CaCCs), and TRP channels [47,48,49]. Alterations of ion channels in vascular endothelial cells during hypertension have been reported [50], and several studies suggest that alterations of the function and/or expression of endothelial ion channels underpin the impaired EDH in animal models of hypertension [46,51,52,53]. In addition, deletion of some of these endothelial ion channels impairs EDH-mediated responses and increases systemic blood pressure in genetically modified mice [54,55].
These findings strongly suggest that alterations of endothelial ion channels greatly contribute to the impaired EDH-mediated responses during hypertension. Such alterations would represent potential therapeutic targets for the prevention of the endothelial dysfunction associated with hypertension. In this review, we discuss the accumulating evidence regarding alterations of endothelial ion channels, focusing on these channels’ relationship with reduced EDH during hypertension.
2. Endothelium-Dependent Hyperpolarization (EDH) in Animal Models of Hypertension
In 1992, Van de Voorde et al. revealed that endothelium-dependent hyperpolarization in response to acetylcholine (ACh), which appears to be mediated by the opening of KCa channels, is impaired in the aorta from two-kidney, one-clip renal hypertensive rats, and they noted that this impairment contributes to the reduced endothelium-dependent relaxation in this model [56]. However, because their study was conducted without inhibiting the synthesis of NO and prostaglandins (both of which produce endothelium-dependent smooth muscle hyperpolarization in certain vascular beds [57,58]), it was difficult to accurately evaluate the relative contribution of EDH to impaired ACh-induced hyperpolarization during hypertension in their rat hypertension model. In a study using the superior mesenteric arteries of SHR, Fujii et al. demonstrated that ACh-induced, endothelium-dependent hyperpolarization and relaxation resistant to inhibitors of NO and prostaglandin synthesis (and thus EDH-mediated responses), were decreased in SHR compared with normotensive Wistar-Kyoto (WKY) rats [40]. Such alterations in EDH during hypertension appear to be secondary to hypertension because EDH-mediated responses are preserved at the pre-hypertensive stage of genetically hypertensive rats [46,59]. Subsequent studies similarly observed impaired EDH-mediated responses in mesenteric [41,46,52,59,60,61,62], renal [63,64,65], coronary [66], femoral [67], and ocular ciliary [68] arteries of genetically hypertensive rats. Reduced EDH-mediated responses have also been reported in arteries from different types of hypertensive rats such as deoxycorticosterone acetate (DOCA)-salt-induced [69,70] and angiotensin II-induced hypertensive rats [71].
In some circumstances, in particular, when NO-mediated vasorelaxation is compromised, EDH may function as a back-up system to maintain overall endothelial function [72]. Indeed, upregulation of EDH-mediated responses in conjunction with reduced NO-mediated responses has been reported in certain vascular beds from animal models of hypertension [73,74,75,76,77]. Further support for this notion is provided by the observation that EDH-mediated responses are upregulated in endothelial nitric oxide synthase (eNOS) knockout mice [78].
The mechanism by which NO inhibits EDH activity remains an open question. However, several possible mechanisms have been suggested including inhibition of cytochrome P-450 enzyme activity by NO [72], reduced permeability of gap junctions by NO [79], and an inhibitory effect of NO on Ca2+-permeable nonselective cation channels via a protein kinase G (PKG)-dependent phosphorylation pathway [80]. Nevertheless, it was reported that such an upregulation of EDH during hypertension disappeared when hypertension was sustained over a long period of time [65,81]. Thus, in general, prolonged hypertension produces an impairment of EDH, and this impairment appears to underpin the endothelial dysfunction associated with hypertension. In the following sections, we summarize the ionic mechanisms in vascular endothelial cells contributing to the reduced EDH during hypertension.
3. Role of Endothelial Ion Channels in Reduced EDH during Hypertension
3.1. Ca2+-Activated K+ (KCa) Channels
In vascular endothelial cells, both small and intermediate conductance Ca2+-activated K+ channels (SKCa and IKCa) are expressed [47,48], and there is a consensus that the activation of the SKCa and IKCa channels in the endothelium results in the generation of EDH in a number of vascular beds [5,6,7]. Indeed, the involvement of both SKCa and IKCa channels in the generation of EDH has now been supported on the basis of the results from mice deficient in these channels [54,55,82]. In addition, KCa channel-deficient mice show high blood pressure, suggesting that endothelial KCa channels play an important role in blood pressure regulation as well [54,55,82]. Although large conductance Ca2+-activated K+ (BKCa) channels are present in the endothelial cells of some vascular beds [83], there is little evidence showing the involvement of endothelial BKCa channels in the generation of EDH; or of the presence of endothelial BKCa channels in intact vessels [5,7].
Changes in the function and/or expression of endothelial SKCa and IKCa channels during hypertension, in particular, those of SKCa channels, have been reported in various types of animal models of hypertension. Thus in mesenteric arteries of SHR and stroke-prone spontaneously hypertensive rats (SHRSP), the function and/or expression of SKCa channels are reduced and such reduction appears to underpin the impaired EDH-mediated responses in this vascular bed [46,52,84,85]. A contribution of reduced SKCa channels’ function and/or expression to impaired EDH-mediated responses has also been suggested in mesenteric arteries from angiotensin II-induced hypertensive rats [86], testosterone-induced hypertensive rats [87], and endothelial connexin40 mutant mice that exhibit hypertension [88].
By contrast, preserved [46,86,87,88] or even enhanced [77,85] function and/or expression of IKCa channels have been reported in hypertensive rats and mice. The reason why SKCa and IKCa channels are differentially regulated during hypertension is not clear. The preserved or enhanced function and/or expression of IKCa channels might be due to the downregulation of the repressor element 1-silencing transcription factor (a negative regulator of IKCa channels) during hypertension, as has been suggested in mesenteric arteries of SHRSP [85]. In addition, the differential regulation of the two endothelial KCa channels might be related to the difference in the subcellular localization of the two channels, i.e., SKCa channels are localized to the endothelial cell membrane (in particular to adjacent endothelial cell borders where connexins, the proteins that compose gap junctions, are also abundantly expressed [89,90,91]), whereas IKCa channels are, in general, localized to myoendothelial projections which bring endothelial cells into contact with smooth muscle cells through the internal elastic lamina [91,92]. Further studies are required to understand the molecular mechanisms underlying these alterations of endothelial KCa channels during hypertension.
Although these observations suggest a causative relationship between a reduction in SKCa channels and the impairment of EDH during hypertension, our recent study indicates that the reduction in SKCa channels alone is not sufficient to explain the impaired EDH in superior mesenteric arteries of SHRSP [46]. In that study, we observed a significant reduction in SKCa but not IKCa channels’ function and expression in superior mesenteric arteries of SHRSP in which the ACh-induced, EDH-mediated responses were substantially impaired compared to those of WKY rats [46]. However, we also found that the EDH-type relaxations induced by simultaneous activators of SKCa and IKCa channels (NS309 or SKA-31) did not differ significantly between the two rat strains, suggesting that the preserved function of IKCa channels together with residual SKCa channels may help maintain EDH-type relaxation in this vascular bed [46]. Indeed, this prediction is in agreement with the observation that the blockade of SKCa channels with the selective inhibitor apamin only marginally inhibited ACh-induced, EDH-mediated responses in rat mesenteric arteries [46,93]. These findings indicate that, at least in rat mesenteric arteries, mechanisms other than a reduction in SKCa channels would also contribute to the impaired EDH during hypertension, as discussed in the following sections.
3.2. Transient Receptor Potential (TRP) Channels
Changes in the membrane potential evoked by EDH are composed of two phases: an initial rapid phase followed by a sustained phase [94,95]. The initial rapid phase appears to be mediated by the Ca2+ released from intracellular Ca2+ stores, and the sustained phase seems to be due to the Ca2+ influx through the opening of nonselective cation channels activated by intracellular Ca2+ depletion [94,95]. Interestingly, several studies demonstrated that Ca2+ influx through endothelial nonselective cation channels of the transient receptor potential (TRP) family plays a critical role in EDH generation in certain vascular beds [26,27,28,29,30].
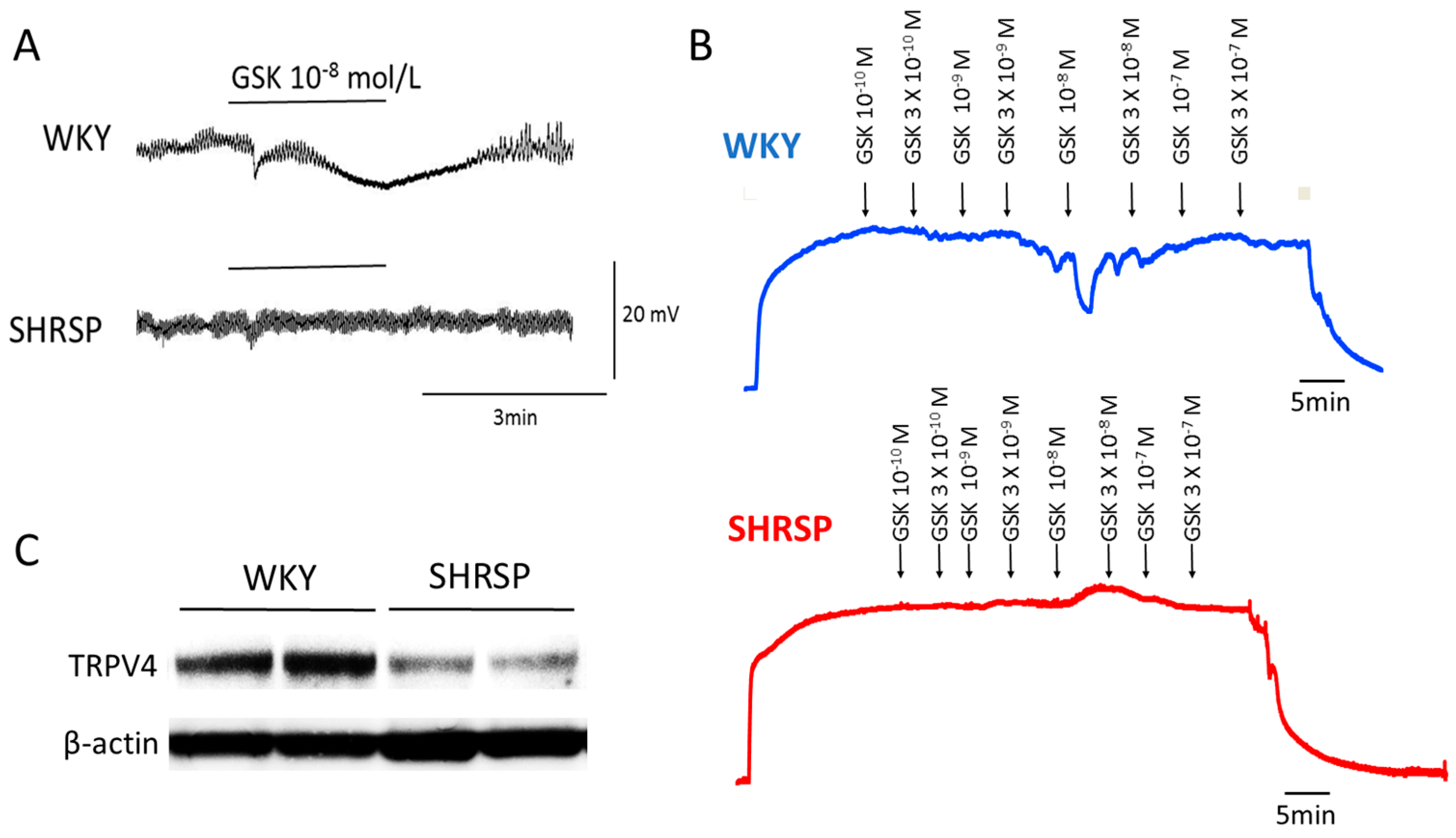
Although limited information is currently available about the role of TRP channels in altered EDH-mediated responses during hypertension, several recent studies shed light on the endothelial TRP vanilloid type 4 (TRPV4) channels as a potential target for this alteration. We recently demonstrated that the opening of endothelial TRPV4 channels and the downstream activation of SKCa and IKCa are involved in ACh-induced, EDH-mediated responses in superior mesenteric arteries of rats [46]. Our study further demonstrated that endothelial TRPV4 and SKCa channels are downregulated in SHRSP but not in WKY rats, contributing to the reduced EDH-mediated responses in superior mesenteric arteries of SHRSP [46] (Figure 2). The downregulation of endothelial TRPV4 seems to be secondary to hypertension because the function and expression of TRPV4 were preserved in pre-hypertensive SHRSP [46].
A decrease in the expression of endothelial TRPV4 and a concomitant impairment of endothelium-dependent relaxation has also been reported in mesenteric arteries of SHR [96]. It thus seems likely that a downregulation of endothelial TRPV4 underpins the impaired EDH in genetically hypertensive rats. Indeed, the causative link between the loss of TRPV4 and the impairment of EDH-mediated responses has been observed in mesenteric arteries of TRPV4 knockout mice [97]. Although another member of the TRP channel family, TRP canonical type 3 (TRPC3), has also been shown to be critical for EDH-mediated responses in rat third-order branch of the superior mesenteric arteries [29], we did not detect any alterations of the function or expression of TRPC3 in superior mesenteric arteries of SHRSP [46].
In contrast, in mesenteric resistance arteries of Wistar rats fed a high-salt diet, the expression of TRPV4 was upregulated despite the significant increase in arterial blood pressure [98]. The reason for the discrepancy between the two studies with respect to the expression of TRPV4 during hypertension is not known. One possible explanation is that the upregulation of TRPV4 may be a compensatory mechanism to maintain endothelial function in salt-induced hypertension. This scenario agrees well with the studies demonstrating that a high-salt diet upregulates EDH to compensate for the loss of NO in mesenteric arteries of rats [74,76].
In the myoendothelial projections (MEPs) of mouse mesenteric arteries, the muscarinic receptor activation of TRPV4 is achieved through the following signaling cascade: muscarinic receptors/diacylglycerol/protein kinase C (PKC) and the PKC anchoring protein A-kinase anchoring protein 150 (AKAP150)/TRPV4 channels, where the AKAP150-dependent clustering of TRPV4 channels plays a critical role in the muscarinic receptor activation of TRPV4 [53]. Moreover, in mesenteric arteries of angiotensin II-induced hypertensive mice, this signaling cascade was dysfunctional due to a loss of AKAP150 at MEPs, resulting in reduced EDH-mediated relaxation [53]. The current density of TRPV4 measured by the patch-clamp technique did not differ in the mesenteric endothelial cells of normotensive versus angiotensin II-induced hypertensive mice, indicating that the function and channel numbers of TRPV4 in endothelial cells per se were similar between normotensive and angiotensin II-induced hypertensive mice [53]. Another study also reported that the protein expression of TRPV4 did not differ in mesenteric arteries of normotensive versus angiotensin II-induced hypertensive mice [99]. Taken together, these findings suggest that a reduced expression of AKAP150 at MEPs underpins impaired EDH in mesenteric arteries of angiotensin II-induced hypertensive mice.
More recently, it was reported that an impairment of the physical and functional interaction of TRPV4-SKCa channels underlies the reduced EDH-mediated responses in small arteries from mice with hypertension induced by a high-salt diet [100]. Of particular interest is that both endothelial SKCa and TRPV4 channels seem to be compromised during hypertension because these two channels are preferentially localized in caveolae, which are specialized lipid rafts on which a number of transduction proteins are located [101]. The issue of whether or not the loss of endothelial SKCa and TRPV4 channels during hypertension is associated with any changes in caveolae warrants further investigation.
3.3. Inward Rectifier K+ (Kir) Channels
A modest increase in the extracellular K+ induces vasodilation through the activation of Na+/K+ ATPase and/or inward rectifier K+ (Kir) channels [102]. Although a few studies have shown impaired K+-induced vasodilator responses in arteries from animal models of hypertension [103,104] and patients with essential hypertension [105], the underlying mechanisms remain poorly understood.
In blood vessels, Kir channels are expressed in both smooth muscle cells and endothelial cells [106,107,108]. However, the specific cellular location of Kir channels appears to differ depending on the vascular beds and vessel size involved [31,107]. In mesenteric resistance arteries of rats and mice, several studies have shown that functional Kir channels are localized to the endothelium [90,108,109,110,111,112]. The endothelial Kir channels are activated not only by extracellular K+ but also by shear stress [112]. Thus, the absence of endothelial Kir channel activation could increase peripheral resistance and hence contribute to blood pressure elevation. In fact, Kir2.1+/− mice showed increased mean blood pressure probably due to the increased microvascular resistance [112].
We have shown that in mesenteric resistance arteries of WKY rats, after the blockade of NO and prostanoids, the overall hyperpolarization to iontophoresed ACh mediated by EDH was significantly reduced by the inhibition of endothelial Kir channels with barium, whereas barium was without effect in SHR arteries [90]. These findings suggest that ACh-induced EDH is augmented by the activation of endothelial Kir channels in WKY but not in SHR mesenteric resistance arteries. The lack of endothelial Kir channels’ involvement in ACh-induced EDH in the SHR in that study does not appear to be related to the alteration of the function or expression of endothelial Kir channels per se, because barium-sensitive hyperpolarizing responses to exogenously applied K+ and the mRNA expression of the Kir2.1 gene in mesenteric resistance arteries did not differ between the WKY rats and SHR [90].
Unaltered K+-induced vasodilator responses have also been reported in mesenteric arteries of angiotensin II-induced hypertensive mice [111] and in cortical parenchymal arterioles of SHR [113], where endothelial Kir2.1 channels play a dominant role in K+-induced vasodilation [111,114]. In addition, in the smooth muscle cells from mesenteric arteries, the protein expression level of the Kir2.1 did not differ significantly between normotensive blood pressure (BPN) and hypertensive blood pressure (BPH) mice [115]. Together, the above-described findings suggest that the function and expression of endothelial Kir channels per se appear to be preserved during hypertension. Thus, the underlying mechanism of the lack of endothelial Kir channels’ involvement in ACh-induced EDH in mesenteric resistance arteries of the SHR in our study is yet to be determined. Nevertheless, ACh-evoked depolarization mediated by the opening of endothelial Ca2+-activated Cl− channels (CaCCs) may result in a failure to activate endothelial Kir channels in this vascular bed [90,116]. This scenario is further supported by the observations that the inhibition of Cl− channels potentiates the EDH-mediated relaxation through the activation of Kir channels in rat mesenteric arteries [108,117].
By contrast, Weston et al. reported that a functional loss of Kir channels in mesenteric resistance arteries of SHR is due to a significant downregulation in the expression of Kir2.1 protein [52]. The discrepancy between the two studies of Kir channel expression cannot be attributable to the difference in the vessel size or the age of the animals used, because the two studies were conducted in mesenteric resistance arteries of rats at about the same age (12 weeks in the Goto et al. study vs. 12–16 weeks in the Weston et al. study) [52,90]. The difference in the expression of Kir channels might be due to the difference in the systolic blood pressure levels of the SHR used (199 ± 5 mmHg in the Goto et al. study vs. 235 ± 5 mmHg in the Weston et al. study) [52,90]. Further studies are required to elucidate the mechanisms underlying the lack of endothelial Kir channels involvement in EDH in the mesenteric resistance arteries of SHR.
3.4. Voltage-Gated K+ (Kv) Channels and ATP-Sensitive K+ (KATP) Channels
Voltage-gated K+ (Kv) channels are activated by membrane depolarization so that the resting membrane potential becomes more negative, and thus these channels contribute to the regulation of vascular smooth muscle tone [118]. Kv channels are highly expressed in vascular smooth muscle cells, and a number of studies have reported the reduced function and expression of smooth muscle Kv channels during hypertension [119]. Several studies have suggested the expression of Kv channels in vascular endothelial cells; however, the physiological role of endothelial Kv channels is poorly understood [48]. The involvement of endothelial Kv channels in EDH generation in guinea-pig carotid artery endothelial cells has been suggested [120]. Endothelial cells of the rat aorta from SHRSP have been reported to show lower levels of Kv1.5 function and expression compared to those from WKY rats [121]. However, it is not yet known whether a decreased expression of endothelial Kv channels contributes to the impaired EDH during hypertension.
ATP-sensitive K+ (KATP) channels are abundantly expressed in vascular smooth muscle cells [106], and several studies have reported the reduced function of KATP channels during hypertension [122,123]. The expression of endothelial KATP channels was also reported in some vascular beds [48]. Although endothelial KATP channels appear to contribute to vasodilation induced by hyperosmolality [124] or hypoxia [125], it is not known whether endothelial KATP channels contribute to EDH and whether function and expression of endothelial KATP channels are altered during hypertension.
As stated above, little is currently known about the role of endothelial Kv and KATP channels in the regulation of EDH during hypertension. Nevertheless, it has been shown that both H2O2 [126,127] and H2S [13,128] produce hyperpolarization of the vascular smooth muscle cells through the activation of Kv and/or KATP channels in certain vascular beds. Interestingly, in the mesenteric arteries of SHR, relaxation response to exogenously applied H2O2 is reduced due to an impaired Kv channels activity [129]. In addition, reduction in endogenous H2S levels and H2S-induced vasorelaxation have been shown in animal models of hypertension as well as hypertensive patients [130,131]. Whether or not endothelial Kv and/or KATP channels contribute to the impaired EDH during hypertension in arteries where H2O2 and/or H2S act as EDHF requires further investigation.
3.5. Ca2+-Activated Chloride Channels (CaCCs)
In addition to their expression in vascular smooth muscle cells, Ca2+-activated Cl− channels (CaCCs) have been shown to be present in vascular endothelial cells [47]. Because the resting membrane potential of vascular smooth muscle cells is more negative than the reversal potential for chloride ion (Cl−), the opening of Cl− channels leads to an efflux of Cl− ions and depolarizes the membrane potential [132]. The activation of endothelial CaCCs would also evoke membrane depolarization, and the depolarization could counteract EDH induced by the activation of endothelial SKCa and IKCa channels because the vascular smooth muscle and endothelial cells behave as an electrical syncytium due to the presence of gap junctions [133]. Indeed, the effects of CaCCs have been suggested to counteract those of KCa channels in vascular endothelial cells [134,135,136].
Several studies identified that the transmembrane member 16A (TMEM16A; also known as Ano1) protein is a molecular counterpart for CaCCs [137,138,139]. In arteries of SHR, the TMEM16A expression level is increased and the systemic blockade of TMEA16A by peripheral administration of a TMEA16A inhibitor, T16Ainh-A01, reduced blood pressure in this model [140]. In angiotensin II-induced hypertensive mice, the knockout of endothelial-specific TMEM 16A lowered the systolic blood pressure and ameliorated endothelial function, whereas the overexpression of endothelial-specific TMEM 16A elevated the systolic blood pressure and deteriorated endothelial function [141]. These findings suggest that CaCC TMEM 16A located on the endothelium contributes to the impairment of endothelial function and thus blood pressure elevation in hypertension.
In mesenteric resistance arteries of SHR, after the blockade of EDH with KCa channel inhibitors, iontophoresed ACh evoked an endothelium-dependent depolarization through the opening of CaCCs that was effectively absent in WKY arteries [116]. In that study, the inhibition of the ACh-evoked depolarization by CaCC inhibitors improved the ACh-induced EDH, suggesting that the upregulation of endothelial CaCCs at least partly underpins the impaired EDH in the mesenteric arteries of SHR [116] (Figure 3). Although the molecular identity for the CaCC observed in that study was unknown at the time [116], the subsequent findings described above strongly suggest that TMEM 16A is the molecular counterpart for CaCCs in mesenteric arteries of SHR. Thus, the impact of endothelial CaCC TMEM 16A on altered EDH during hypertension and therefore on blood pressure regulation warrants further investigation.
4. Therapeutic Implications
A number of studies (including our group’s) have demonstrated that antihypertensive treatment improves the impairment of EDH-mediated responses associated with hypertension [41,61,142,143,144,145,146,147]. As several classes of antihypertensive drugs such as angiotensin-converting enzyme (ACE) inhibitors [61,142,143,144,145,146], angiotensin II type 1 receptor blockers [144,145,147], angiotensin II receptor–neprilysin inhibitor [147], and diuretics [143] were effective in improving reduced EDH-mediated responses associated with hypertension, blood pressure lowering per se appears to play a crucial role in these improvements. However, the inhibition of renin–angiotensin system (RAS) by a RAS inhibitor may have an additional benefit against EDH beyond, or in combination with, blood pressure lowering. Indeed, in the mesenteric arteries of WKY rats, treatment with RAS inhibitors but not diuretics prevented the age-related decline in EDH-mediated responses despite similar effects on blood pressure [148,149].
The underlying mechanisms by which antihypertensive treatments improve the reduced EDH-mediated responses associated with hypertension are currently far from clear. In mesenteric arteries of SHR, a reduction of blood pressure with enalapril (an ACE inhibitor) augmented EDH with no significant changes in the endothelial connexin expression or the number of MEGJs, suggesting that an upregulation of gap junctions was not involved in the restoration of EDH in that study [44]. Several studies have demonstrated that ACE inhibitors restore the impairment of EDH-mediated responses through the upregulation of SKCa and/or IKCa channels in hypertensive rats [150,151]. In line with these observations, it has been reported that angiotensin II inhibits KCa channels in isolated vascular smooth muscle cells [152,153].
Another possible explanation is that the improvement of EDH-mediated responses results from the reduced oxidative stress during antihypertensive treatments. This possibility is based on the report that oxidative stress impairs both SKCa and IKCa channel functions via a downregulation of these two endothelial KCa channels in rat mesenteric arteries [154]. However, caution should be exercised in generalizing the results of that report [154], because other studies have demonstrated that EDH-mediated responses are insensitive to oxidative stress in other vascular beds [155,156]. Further studies are needed to determine whether an upregulation of endothelial KCa channels contributes to the improvement of EDH-mediated responses during antihypertensive treatments, and if so, to elucidate the mechanisms underlying the regulation of endothelial KCa channels in hypertension.
5. Endothelium-Dependent Hyperpolarization (EDH) in Human Hypertension
As discussed in the preceding paragraphs, EDH-mediated responses are reduced in animal models of hypertension. However, caution should be exercised in extrapolating data from animals to humans. Although many studies mentioned above were conducted using SHR strain, it has been suggested that this strain has significant genetic diversity between laboratories [157]. Such genetic diversity might cause difference in EDH-mediated responses independently of blood pressure. Caution should also be taken when interpreting the role of ion channels for EDH in genetically manipulated mice because the difference in genetic background and genetic compensation could influence the phenotype [158]. Thus, it is of particular importance to gather human data.
Although the contribution of EDH to the regulation of vasomotor tone has been demonstrated in a number of human arteries [159], only a few studies have investigated the role of EDH in human hypertension. In patients with essential hypertension, the upregulation of EDH-mediated responses compensates for the loss of NO to maintain endothelium-dependent vasodilation in the forearm microcirculation [160,161] and in the small arteries dissected from gluteal biopsies [162].
By contrast, both decreased NO-mediated and decreased EDH-mediated responses have been reported in the great omental arteries of essential hypertensive patients [163]. Decreased EDH-mediated responses have also been shown in small myometrial arteries from patients with preeclampsia, a condition in pregnancy characterized by hypertension and proteinuria [164,165,166]. The compromised myoendothelial communication [165] and/or downregulation of endothelial IKCa channels due to the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2-derived superoxide [167] may be, at least in part, associated with impaired EDH-mediated responses in patients with preeclampsia. Although treatment with ACE inhibitors improved endothelial function through the upregulation of EDH-mediated responses in the internal thoracic artery of hypertensive patients [168], the underlying mechanisms of this improvement are not known.
6. Conclusions
Endothelium-dependent hyperpolarization (EDH) and EDH-mediated relaxation are impaired during prolonged hypertension. Accumulating evidence from animal models of hypertension suggests that alterations of endothelial ion channels contribute to the impaired EDH-mediated responses during hypertension. A number of studies have demonstrated that the reduced function and/or expression of endothelial SKCa channels play a causative role in this impairment. In addition, emerging evidence reveals that the decrease in EDH-mediated responses in hypertension is accompanied by a loss of function and/or expression of endothelial TRPV4 channels. The membrane depolarization evoked by the activation of endothelial CaCCs appears to counteract the hyperpolarizing influence of endothelial KCa and Kir channels, thereby leading to a reduction in EDH-mediated responses during hypertension. Thus, it is reasonable to suggest that alterations of these endothelial ion channels could have a substantial impact on EDH during hypertension.
Although some studies have suggested a causative relationship between oxidative stress and the loss of endothelial KCa channels, the mechanisms underlying these alterations of endothelial ion channels during hypertension remain largely unknown. The existence of EDH has been demonstrated in a number of human arteries. Moreover, EDH-mediated responses are decreased in some but not all arteries from patients with hypertension. As EDH is a dominant vasodilator in resistance arteries (that play an important role in blood pressure control), an in-depth understanding of the mechanisms that regulate the alteration of endothelial ion channels during hypertension could pave the way for new treatments for hypertension and related cardiovascular diseases.
Acknowledgments
This work was supported in part by a Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (16K09645), Tokyo, Japan.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACE | Angiotensin converting enzyme |
| ACh | Acetylcholine |
| AKAP150 | A-kinase anchoring protein 150 |
| BKCa | Large conductance Ca2+-activated K+ |
| CaCC | Ca2+-activated Cl− channel |
| CYP | Cytochrome P450 |
| DOCA | Deoxycorticosterone acetate |
| EDH | Endothelium-dependent hyperpolarization |
| EDHF | Endothelium-derived hyperpolarizing factor |
| EETs | Epoxyeicosatrienoic acids |
| eNOS | endothelial nitric oxide synthase |
| GSK | GSK1016790A |
| H2O2 | Hydrogen peroxide |
| H2S | Hydrogen sulfide |
| IKCa | Intermediate–conductance Ca2+-activated K+ |
| KATP | ATP-sensitive K+ |
| Kir | Inward rectifier K+ |
| Kv | Voltage-gated K+ |
| l-NAME | Nω-nitro-l-arginine |
| MEGJs | Myoendothelial gap junctions |
| MEPs | Myoendothelial projections |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NO | Nitric oxide |
| PKC | Protein kinase C |
| RAS | Renin–angiotensin system |
| SHR | Spontaneously hypertensive rats |
| SHRSP | Stroke-prone spontaneously hypertensive rats |
| SKCa | Small-conductance Ca2+-activated K+ |
| TMEM16A | Transmembrane member 16A |
| TRP | Transient receptor potential |
| TRPC3 | TRP canonical type 3 |
| TRPV4 | TRP vanilloid type 4 |
| WKY | Wistar-Kyoto |
References
- Hill, C.E.; Phillips, J.K.; Sandow, S.L. Heterogeneous control of blood flow amongst different vascular beds. Med. Res. Rev. 2001, 21, 1–60. [Google Scholar] [CrossRef]
- Feletou, M.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988, 93, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Suzuki, H.; Weston, H.A. Acetylcholine releases endothelium-derived hyperpolarizing factors and EDRF from rat blood vessels. Br. J. Pharmacol. 1988, 95, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; McPherson, G.A. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br. J. Pharmacol. 1992, 105, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Busse, R.; Edwards, G.; Feletou, M.; Fleming, I.; Vanhoutte, P.M.; Weston, A.H. EDHF: Bringing the concepts together. Trends Pharmacol. Sci. 2002, 23, 374–380. [Google Scholar] [CrossRef]
- Sandow, S.L. Factors, fiction and endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 2004, 31, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 2017, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B.; Gebremedhin, D.; Pratt, P.F.; Harder, D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996, 78, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [PubMed]
- Edwards, G.; Dora, K.A.; Gardener, M.J.; Garland, C.J.; Weston, A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998, 396, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.D.; Nilsson, H.; Ahluwalia, A.; Hobbs, A.J. Release of C-type natriuretic peptide accounts for the biological activity of endothelium-derived hyperpolarizing factor. Proc. Natl. Acad. Sci. USA 2003, 100, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Matoba, T.; Shimokawa, H.; Nakashima, M.; Hirakawa, Y.; Mukai, Y.; Hirano, K.; Kanaide, H.; Takeshita, A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Investig. 2000, 106, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Campbell, W.B.; Fleming, I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers. Arch. 2010, 459, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.T.; Campbell, W.B.; Gutterman, D.D. Beyond vasodilatation: Non-vasomotor roles of epoxyeicosatrienoic acids in the cardiovascular system. Trends Pharmacol. Sci. 2007, 28, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Hashitani, H.; Suzuki, H. K+ channels which contribute to the acetylcholine-induced hyperpolarization in smooth muscle of the guinea-pig submucosal arteriole. J. Physiol. 1997, 501, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.J.; Gallagher, N.; Dora, K.A.; Garland, C.J. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J. Physiol. 2003, 553, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Chaytor, A.T.; Evans, W.H.; Griffith, T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998, 508, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Imaeda, K.; Suzuki, H. Endothelium-dependent hyperpolarization and intracellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999, 514, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Hill, C.E. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 2000, 86, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.A.; Tare, M.; Parkington, H.C. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J. Physiol. 2001, 531, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Tare, M.; Coleman, H.A.; Hill, C.E.; Parkington, H.C. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ. Res. 2002, 90, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Kansui, Y.; Abe, I.; Iida, M. Critical role of gap junctions in endothelium-dependent hyperpolarization in rat mesenteric arteries. Clin. Exp. Pharmacol. Physiol. 2002, 29, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Sandow, S.L.; Gallagher, N.T.; Takano, H.; Rummery, N.M.; Hill, C.E.; Garland, C.J. Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J. Vasc. Res. 2003, 40, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Gonzales, A.L.; Crnich, R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 2009, 104, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Gonzales, A.L.; Garcia, Z.I. A dietary agonist of transient receptor potential cation channel V3 elicits endothelium-dependent vasodilation. Mol. Pharmacol. 2010, 77, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Senadheera, S.; Kim, Y.; Grayson, T.H.; Toemoe, S.; Kochukov, M.Y.; Abramowitz, J.; Housley, G.D.; Bertrand, R.L.; Chadha, P.S.; Bertrand, P.P.; et al. Transient receptor potential canonical type 3 channels facilitate endothelium-derived hyperpolarization-mediated resistance artery vasodilator activity. Cardiovasc. Res. 2012, 95, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Kochukov, M.Y.; Balasubramanian, A.; Abramowitz, J.; Birnbaumer, L.; Marrelli, S.P. Activation of endothelial transient receptor potential C3 channel is required for small conductance calcium-activated potassium channel activation and sustained endothelial hyperpolarization and vasodilation of cerebral artery. J. Am. Heart Assoc. 2014, 3, E000913. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. Boosting the signal: Endothelial inward rectifier K+ channels. Microcirculation 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Illiano, S.; Vanhoutte, P.M. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-l-arginine in rats. Am. J. Physiol. 1992, 263, H1090–H1094. [Google Scholar] [CrossRef] [PubMed]
- Hwa, J.J.; Ghibaudi, L.; Williams, P.; Chatterjee, M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. Am. J. Physiol. 1994, 266, H952–H958. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Yasutake, H.; Fujii, K.; Owada, M.K.; Nakaike, R.; Fukumoto, Y.; Takayanagi, T.; Nagao, T.; Egashira, K.; Fujishima, M.; et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 1996, 28, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Lawers, C.M.; Hoorn, S.V.; Rodgers, A. Global burden of blood-pressure-related disease. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Vanhoutte, P.M. Endothelial dysfunction and atherosclerosis. Eur. Heart J. 1997, 18, E19–E29. [Google Scholar] [CrossRef]
- Schiffrin, E.L. Beyond blood pressure: The endothelium and atherosclerosis progression. Am. J. Hypertens. 2002, 15 (Suppl. 1), S115–S122. [Google Scholar] [CrossRef]
- Halcox, J.P.; Schenke, W.H.; Zalos, G.; Mincemoyer, R.; Prasad, A.; Waclawiw, M.A.; Nour, K.R.; Quyyumi, A.A. Prognostic value of coronary vascular endothelial dysfunction. Circulation 2002, 106, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Feletou, M.; Taddei, S. Endothelium-dependent contractions in hypertension. Br. J. Pharmacol. 2005, 144, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Tominaga, M.; Ohmori, S.; Kobayashi, K.; Koga, T.; Takata, Y.; Fujishima, M. Decreased endothelium-dependent hyperpolarization to acetylcholine in smooth muscle of the mesenteric artery of spontaneously hypertensive rats. Circ. Res. 1992, 70, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Kansui, Y.; Iida, M. Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: Response to chronic treatment with renin-angiotensin system inhibitors. Clin. Exp. Pharmacol. Physiol. 2004, 31, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Nabika, T.; Nara, Y.; Ikeda, K.; Endo, J.; Yamori, Y. Genetic heterogeneity of the spontaneously hypertensive rat. Hypertension 1991, 18, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Bramich, N.J.; Bandi, H.P.; Rummery, N.M.; Hill, C.E. Structure, function, and endothelium-derived hyperpolarizing factor in the caudal artery of the SHR and WKY rat. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.; Goto, K.; Chaston, D.J.; Brackenbury, T.D.; Meaney, K.R.; Falck, J.R.; Wojcikiewicz, R.J.; Hill, C.E. Enalapril treatment alters the contribution of epoxyeicosatrienoic acids but not gap junctions to endothelium-derived hyperpolarizing factor activity in mesenteric arteries of spontaneously hypertensive rats. J. Pharmacol. Exp. Ther. 2009, 330, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.D.; Dora, K.A.; Ings, N.T.; Crane, G.J.; Garland, C.J. Activation of endothelial cell IK(Ca) with 1-ethyl-2-benzimidazolinone evokes smooth muscle hyperpolarization in rat isolated mesenteric artery. Br. J. Pharmacol. 2001, 134, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Goto, K.; Kiyohara, K.; Kansui, Y.; Murakami, N.; Haga, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Downregulation of endothelial transient receptor potential vanilloid type 4 channel and small-conductance of Ca2+-activated K+ channels underpins impaired endothelium-dependent hyperpolarization in hypertension. Hypertension 2017, 69, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. Potassium channels in the peripheral microcirculation. Microcirculation 2005, 12, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Ohya, Y.; Fujishima, M. Alterations of ion channels in vascular muscle cells and endothelial cells during hypertension and aging. In Advances in Cell. Aging and Gerontology; Hagen, T., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2002; Volume 11, pp. 165–182. ISBN 978-0-08-093020-6. [Google Scholar]
- Grgic, I.; Kaistha, B.P.; Hoyer, J.; Köhler, R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses-relevance to cardiovascular pathologies and drug discovery. Br. J. Pharmacol. 2009, 157, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.H.; Porter, E.L.; Harno, E.; Edwards, G. Important of endothelial SKCa channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br. J. Pharmacol. 2010, 160, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Dalsgaard, T.; Bonev, A.D.; Hill-Eubanks, D.C.; Kotlikoff, M.I.; Scott, J.D.; Santana, L.F.; Nelson, M.T. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci. Signal. 2014, 7, ra66. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Bonev, A.D.; Gross, T.P.; Eckman, D.M.; Brayden, J.E.; Bond, C.T.; Adelman, J.P.; Nelson, M.T. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ. Res. 2003, 93, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Brähler, S.; Kaistha, A.; Schmidt, V.J.; Wölfle, S.E.; Busch, C.; Kaistha, B.P.; Kacik, M.; Hasenau, A.L.; Grgic, I.; Si, H.; et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 2009, 119, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Van de Voorde, J.; Vanheel, B.; Leusen, I. Endothelium-dependent relaxation and hyperpolarization in aorta from control and renal hypertensive rats. Circ. Res. 1992, 70, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tare, M.; Parkington, H.C.; Coleman, H.A.; Neild, T.O.; Dusting, G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature 1990, 346, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Parkington, H.C.; Tare, M.; Tonta, M.A.; Coleman, H.A. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993, 465, 459–476. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ohmori, S.; Tominaga, M.; Abe, I.; Takata, Y.; Ohya, Y.; Kobayashi, K.; Fujishima, M. Age-related changes in endothelium-dependent hyperpolarization in the rat mesenteric artery. Am. J. Physiol. 1993, 265, H509–H516. [Google Scholar] [CrossRef] [PubMed]
- Mantelli, L.; Amerini, S.; Ledda, F. Roles of nitric oxide and endothelium-derived hyperpolarizing factor in vasorelaxant effect of acetylcholine as influenced by aging and hypertension. J. Cardiovasc. Pharmacol. 1995, 25, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Kähönen, M.; Mäkynen, H.; Wu, X.; Arvola, P.; Pörsti, I. Endothelial function in spontaneously hypertensive rats: Influence of quinapril treatment. Br. J. Pharmacol. 1995, 115, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Sunano, S.; Watanabe, H.; Tanaka, S.; Sekiguchi, F.; Shimamura, K. Endothelium-derived relaxing, contracting and hyperpolarizing factors of mesenteric arteries of hypertensive and normotensive rats. Br. J. Pharmacol. 1999, 126, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, H.; Hirata, Y.; Suzuki, E.; Sugimoto, T.; Matsuoka, H.; Kikuchi, K.; Nagano, T.; Hirobe, M.; Sugimoto, T. Mechanisms for altered endothelium-dependent vasorelaxation in isolated kidneys from experimental hypertensive rats. Am. J. Physiol. 1993, 264, H1535–H1541. [Google Scholar] [CrossRef] [PubMed]
- Dohi, Y.; Kojima, M.; Sato, K. Benidipine improves endothelial function in renal resistance arteries of hypertensive rats. Hypertension 1996, 28, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Büssemaker, E.; Popp, R.; Fisslthaler, B.; Larson, C.M.; Fleming, I.; Busse, R.; Brandes, R.P. Aged spontaneously hypertensive rats exhibit a selective loss of EDHF-mediated relaxation in the renal artery. Hypertension 2003, 42, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Pérez, S.; Navarro-Cid, J.; de las Heras, N.; Cediel, E.; Sanz-Rosa, D.; Ruilope, L.M.; Cachofeiro, V.; Lahera, V. Relevance of endothelium-derived hyperpolarizing factor in the effects of hypertension on rat coronary relaxations. J. Hypertens. 2001, 19, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ohyanagi, M.; Koida, S.; Ueda, A.; Ishiko, K.; Iwasaki, T. Effects of endothelium-derived hyperpolarizing factor and nitric oxide on endothelial function in femoral resistance arteries of spontaneously hypertensive rats. Hypertens. Res. 2006, 29, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Watabe, H.; Cui, J.; Abe, S.; Sato, N.; Ishikawa, H.; Yoshitomi, T. Reduced effects of endothelium-derived hyperpolarizing factor in ocular ciliary arteries from spontaneous hypertensive rats. Exp. Eye Res. 2010, 90, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Mäkynen, H.; Kähönen, M.; Wu, X.; Arvola, P.; Pörsti, I. Endothelial function in deoxycorticosterone-NaCl hypertension: Effect of calcium supplementation. Circulation 1996, 93, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Adeagbo, A.S.; Joshua, I.G.; Falkner, C.; Matheson, P.J. Tempol, an antioxidant, restores endothelium-derived hyperpolarizing factor-mediated vasodilation during hypertension. Eur. J. Pharmacol. 2003, 481, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Dal-Ros, S.; Bronner, C.; Schott, C.; Kane, M.O.; Chataigneau, M.; Schini-Kerth, V.B.; Chataigneau, T. Angiotensin II-induced hypertension is associated with a selective inhibition of endothelium-derived hyperpolarizing factor-mediated responses in the rat mesenteric artery. J. Pharmacol. Exp. Ther. 2009, 328, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Stepp, D.W.; Chilian, W.M. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H459–H465. [Google Scholar] [CrossRef] [PubMed]
- Bund, S.J. Influence of mode of contraction on the mechanism of acetylcholine-mediated relaxation of coronary arteries from normotensive and spontaneously hypertensive rats. Clin. Sci. 1998, 94, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Sofola, O.A.; Knill, A.; Hainsworth, R.; Drinkhill, M. Change in endothelial function in mesenteric arteries of Sprague-Dawley rats fed a high salt diet. J. Physiol. 2002, 543, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Sendao, O.; Liveira, A.P.; Bendhack, L.M. Relaxation induced by acetylcholine involves endothelium-derived hyperpolarizing factor in 2-kidney 1-clip hypertensive rat carotid arteries. Pharmacology 2004, 72, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Kansui, Y.; Oniki, H.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Upregulation of endothelium-derived hyperpolarizing factor compensates for the loss of nitric oxide in mesenteric arteries of dahl salt-sensitive hypertensive rats. Hypertens. Res. 2012, 35, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Simonet, S.; Isabelle, M.; Bousquenaud, M.; Clavreul, N.; Félétou, M.; Vayssettes-Courchay, C.; Verbeuren, T.J. KCa 3.1 channels maintain endothelium-dependent vasodilatation in isolated perfused kidneys of spontaneously hypertensive rats after chronic inhibition of NOS. Br. J. Pharmacol. 2012, 167, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Waldron, G.J.; Ding, H.; Lovren, F.; Kubes, P.; Triggle, C.R. Acetylcholine-induced relaxation of peripheral arteries isolated from mice lacking endothelial nitric oxide synthase. Br. J. Pharmacol. 1999, 128, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; McMahon, D.G. Modulation of hybrid bass retinal gap junctional channel gating by nitric oxide. J. Physiol. 1997, 499, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Garland, C.J.; Kwan, H.Y.; Yao, X. Endothelial cell protein kinase G inhibits release of EDHF through a PKG-sensitive cation channel. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1272–H1277. [Google Scholar] [CrossRef] [PubMed]
- Onaka, U.; Fujii, K.; Abe, I.; Fujishima, M. Antihypertensive therapy improves endothelium dependent hyperpolarization. In Endothelium-Dependent Hyperpolarization; Vanhoutt, P.M., Ed.; Harwood Academic Publishers: Amsterdam, The Netherlands, 1999; pp. 305–312. [Google Scholar]
- Köhler, R.; Ruth, P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflugers. Arch. 2010, 459, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Grayson, T.H. Limits of isolation and culture: Intact vascular endothelium and BKCa. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1–H7. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.W.; Man, R.Y.; Gao, Y.; Vanhoutte, P.M.; Leung, S.W. Reduced activity of SKC a and Na-K ATPase underlies the accelerated impairment of EDH-type relaxations in mesenteric arteries of aging spontaneously hypertensive rats. Pharmacol. Res. Perspect. 2015, 3, e00150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachini, F.R.; Carneiro, F.S.; Lima, V.V.; Carneiro, Z.N.; Dorrance, A.; Webb, R.C.; Tostes, R.C. Upregulation of intermediate calcium-activated potassium channels counterbalance the impaired endothelium-dependent vasodilation in stroke-prone spontaneously hypertensive rats. Transl. Res. 2009, 154, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, R.H.; Webb, R.C. Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2275–H2284. [Google Scholar] [CrossRef] [PubMed]
- Chinnathambi, V.; Yallampalli, C.; Sathishkumar, K. Prenatal testosterone induces sex-specific dysfunction in endothelium-dependent relaxation pathways in adult male and female rats. Biol. Reprod. 2013, 89, 97. [Google Scholar] [CrossRef] [PubMed]
- Chaston, D.J.; Haddock, R.E.; Howitt, L.; Morton, S.K.; Brown, R.D.; Matthaei, K.I.; Hill, C.E. Perturbation of chemical coupling by an endothelial Cx40 mutant attenuates endothelium-dependent vasodilation by KCa channels and elevates blood pressure in mice. Pflugers. Arch. 2015, 467, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Kansui, Y.; Fujii, K.; Nakamura, K.; Goto, K.; Oniki, H.; Abe, I.; Shibata, Y.; Iida, M. Angiotensin II receptor blockade corrects altered expression of gap junctions in vascular endothelial cells from hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H216–H224. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Rummery, N.M.; Grayson, T.H.; Hill, C.E. Attenuation of conducted vasodilatation in rat mesenteric arteries during hypertension: Role of inwardly rectifying potassium channels. J. Physiol. 2004, 561, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Neylon, C.B.; Chen, M.X.; Garland, C.J. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (K(Ca)) and connexins: Possible relationship to vasodilator function? J. Anat. 2006, 209, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Gallagher, N.T.; McNeish, A.; Garland, C.J. Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ. Res. 2008, 102, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.A.; Tare, M.; Parkington, H.C. Nonlinear effects of potassium channel blockers on endothelium-dependent hyperpolarization. Acta Physiol. 2017, 219, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Suzuki, H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J. Physiol. 1990, 421, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Fukao, M.; Hattori, Y.; Kanno, M.; Sakuma, I.; Kitabatake, A. Sources of Ca2+ in relation to generation of acetylcholine-induced endothelium-dependent hyperpolarization in rat mesenteric artery. Br. J. Pharmacol. 1997, 120, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Boudaka, A.; Al-Suleimani, M.; BaOmar, H.; Al-Lawati, I.; Zadjali, F. Impairment of transient receptor potential Vanilloid 4-Mediated dilation in Mesenteric arteries of spontaneously hypertensive rats. Proc. Physiol. Soc. 2016, 37, PCA124. [Google Scholar]
- Zhang, D.X.; Mendoza, S.A.; Bubolz, A.H.; Mizuno, A.; Ge, Z.D.; Li, R.; Warltier, D.C.; Suzuki, M.; Gutterman, D.D. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 2009, 53, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sui, D.; Garavito, R.M.; Worden, R.M.; Wang, D.H. Salt intake augments hypotensive effects of transient receptor potential vanilloid 4: Functional significance and implication. Hypertension 2009, 53, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, Y.; Zheng, X.; Lund, H.; Suzuki, M.; Mattson, D.L.; Zhang, D.X. Characterization of blood pressure and endothelial function in TRPV4-deficient mice with l-NAME- and angiotensin II-induced hypertension. Physiol. Rep. 2014, 2, e00199. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Pan, Q.; Chen, Z.; Sun, C.; Zhang, P.; Mao, A.; Zhu, Y.; Li, H.; Lu, C.; Xie, M.; et al. Treatment of hypertension by increasing impaired endothelial TRPV4-KCa2.3 interaction. EMBO Mol. Med. 2017, 9, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Rath, G.; Dessy, C.; Feron, O. Caveolae, caveolin and control of vascular tone: Nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J. Physiol. Pharmacol. 2009, 60 (Suppl. 4), 105–109. [Google Scholar] [PubMed]
- Haddy, F.J.; Vanhoutte, P.M.; Feletou, M. Role of potassium in regulating blood flow and blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R546–R552. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Halpern, W. Impaired potassium-induced dilation in hypertensive rat cerebral arteries does not reflect altered Na+,K(+)-ATPase dilation. Circ. Res. 1990, 67, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S.; Sobey, C.G. Inwardly rectifying potassium channels in the regulation of vascular tone. Curr. Drug Targets 2003, 4, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Overbeck, H.W.; Derifield, R.S.; Pamnani, M.B.; Sözen, T. Attenuated vasodilator responses to K+ in essential hypertensive men. J. Clin. Investig. 1974, 53, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Quayle, J.M.; Nelson, M.T.; Standen, N.B. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev. 1997, 77, 1165–1232. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.E. Inward rectification and vascular function: As it was in the beginning. J. Physiol. 2008, 586, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Doughty, J.M.; Boyle, J.P.; Langton, P.D. Blockade of chloride channels reveals relaxations of rat small mesenteric arteries to raised potassium. Br. J. Pharmacol. 2001, 132, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.J.; Walker, S.D.; Dora, K.A.; Garland, C.J. Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J. Vasc. Res. 2003, 40, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Garland, C.J. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2424–H2429. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Dalsgaard, T.; Bonev, A.D.; Nelson, M.T. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J. Physiol. 2016, 594, 3271–3285. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Fancher, I.S.; Bian, J.T.; Zhang, C.X.; Schwab, S.; Gaffin, R.; Phillips, S.A.; Levitan, I. Inwardly rectifying K+ channels are major contributors to flow-induced vasodilatation in resistance arteries. J. Physiol. 2017, 595, 2339–2364. [Google Scholar] [CrossRef] [PubMed]
- Iddings, J.A.; Kim, K.J.; Zhou, Y.; Higashimori, H.; Filosa, J.A. Enhanced parenchymal arteriole tone and astrocyte signaling protect neurovascular coupling mediated parenchymal arteriole vasodilation in the spontaneously hypertensive rat. J. Cereb. Blood Flow Metab. 2015, 35, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.A.; Nelson, M.T. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 2015, 22, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Tajada, S.; Cidad, P.; Moreno-Domínguez, A.; Pérez-García, M.T.; López-López, J.R. High blood pressure associates with the remodelling of inward rectifier K+ channels in mice mesenteric vascular smooth muscle cells. J. Physiol. 2012, 590, 6075–6091. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Edwards, F.R.; Hill, C.E. Depolarization evoked by acetylcholine in mesenteric arteries of hypertensive rats attenuates endothelium-dependent hyperpolarizing factor. J. Hypertens. 2007, 25, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kwan, Y.W.; Chan, S.W.; Lee, S.M.; Leung, G.P. Potentiation of EDHF-mediated relaxation by chloride channel blockers. Acta Pharmacol. Sin. 2010, 31, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. KV channels and the regulation of vascular smooth muscle tone. Microcirculation 2018, 25, e12421. [Google Scholar] [CrossRef] [PubMed]
- Nieves-Cintrón, M.; Syed, A.U.; Nystoriak, M.A.; Navedo, M.F. Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation 2018, 25, e12423. [Google Scholar] [CrossRef] [PubMed]
- Quignard, J.F.; Feletou, M.; Edwards, G.; Duhault, J.; Weston, A.H.; Vanhoutte, P.M. Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br. J. Pharmacol. 2000, 129, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Sadanaga, T.; Ohya, Y.; Ohtsubo, T.; Goto, K.; Fujii, K.; Abe, I. Decreased 4-aminopyridine sensitive K+ currents in endothelial cells from hypertensive rats. Hypertens. Res. 2002, 25, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Kitazono, T.; Heistad, D.D.; Faraci, F.M. ATP-sensitive potassium channels in the basilar artery during chronic hypertension. Hypertension 1993, 22, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Ohya, Y.; Setoguchi, M.; Fujii, K.; Nagao, T.; Abe, I.; Fujishima, M. Impaired action of levcromakalim on ATP-sensitive K+ channels in mesenteric artery cells from spontaneously hypertensive rats. Hypertension 1996, 27, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, H.; Kuo, L. Endothelial, ATP-sensitive potassium channels mediate coronary microvascular dilation to hyperosmolarity. Am. J. Physiol. 1997, 273, H104–H112. [Google Scholar] [CrossRef] [PubMed]
- Aziz, Q.; Li, Y.; Anderson, N.; Ojake, L.; Tsisanova, E.; Tinker, A. Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J. Biol. Chem. 2017, 292, 17587–17597. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.A.; Dick, G.M.; Knudson, J.D.; Focardi, M.; Bratz, I.N.; Swafford, A.N., Jr.; Saitoh, S.; Tune, J.D.; Chilian, W.M. H2O2-induced redox-sensitive coronary vasodilation is mediated by 4-aminopyridine-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2473–H2482. [Google Scholar] [CrossRef] [PubMed]
- Lacza, Z.; Puskar, M.; Kis, B.; Perciaccante, J.V.; Miller, A.W.; Busija, D.W. Hydrogen peroxide acts as an EDHF in the piglet pial vasculature in response to bradykinin. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H406–H411. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Wong, W.T.; Shen, B.; Lau, C.W.; Tian, X.Y.; Tsang, S.Y.; Yao, X.; Chen, Z.Y.; Huang, Y. 4-aminopyridine-sensitive K+ channels contributes to NaHS-induced membrane hyperpolarization and relaxation in the rat coronary artery. Vascul. Pharmacol. 2010, 53, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.J.; Zhang, Y.; Hirota, S.; Janssen, L.J.; Lee, R.M. Vascular relaxation response to hydrogen peroxide is impaired in hypertension. Br. J. Pharmacol. 2004, 142, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Ma, Y.; Xie, L.; Ferro, A.; Ji, Y. Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br. J. Pharmacol. 2015, 172, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Greaney, J.L.; Kutz, J.L.; Shank, S.W.; Jandu, S.; Santhanam, L.; Alexander, L.M. Impaired hydrogen sulfide-mediated vasodilation contributes to microvascular endothelial dysfunction in hypertensive adults. Hypertension 2017, 69, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Large, W.A.; Wang, Q. Characteristics and physiological role of the Ca(2+)-activated Cl- conductance in smooth muscle. Am. J. Physiol. 1996, 271, C435–C454. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Suzuki, H. Dependency of endothelial cell function on vascular smooth muscle cells in guinea-pig mesenteric arteries and arterioles. J. Smooth Muscle Res. 2005, 41, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K.; Graier, W.F.; Kukovetz, W.R. Histamine induces K+, Ca2+, and Cl- currents in human vascular endothelial cells. Role of ionic currents in stimulation of nitric oxide biosynthesis. Circ. Res. 1994, 75, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Prenen, J.; Szücs, G.; Wei, L.; Tanzi, F.; Voets, T.; Droogmans, G. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J. Physiol. 1997, 498, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Suzuki, H. Effects of increased intracellular Cl- concentration on membrane responses to acetylcholine in the isolated endothelium of guinea pig mesenteric arteries. J. Physiol. Sci. 2007, 57, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, C.; Huai, R.; Qu, Z. Overexpression of ANO1/TMEM16A, an arterial Ca2+-activated Cl- channel, contributes to spontaneous hypertension. J. Mol. Cell. Cardiol. 2015, 82, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.M.; Gao, M.; Guo, K.M.; Wang, M.; Li, X.Y.; Zeng, X.L.; Sun, L.; Lv, X.F.; Du, Y.H.; Wang, G.L.; et al. TMEM16A Contributes to Endothelial Dysfunction by Facilitating Nox2 NADPH Oxidase-Derived Reactive Oxygen Species Generation in Hypertension. Hypertension 2017, 69, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Hutri-Kähönen, N.; Kähönen, M.; Tolvanen, J.P.; Wu, X.; Sallinen, K.; Pörsti, I. Ramipril therapy improves arterial dilation in experimental hypertension. Cardiovasc. Res. 1997, 33, 188–195. [Google Scholar] [CrossRef]
- Onaka, U.; Fujii, K.; Abe, I.; Fujishima, M. Antihypertensive treatment improves endothelium-dependent hyperpolarization in the mesenteric artery of spontaneously hypertensive rats. Circulation 1998, 98, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kähönen, M.; Tolvanen, J.P.; Kalliovalkama, J.; Wu, X.; Karjala, K.; Mäkynen, H.; Pörsti, I. Losartan and enalapril therapies enhance vasodilatation in the mesenteric artery of spontaneously hypertensive rats. Eur. J. Pharmacol. 1999, 368, 213–222. [Google Scholar] [CrossRef]
- Goto, K.; Fujii, K.; Onaka, U.; Abe, I.; Fujishima, M. Renin-angiotensin system blockade improves endothelial dysfunction in hypertension. Hypertension 2000, 36, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Vettoretti, S.; Ochodnicky, P.; Buikema, H.; Henning, R.H.; Kluppel, C.A.; de Zeeuw, D.; van Dokkum, R.P. Altered myogenic constriction and endothelium-derived hyperpolarizing factor-mediated relaxation in small mesenteric arteries of hypertensive subtotally nephrectomized rats. J. Hypertens. 2006, 24, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Goto, K.; Kansui, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Angiotensin II Receptor-Neprilysin Inhibitor Sacubitril/Valsartan Improves Endothelial Dysfunction in Spontaneously Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, E006617. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Onaka, U.; Abe, I.; Fujishima, M. Angiotensin-converting enzyme inhibitor prevents age-related endothelial dysfunction. Hypertension 2000, 36, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Kansui, Y.; Fujii, K.; Goto, K.; Abe, I.; Iida, M. Angiotensin II receptor antagonist improves age-related endothelial dysfunction. J. Hypertens. 2002, 20, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Albarwani, S.; Al-Siyabi, S.; Al-Husseini, I.; Al-Ismail, A.; Al-Lawati, I.; Al-Bahrani, I.; Tanira, M.O. Lisinopril alters contribution of nitric oxide and K(Ca) channels to vasodilatation in small mesenteric arteries of spontaneously hypertensive rats. Physiol. Res. 2015, 64, 39–49. [Google Scholar] [PubMed]
- More, A.S.; Mishra, J.S.; Hankins, G.D.; Yallampalli, C.; Sathishkumar, K. Enalapril normalizes endothelium-derived hyperpolarizing factor-mediated relaxation in mesenteric artery of adult hypertensive rats prenatally exposed to testosterone. Biol. Reprod. 2015, 92, 155. [Google Scholar] [CrossRef] [PubMed]
- Toro, L.; Amador, M.; Stefani, E. ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. Am. J. Physiol. 1990, 258, H912–H915. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Hirata, Y.; Tokumura, A.; Nakaya, Y.; Fukuzawa, K. Protein kinase C-independent inhibition of the Ca(2+)-activated K+ channel by angiotensin II and endothelin-1. Biochem. Pharmacol. 1995, 49, 1051–1056. [Google Scholar] [CrossRef]
- Zhao, L.M.; Wang, Y.; Ma, X.Z.; Wang, N.P.; Deng, X.L. Advanced glycation end products impair K(Ca)3.1- and K(Ca)2.3-mediated vasodilatation via oxidative stress in rat mesenteric arteries. Pflugers. Arch. 2014, 466, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kaw, S.; Hecker, M. Endothelium-derived hyperpolarizing factor, but not nitric oxide or prostacyclin release, is resistant to menadione-induced oxidative stress in the bovine coronary artery. Naunyn. Schmiedebergs. Arch. Pharmacol. 1999, 359, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; McPhaden, A.R.; Berg, G.; Pathi, V.; Dominiczak, A.F. Is hydrogen peroxide an EDHF in human radial arteries? Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2451–H2455. [Google Scholar] [CrossRef] [PubMed]
- Louis, W.J.; Howes, L.G. Genealogy of the spontaneously hypertensive rat and Wistar-Kyoto rat strains: Implications for studies of inherited hypertension. J. Cardiovasc. Pharmacol. 1990, 16 (Suppl. 7), S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, K.I. Identification of therapeutic drug targets through genetically manipulated mice: Are we getting it right? Pharmacol. Ther. 2009, 123, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Bellien, J.; Thuillez, C.; Joannides, R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam. Clin. Pharmacol. 2008, 22, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Ghiadoni, L.; Virdis, A.; Buralli, S.; Salvetti, A. Vasodilation to bradykinin is mediated by an ouabain-sensitive pathway as a compensatory mechanism for impaired nitric oxide availability in essential hypertensive patients. Circulation 1999, 100, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Versari, D.; Cipriano, A.; Ghiadoni, L.; Galetta, F.; Franzoni, F.; Magagna, A.; Virdis, A.; Salvetti, A. Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients. J. Am. Coll. Cardiol. 2006, 48, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, C.A.; Coleman, J.; Brady, A.J.; Connell, J.M.; Hillier, C.; Petrie, J.R. Endothelium-dependent relaxation is resistant to inhibition of nitric oxide synthesis, but sensitive to blockade of calcium-activated potassium channels in essential hypertension. J. Hum. Hypertens. 2007, 21, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, Z.; Jiang, D.J.; Li, D.; Tan, B.; Liu, H.; Li, Y.J. Reduction of NO- and EDHF-mediated vasodilatation in hypertension: Role of asymmetric dimethylarginine. Clin. Exp. Hypertens. 2007, 29, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Kenny, L.C.; Baker, P.N.; Kendall, D.A.; Randall, M.D.; Dunn, W.R. Differential mechanisms of endothelium-dependent vasodilator responses in human myometrial small arteries in normal pregnancy and pre-eclampsia. Clin. Sci. 2002, 103, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Luksha, L.; Luksha, N.; Kublickas, M.; Nisell, H.; Kublickiene, K. Diverse mechanisms of endothelium-derived hyperpolarizing factor-mediated dilatation in small myometrial arteries in normal human pregnancy and preeclampsia. Biol. Reprod. 2010, 83, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Goulopoulou, S. Maternal Vascular Physiology in Preeclampsia. Hypertension 2017, 70, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, J.A.; Na, H.Y.; Kim, J.E.; Park, S.; Han, K.H.; Kim, Y.J.; Suh, S.H. NADPH oxidase 2-derived superoxide downregulates endothelial KCa3.1 in preeclampsia. Free Radic. Biol. Med. 2013, 57, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Deja, M.A.; Gołba, K.S.; Widenka, K.; Mrozek, R.; Biernat, J.; Kolowca, M.; Malinowski, M.; Woś, S. Angiotensin-converting enzyme inhibitors reveal non-NO-, non-prostacycline-mediated endothelium-dependent relaxation in internal thoracic artery of hypertensive patients. Int. J. Cardiol. 2005, 102, 455–460. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diffusible and contact-mediated mechanisms of endothelium-dependent smooth muscle hyperpolarization. In certain vascular beds in specific conditions, diffusible factors such as epoxyeicosatrienoic acids (EETs), K+ ions, and hydrogen peroxide (H2O2) hyperpolarize smooth muscle cells through the opening of potassium channels and/or Na+/K+-ATPase. In addition, endothelium-dependent hyperpolarization initiated in endothelial cells with a rise in intracellular calcium and the subsequent activation of small (SKCa) and intermediate conductance (IKCa) Ca2+-activated K+ channels spreads to adjacent smooth muscle cells via myoendothelial gap junctions (MEGJs) in a number of vascular beds. In some vascular beds, combination of diffusible and contact-mediated mechanisms underpin smooth muscle hyperpolarization.
Figure 1.
Diffusible and contact-mediated mechanisms of endothelium-dependent smooth muscle hyperpolarization. In certain vascular beds in specific conditions, diffusible factors such as epoxyeicosatrienoic acids (EETs), K+ ions, and hydrogen peroxide (H2O2) hyperpolarize smooth muscle cells through the opening of potassium channels and/or Na+/K+-ATPase. In addition, endothelium-dependent hyperpolarization initiated in endothelial cells with a rise in intracellular calcium and the subsequent activation of small (SKCa) and intermediate conductance (IKCa) Ca2+-activated K+ channels spreads to adjacent smooth muscle cells via myoendothelial gap junctions (MEGJs) in a number of vascular beds. In some vascular beds, combination of diffusible and contact-mediated mechanisms underpin smooth muscle hyperpolarization.

Figure 2.
Downregulation of transient receptor potential vanilloid type 4 channel (TRPV4) in hypertension. Representative tracing of GSK1016790A (GSK), a selective TRPV4 activator, induced hyperpolarization (A) and relaxation (B) in mesenteric arteries of Wistar–Kyoto (WKY) rats and stroke-prone spontaneously hypertensive rats (SHRSP). GSK evoked hyperpolarization and relaxation in WKY but not in SHRSP arteries. Arteries were depolarized (A) or pre-contracted (B) with phenylephrine (10−5 mol/L). Indomethacin (10−5 mol/L) and Nω-nitro-l-arginine (l-NAME, 10−4 mol/L) were present throughout the experiments. (C) Representative immunoblots of the expression of TRPV4 in mesenteric arteries from WKY and SHRSP. The expression of the TRPV4 protein was significantly decreased in the SHRSP mesenteric arteries compared with that from WKY. Modified from Seki et al. [46].
Figure 2.
Downregulation of transient receptor potential vanilloid type 4 channel (TRPV4) in hypertension. Representative tracing of GSK1016790A (GSK), a selective TRPV4 activator, induced hyperpolarization (A) and relaxation (B) in mesenteric arteries of Wistar–Kyoto (WKY) rats and stroke-prone spontaneously hypertensive rats (SHRSP). GSK evoked hyperpolarization and relaxation in WKY but not in SHRSP arteries. Arteries were depolarized (A) or pre-contracted (B) with phenylephrine (10−5 mol/L). Indomethacin (10−5 mol/L) and Nω-nitro-l-arginine (l-NAME, 10−4 mol/L) were present throughout the experiments. (C) Representative immunoblots of the expression of TRPV4 in mesenteric arteries from WKY and SHRSP. The expression of the TRPV4 protein was significantly decreased in the SHRSP mesenteric arteries compared with that from WKY. Modified from Seki et al. [46].

Figure 3.
Endothelial ion channels in normotension and hypertension. In normotension, in response to agonist stimulation of endothelial cells, a rise in intracellular Ca2+ occurs due to the release from intracellular Ca2+ stores and Ca2+ entry via transient potential vanilloid type 4 channel (TRPV4). A rise in intracellular Ca2+ subsequently generates endothelium-dependent hyperpolarization (EDH) through the activation of both small (SKCa) and intermediate conductance (IKCa) Ca2+-activated K+ channels. In some arteries, K+ released from endothelial KCa channels activates endothelial Kir channels, which in turn amplifies EDH. EDH spreads to adjacent smooth muscle cells via myoendothelial gap junctions (MEGJs), resulting in vascular relaxation. In hypertension, alterations of endothelial ion channels additively reduce EDH; these alterations include downregulation of endothelial SKCa and TRPV4 channels, upregulation of endothelial Ca2+-activated chloride channels (CaCCs), and functional loss of endothelial Kir channels.
Figure 3.
Endothelial ion channels in normotension and hypertension. In normotension, in response to agonist stimulation of endothelial cells, a rise in intracellular Ca2+ occurs due to the release from intracellular Ca2+ stores and Ca2+ entry via transient potential vanilloid type 4 channel (TRPV4). A rise in intracellular Ca2+ subsequently generates endothelium-dependent hyperpolarization (EDH) through the activation of both small (SKCa) and intermediate conductance (IKCa) Ca2+-activated K+ channels. In some arteries, K+ released from endothelial KCa channels activates endothelial Kir channels, which in turn amplifies EDH. EDH spreads to adjacent smooth muscle cells via myoendothelial gap junctions (MEGJs), resulting in vascular relaxation. In hypertension, alterations of endothelial ion channels additively reduce EDH; these alterations include downregulation of endothelial SKCa and TRPV4 channels, upregulation of endothelial Ca2+-activated chloride channels (CaCCs), and functional loss of endothelial Kir channels.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Goto, K.; Ohtsubo, T.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. Int. J. Mol. Sci. 2018, 19, 315. https://doi.org/10.3390/ijms19010315
AMA Style
Goto K, Ohtsubo T, Kitazono T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. International Journal of Molecular Sciences. 2018; 19(1):315. https://doi.org/10.3390/ijms19010315
Chicago/Turabian StyleGoto, Kenichi, Toshio Ohtsubo, and Takanari Kitazono. 2018. "Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels" International Journal of Molecular Sciences 19, no. 1: 315. https://doi.org/10.3390/ijms19010315
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.