Src Cooperates with Oncogenic Ras in Tumourigenesis via the JNK and PI3K Pathways in Drosophila epithelial Tissue


Abstract
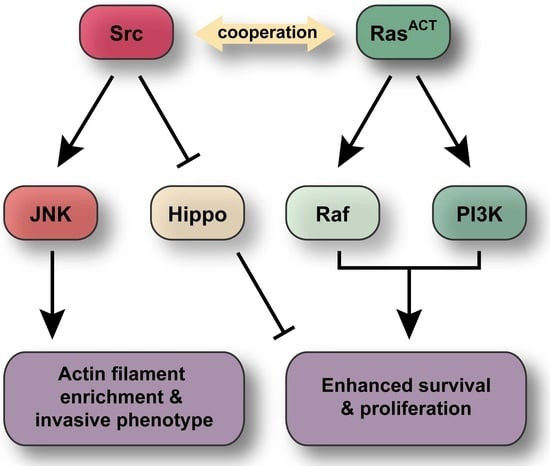
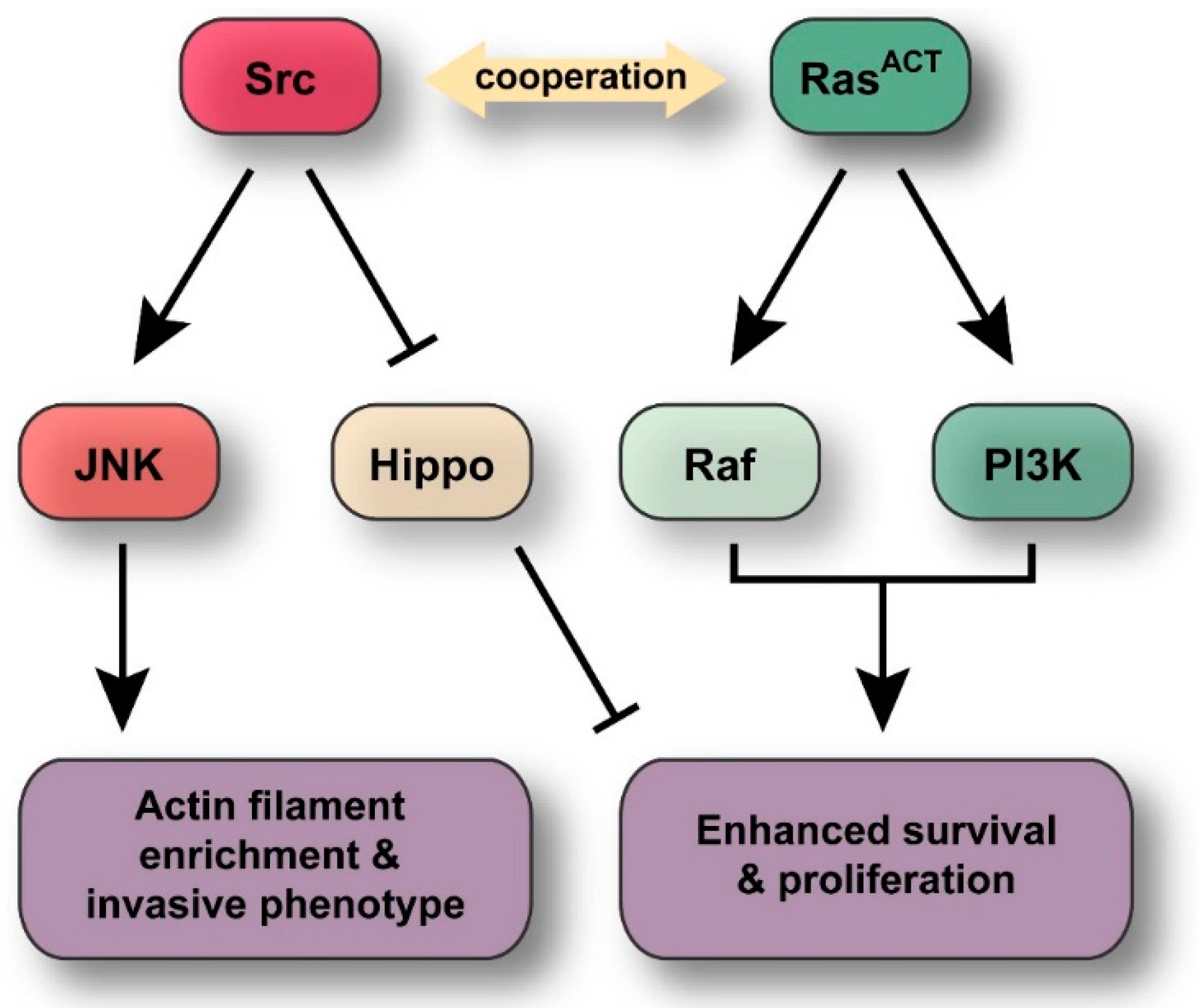
:
1. Introduction
2. Results
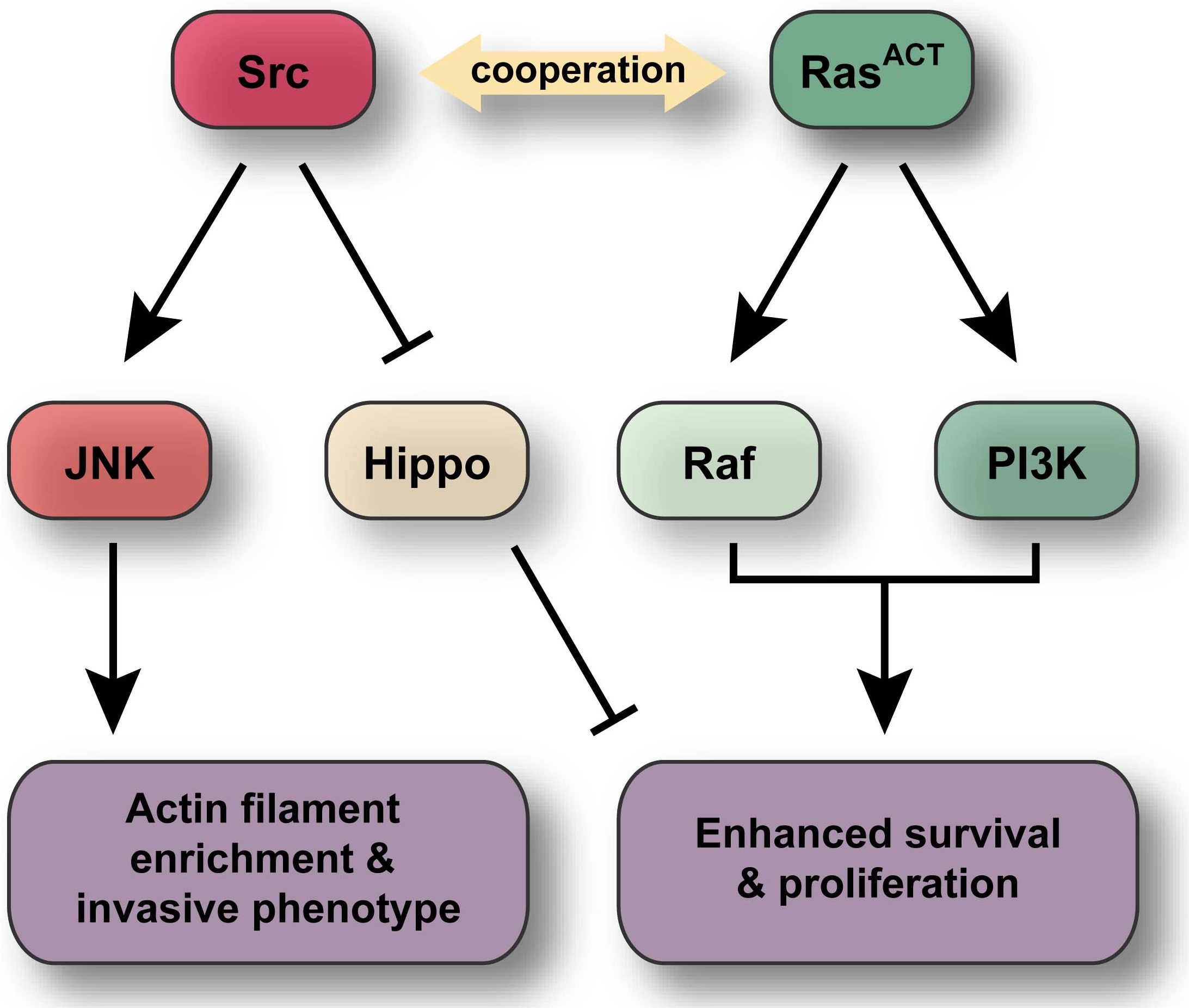
2.1. Src42A Overexpression Enhances the Eyeless-Driven RasACT Hyperplastic Eye Phenotype
2.2. Src64B Overexpression Also Enhances the ey > RasACT Hyperplastic Eye Phenotype
2.3. RasACT Signalling Contributes more than just Survival Signals in Cooperation with Src
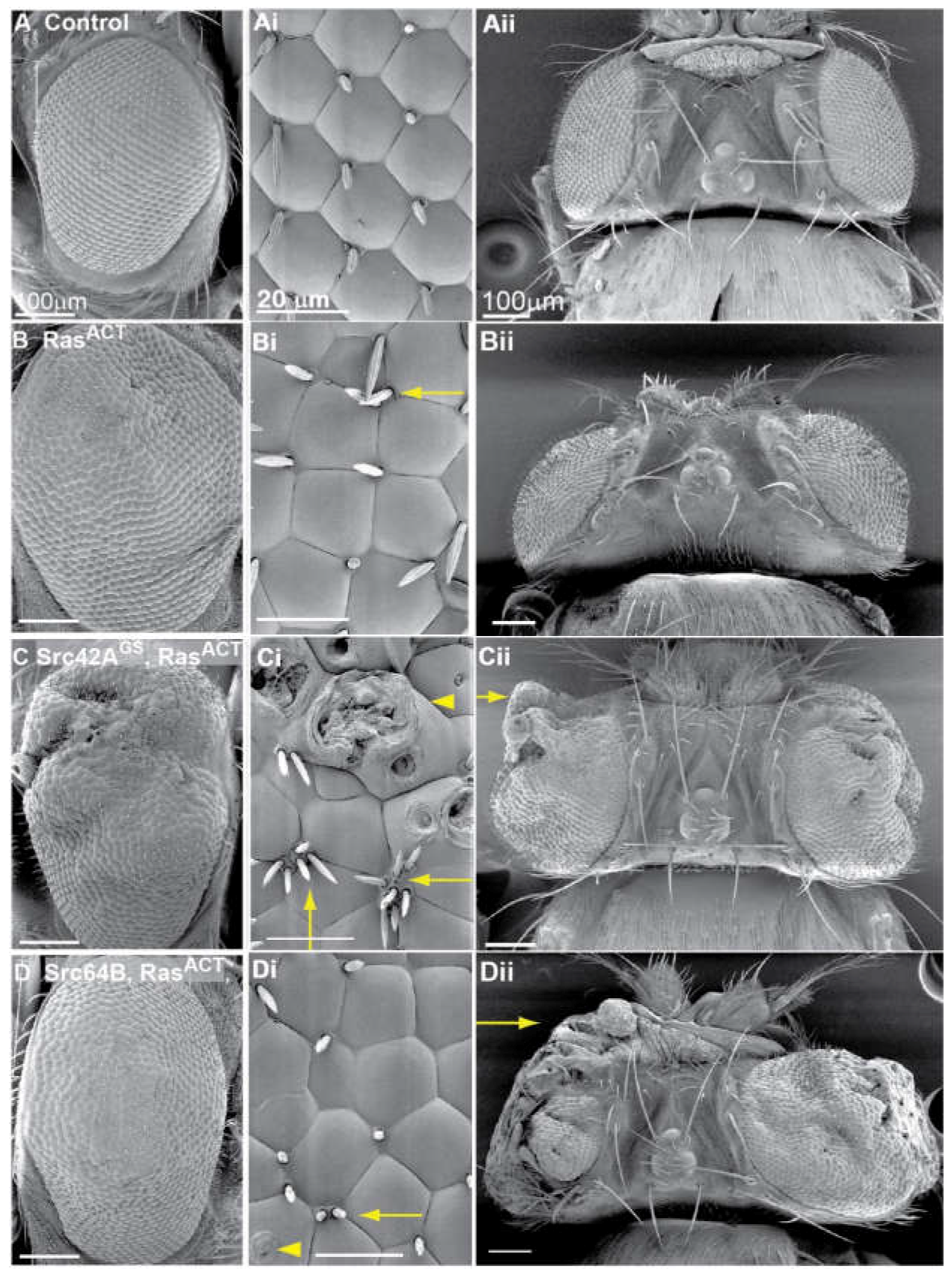
2.4. Src and RasACT Cooperate to form Overgrown Neoplastic Tumours in the Eye Epithelium
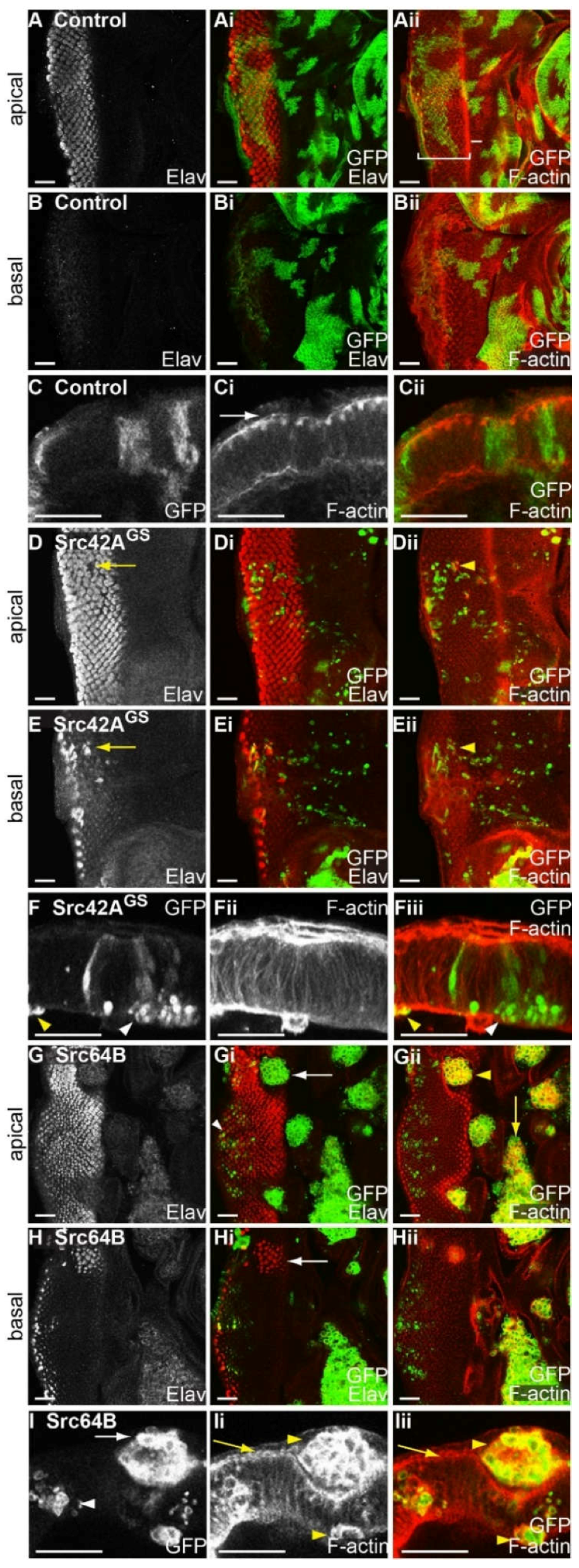
2.5. Expression of Drosophila Src42A and Src64B Results in Distinct Effects in Eye Epithelial Clones
2.6. Expression of Src64B in Eye Disc Clones Results in A Loss of Cell Polarity
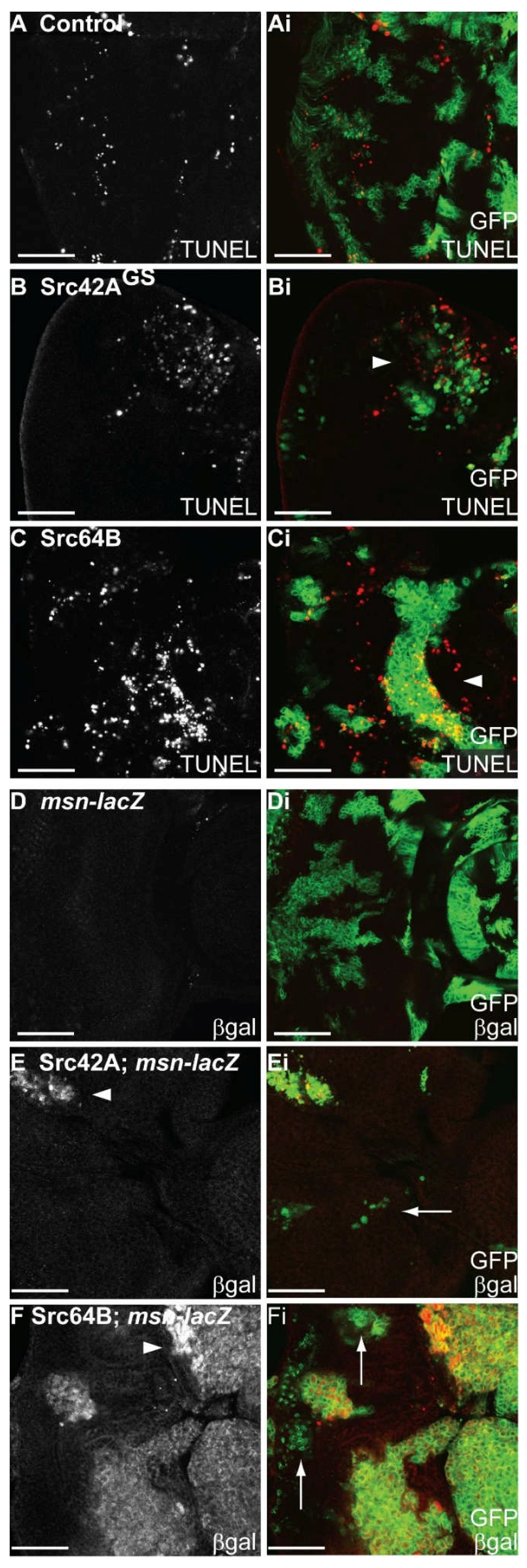
2.7. Expression of Src in Eye Epithelial Clones Promotes Cell Death, but Does Not Reduce Cell Proliferation
2.8. Expression of Src in Eye Disc Clones Promotes JNK Pathway Signalling and Activity
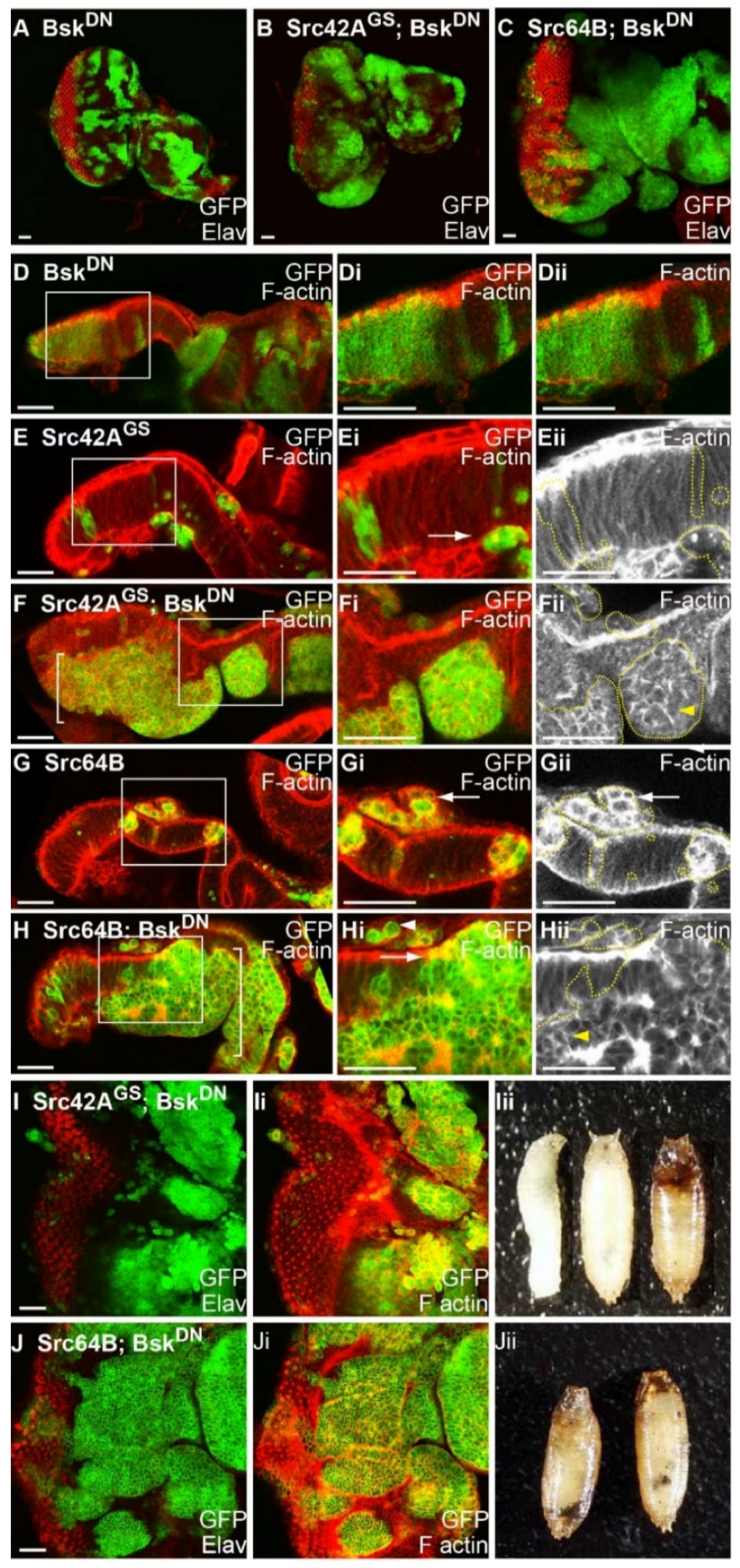
2.9. Blocking JNK Increases Clone Viability of Src-Expressing Clones, Reduces F-actin Accumulation and Results in Basal Extrusion
2.10. JNK is Activated in Src + RasACT Neoplastic Overgrowth, and Blocking JNK Results in Partial Suppression of the Overgrowth and Differentiation Defects
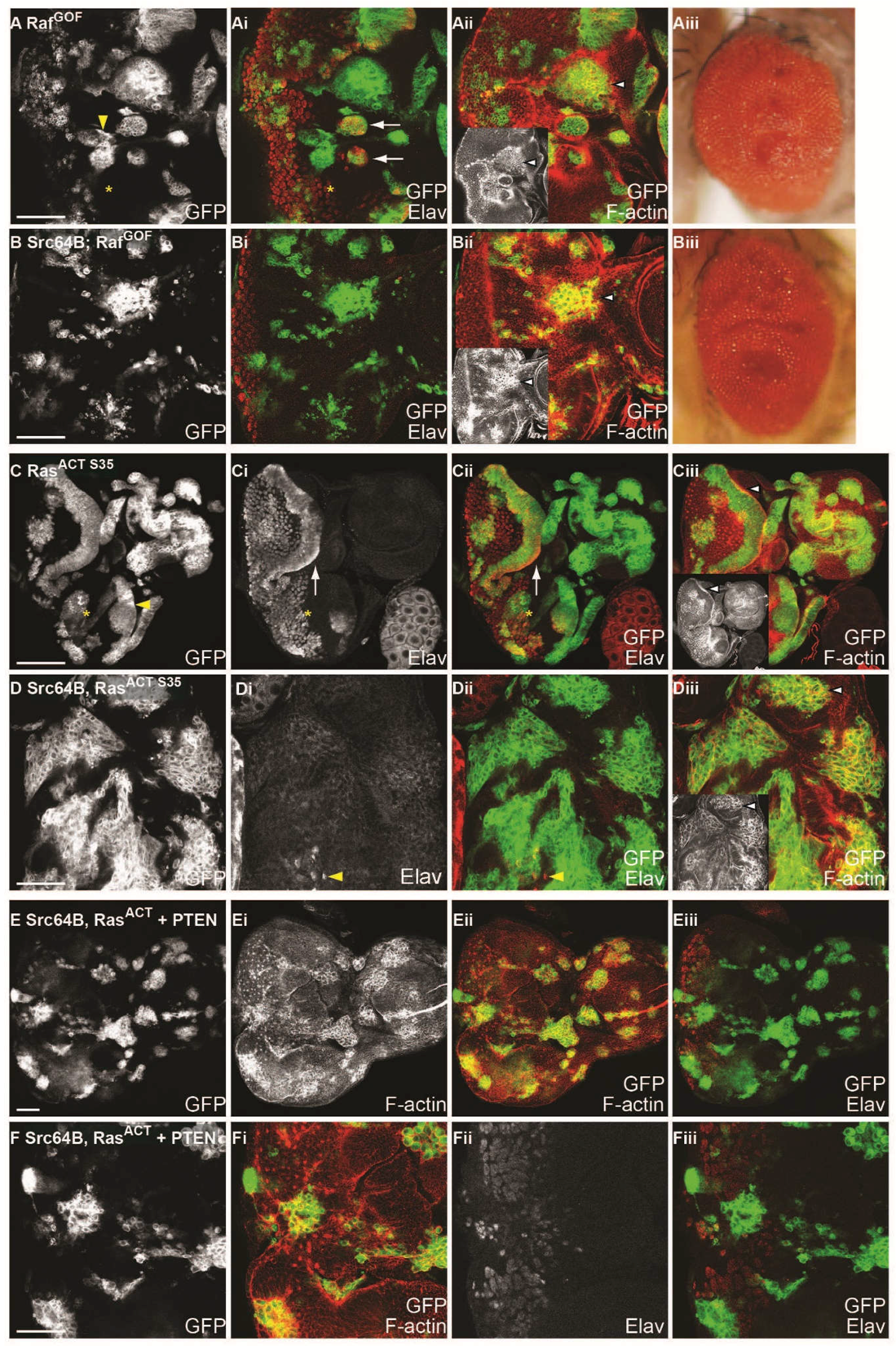
2.11. Ras-Raf-MAPK and Ras-PI3K Pathways are Required with Src for Cooperative Tumourigenesis
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks
4.2. Immunohistochemistry
4.3. Adult Eye Imaging
4.4. Western Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Baz | Bazooka |
| Csk | C-terminal Src-like kinase |
| Dlg | Discs-large |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| JNK | Jun N-terminal Kinase |
| MMP | Metalloproteinase |
| MAPK | Mitogen-activated protein kinase |
| PI3K | PhosphoInoitide 3-kinase |
References
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta Rev. Cancer 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S. The hunting of the Src. Nat. Rev. Mol. Cell Biol 2001, 2, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Yeatman, T.J. A renaissance for Src. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C. Newest findings on the oldest oncogene; how activated Src does it. J. Cell Sci 2004, 117, 989–998. [Google Scholar] [PubMed]
- Talamonti, M.S.; Roh, M.S.; Curley, S.A.; Gallick, G.E. Increase in activity and level of pp60c-Src in progressive stages of human colorectal cancer. J. Clin. Investig. 1993, 91, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Mao, W.; Coppola, D.; Kang, J.; Loubeau, J.M.; Trudeau, W.; Karl, R.; Fujita, D.J.; Jove, R.; Yeatman, T.J. Activating Src mutation in a subset of advanced human colon cancers. Nat. Genet. 1999, 21, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Rosen, N.; Bolen, J.B.; Schwartz, A.M.; Cohen, P.; DeSeau, V.; Israel, M.A. Analysis of pp60c-Src protein kinase activity in human tumor cell lines and tissues. J. Biol. Chem. 1986, 261, 13754–13759. [Google Scholar] [PubMed]
- Barnekow, A.; Schartl, M. Comparative studies on the Src proto-oncogene and its gene product pp60c-Src in normal and neoplastic tissues of lower vertebrates. Comp. Biochem. Physiol. B Comp. Biochem. 1987, 87, 663–670. [Google Scholar] [CrossRef]
- Cartwright, C.A.; Kamps, M.P.; Meisler, A.I.; Pipas, J.M.; Eckhart, W. Pp60c-Src activation in human colon carcinoma. J. Clin. Investig. 1989, 83, 2025–2033. [Google Scholar] [CrossRef] [PubMed]
- Fanning, P.; Bulovas, K.; Saini, K.S.; Libertino, J.A.; Joyce, A.D.; Summerhayes, I.C. Elevated expression of pp60c-Src in low grade human bladder carcinoma. Cancer Res. 1992, 52, 1457–1462. [Google Scholar] [PubMed]
- Barnekow, A.; Paul, E.; Schartl, M. Expression of the c-Src protooncogene in human skin tumors. Cancer Res. 1987, 47, 235–240. [Google Scholar] [PubMed]
- Muthuswamy, S.K.; Muller, W.J. Activation of Src family kinases in neu-induced mammary tumors correlates with their association with distinct sets of tyrosine phosphorylated proteins in vivo. Oncogene 1995, 11, 1801–1810. [Google Scholar] [PubMed]
- Mao, W.; Irby, R.; Coppola, D.; Fu, L.; Wloch, M.; Turner, J.; Yu, H.; Garcia, R.; Jove, R.; Yeatman, T.J. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 1997, 15, 3083–3090. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.R.; Windham, T.C.; Estrella, V.C.; Parikh, N.U.; Thall, P.F.; Deavers, M.T.; Bast, R.C.; Mills, G.B.; Gallick, G.E. Activated Src protein tyrosine kinase is overexpressed in late-stage human ovarian cancers. Gynecol. Oncol. 2003, 88, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Myers, J.N.; Lippman, S.M.; Johnson, F.M.; Williams, M.D.; Rayala, S.; Ohshiro, K.; Rosenthal, D.I.; Weber, R.S.; Gallick, G.E.; et al. Epithelial to mesenchymal transition in head and neck squamous carcinoma: Association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer 2008, 112, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Boyer, B.; Bourgeois, Y.; Poupon, M.F. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene 2002, 21, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.; Mao, W.; Coppola, D.; Jove, R.; Gamero, A.; Cuthbertson, D.; Fujita, D.J.; Yeatman, T.J. Overexpression of normal c-Src in poorly metastatic human colon cancer cells enhances primary tumor growth but not metastatic potential. Cell Growth Differ. 1997, 8, 1287–1295. [Google Scholar] [PubMed]
- Maa, M.C.; Leu, T.H.; McCarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995, 92, 6981–6985. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Chen, H.C. P120Rasgap-mediated activation of c-Src is critical for oncogenic Ras to induce tumor invasion. Cancer Res. 2012, 72, 2405–2415. [Google Scholar] [CrossRef] [PubMed]
- Shields, D.J.; Murphy, E.A.; Desgrosellier, J.S.; Mielgo, A.; Lau, S.K.; Barnes, L.A.; Lesperance, J.; Huang, M.; Schmedt, C.; Tarin, D.; et al. Oncogenic Ras/Src cooperativity in pancreatic neoplasia. Oncogene 2011, 30, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Dimri, M.; Naramura, M.; Duan, L.; Chen, J.; Ortega-Cava, C.; Chen, G.; Goswami, R.; Fernandes, N.; Gao, Q.; Dimri, G.P.; et al. Modeling breast cancer-associated c-Src and Egfr overexpression in human mecs: C-Src and Egfr cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007, 67, 4164–4172. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Choi, S.H.; Egloff, A.M.; Cai, Q.; Suzuki, S.; Freilino, M.; Nozawa, H.; Thomas, S.M.; Gooding, W.E.; SiEgfried, J.M.; et al. Combined inhibition of c-Src and epidermal growth factor receptor abrogates growth and invasion of head and neck squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Iba, H.; Takeya, T.; Cross, F.R.; Hanafusa, T.; Hanafusa, H. Rous sarcoma virus variants that carry the cellular Src gene instead of the viral Src gene cannot transform chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 1984, 81, 4424–4428. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.C.; Varmus, H.E.; Michael Bishop, J. Expression of v-Src and chicken c-Src in rat cells demonstrates qualitative differences between pp60v-Src and pp60c-Src. Cell 1984, 37, 131–139. [Google Scholar] [CrossRef]
- Shalloway, D.; Johnson, P.J.; Freed, E.O.; Coulter, D.; Flood, W.A. Transformation of nih 3t3 cells by cotransfection with c-Src and nuclear oncogenes. Mol. Cell. Biol. 1987, 7, 3582–3590. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.C.; Tice, D.A.; Karaoli, T.; Parsons, S.J. The c terminus of c-Src inhibits breast tumor cell growth by a kinase-independent mechanism. J. Biol. Chem. 2004, 279, 23773–23781. [Google Scholar] [CrossRef] [PubMed]
- Kline, C.L.B.; Jackson, R.; Engelman, R.; Pledger, W.J.; Yeatman, T.J.; Irby, R.B. Src kinase induces tumor formation in the c-SRC C57BL/6 mouse. Int. J. Cancer 2008, 122, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Leroy, C.; Simon, V.; Benistant, C.; Roche, S. Yes oncogenic activity is specified by its SH4 domain and regulates Ras/MAPK signaling in colon carcinoma cells. Am. J. Cancer Res. 2015, 5, 1972–1987. [Google Scholar] [PubMed]
- Fenton, S.E.; Hutchens, K.A.; Denning, M.F. Targeting Fyn in Ras-transformed cells induces F-actin to promote adherens junction-mediated cell-cell adhesion. Mol. Carcinog. 2015, 54, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Denning, M.F. Fyn is induced by Ras/PI3K/Akt signaling and is required for enhanced invasion/migration. Mol. Carcinog. 2011, 50, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P.; Montgomery, C.; Geske, R.; Bradley, A. Targeted disruption of the c-Src proto-oncogene leads to osteopetrosis in mice. Cell 1991, 64, 693–702. [Google Scholar] [CrossRef]
- Stein, P.L.; Vogel, H.; Soriano, P. Combined deficiencies of Src, Fyn, and yes tyrosine kinases in mutant mice. Genes Dev. 1994, 8, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Kussick, S.J.; Basler, K.; Cooper, J.A. Ras1-dependent signaling by ectopically-expressed Drosophila Src gene product in the embryo and developing eye. Oncogene 1993, 8, 2791–2803. [Google Scholar] [PubMed]
- Takahashi, F.; Endo, S.; Kojima, T.; Saigo, K. Regulation of cell-cell contacts in developing Drosophila eyes by dSrc41, a new, close relative of vertebrate c-Src. Genes Dev. 1996, 10, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, L.G.; Stewart, R.A.; Li, D.M.; Xu, T. Drosophila Src-family kinases function with Csk to regulate cell proliferation and apoptosis. Oncogene 2004, 23, 4754–4762. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Warner, S.; Read, R.; Cagan, R.L. Differing Src signaling levels have distinct outcomes in Drosophila. Cancer Res. 2007, 67, 10278–10285. [Google Scholar] [CrossRef] [PubMed]
- Read, R.D.; Bach, E.A.; Cagan, R.L. Drosophila c-terminal Src kinase negatively regulates organ growth and cell proliferation through inhibition of the Src, jun N-terminal kinase, and stat pathways. Mol. Cell. Biol. 2004, 24, 6676–6689. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.A.; Li, D.M.; Huang, H.; Xu, T. A genetic screen for modifiers of the lats tumor suppressor gene identifies c-terminal Src kinase as a regulator of cell proliferation in Drosophila. Oncogene 2003, 22, 6436–6444. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, C.M. The Hippo pathway: A master regulatory network important in development and dysregulated in disease. Curr. Top. Dev. Biol. 2017, 123, 181–228. [Google Scholar] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Igaki, T. Src controls tumorigenesis via JNK-dependent regulation of the Hippo pathway in Drosophila. EMBO Rep. 2013, 14, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Waghmare, I.; Verghese, S.; Singh, A.; Singh, A.; Kango-Singh, M. Drosophila C-terminal Src kinase regulates growth via the Hippo signaling pathway. Dev. Biol. 2015, 397, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Takahashi, F.; Ui-Tei, K.; Kojima, T.; Saigo, K. Requirements of genetic interactions between Src42a, armadillo and shotgun, a gene encoding E-cadherin, for normal development in Drosophila. Development 2005, 132, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Larson, D.E.; Cagan, R.L. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell. 2006, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Salavaggione, L.; Ylagan, L.; Wilkins, M.; Watson, M.; Weilbaecher, K.; Cagan, R. A role for the epithelial microenvironment at tumor boundaries: Evidence from Drosophila and human squamous cell carcinomas. Am. J. Pathol. 2010, 176, 3007–3014. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shao, Y.; Zheng, H.; Li, M.; Li, W.; Xue, L. Src42a modulates tumor invasion and cell death via ben/dUev1a-mediated JNK activation in Drosophila. Cell Death Dis. 2013, 4, e864. [Google Scholar] [CrossRef] [PubMed]
- Tateno, M.; Nishida, Y.; Adachi-Yamada, T. Regulation of JNK by Src during Drosophila development. Science 2000, 287, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Brumby, A.M.; Goulding, K.R.; Schlosser, T.; Loi, S.; Galea, R.; Khoo, P.; Bolden, J.E.; Aigaki, T.; Humbert, P.O.; Richardson, H.E. Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: A RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics 2011, 188, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Bellen, H.J.; Levis, R.W.; Liao, G.; He, Y.; Carlson, J.W.; Tsang, G.; Evans-Holm, M.; Hiesinger, P.R.; Schulze, K.L.; Rubin, G.M.; et al. The BDGP gene disruption project: Single Transposon Insertions Associated With 40% of Drosophila Genes. Genetics 2004, 167, 761–781. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.T.; Bellen, H.J. Transgenesis upgrades for Drosophila melanogaster. Development 2007, 134, 3571–3584. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.C.; O’Neill, E.M.; Rubin, G.M. Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development 1993, 119, 855–865. [Google Scholar] [PubMed]
- Freeman, M. Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell 1996, 87, 651–660. [Google Scholar] [CrossRef]
- Hazelett, D.J.; Bourouis, M.; Walldorf, U.; Treisman, J.E. Decapentaplegic and wingless are regulated by eyes absent and eyegone and interact to direct the pattern of retinal differentiation in the eye disc. Development 1998, 125, 3741–3751. [Google Scholar] [PubMed]
- Hauck, B.; Gehring, W.J.; Walldorf, U. Functional analysis of an eye specific enhancer of the eyeless gene in Drosophila. Proc. Natl. Acad. Sci. USA 1999, 96, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Muda, M.; Worby, C.A.; Simonson-Leff, N.; Clemens, J.C.; Dixon, J.E. Use of double-stranded rna-mediated interference to determine the substrates of protein tyrosine kinases and phosphatases. Biochem. J. 2002, 366, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Gould, K.L.; Cartwright, C.A.; Hunter, T. Tyr527 is phosphorylated in pp60c-Src: Implications for regulation. Science 1986, 231, 1431–1434. [Google Scholar] [CrossRef] [PubMed]
- Jove, R.; Hanafusa, H. Cell transformation by the viral Src oncogene. Annu. Rev. Cell Biol 1987, 3, 31–56. [Google Scholar] [CrossRef] [PubMed]
- Halfar, K.; Rommel, C.; Stocker, H.; Hafen, E. Ras controls growth, survival and differentiation in the Drosophila eye by different thresholds of map kinase activity. Development 2001, 128, 1687–1696. [Google Scholar] [PubMed]
- Karim, F.D.; Rubin, G.M. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 1998, 125, 1–9. [Google Scholar] [PubMed]
- O’Keefe, D.D.; Prober, D.A.; Moyle, P.S.; Rickoll, W.L.; Edgar, B.A. Egfr/Ras signaling regulates DE-cadherin/shotgun localization to control vein morphogenesis in the Drosophila wing. Dev. Biol. 2007, 311, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Prober, D.A.; Edgar, B.A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002, 16, 2286–2299. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Baker, N.E. Role of the Egfr/Ras/Raf pathway in specification of photoreceptor cells in the Drosophila retina. Development 2001, 128, 1183–1191. [Google Scholar] [PubMed]
- Bergmann, A.; Agapite, J.; McCall, K.; Steller, H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 1998, 95, 331–341. [Google Scholar] [CrossRef]
- Kurada, P.; White, K. Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 1998, 95, 319–329. [Google Scholar] [CrossRef]
- Hay, B.; Wolff, T.; Rubin, G. Expression of baculovirus p35 prevents cell death in Drosophila. Development 1994, 120, 2121–2129. [Google Scholar] [PubMed]
- Meier, P.; Silke, J.; Leevers, S.J.; Evan, G.I. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000, 19, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 1999, 22, 451–461. [Google Scholar] [CrossRef]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Brumby, A.M.; Richardson, H.E. Scribble mutants cooperate with oncogenic Ras or notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.A.; Xu, T. A genetic screen in Drosophila for metastatic behavior. Science 2003, 302, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Khoo, P.; Allan, K.; Willoughby, L.; Brumby, A.M.; Richardson, H.E. In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signalling. Dis. Model Mech. 2013, 6, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; McCall, K.; Steller, H. Dcp-1, a Drosophila cell death protease essential for development. Science 1997, 275, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Hanratty, W.P.; Dearolf, C.R. An amino acid substitution in the Drosophila hoptum-l jak kinase causes leukemia-like hematopoietic defects. EMBO J. 1995, 14, 1412–1420. [Google Scholar] [PubMed]
- Arbouzova, N.I.; Zeidler, M.P. Jak/stat signalling in Drosophila: Insights into conserved regulatory and cellular functions. Development 2006, 133, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.F.; Wu, J.-W.; Bryant, P.J. Localization of proteins to the apico-lateral junctions of Drosophila epithelia. Dev. Genet. 1997, 20, 111–118. [Google Scholar] [CrossRef]
- Harris, T.J.C.; Peifer, M. The positioning and segregation of apical cues during epithelial polarity establishment in Drosophila. J. Cell Biol. 2005, 170, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Martín, F.A.; Perez-Garijo, A.; Morata, G. Apoptosis in Drosophila: Compensatory proliferation and undead cells. Int. J. Dev. Biol. 2009, 53, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Adachi-Yamada, T.; Nakamura, M.; Irie, K.; Tomoyasu, Y.; Sano, Y.; Mori, E.; Goto, S.; Ueno, N.; Nishida, Y.; Matsumoto, K. P38 mitogen-activated protein kinase can be involved in transforming growth factor beta superfamily signal transduction in Drosophila wing morphogenesis. Mol. Cell. Biol. 1999, 19, 2322–2329. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Moreno, E.; Yan, M.; Basler, K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002, 12, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Galko, M.J.; KRasnow, M.A. Cellular and genetic analysis of wound healing in Drosophila larvae. PLOS Biol. 2004, 2, e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesgo-Escovar, J.R.; Jenni, M.; Fritz, A.; Hafen, E. The Drosophila jun-N-terminal kinase is required for cell morphogenesis but not for djun-dependent cell fate specification in the eye. Genes Dev. 1996, 10, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Sluss, H.K.; Han, Z.; Barrett, T.; Davis, R.J.; Ip, Y.T. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 1996, 10, 2745–2758. [Google Scholar] [CrossRef] [PubMed]
- Weber, U.; Paricio, N.; Mlodzik, M. Jun mediates frizzled-induced R3/R4 cell fate distinction and planar polarity determination in the Drosophila eye. Development 2000, 127, 3619–3629. [Google Scholar] [PubMed]
- Fernandez, B.; Jezowska, B.; Janody, F. Drosophila actin-capping protein limits JNK activation by the Src proto-oncogene. Oncogene 2014, 33, 2027–2039. [Google Scholar] [CrossRef] [PubMed]
- Rudrapatna, V.A.; Bangi, E.; Cagan, R.L. A JNK-Rho-actin remodeling positive feedback network directs Src-driven invasion. Oncogene 2014, 33, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Uhlirova, M.; Bohmann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006, 25, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Leong, G.R.; Goulding, K.R.; Amin, N.; Richardson, H.E.; Brumby, A.M. Scribble mutants promote APKC and JNK-dependent epithelial neoplasia independently of crumbs. BMC Biol. 2009, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Doggett, K.; Turkel, N.; Willoughby, L.F.; Ellul, J.; Murray, M.J.; Richardson, H.E.; Brumby, A.M. BTB-zinc finger oncogenes are required for Ras and Notch-driven tumorigenesis in Drosophila. PLoS ONE 2015, 10, e0132987. [Google Scholar] [CrossRef] [PubMed]
- Külshammer, E.; Uhlirova, M. The actin cross-linker Filamin/Cheerio mediates tumor malignancy downstream of JNK signaling. J. Cell Sci. 2013, 126, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Külshammer, E.; Mundorf, J.; Kilinc, M.; Frommolt, P.; Wagle, P.; Uhlirova, M. Interplay among Drosophila transcription factors Ets21c, Fos and Ftz-F1 drives JNK-mediated tumor malignancy. Dis. Models Mech. 2015, 8, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, M.; Potier, D.; Romanelli, L.; Jacobs, J.; Mach, J.; Hamaratoglu, F.; Aerts, S.; Halder, G. An ectopic network of transcription factors regulated by Hippo signaling drives growth and invasion of a malignant tumor model. Curr. Biol. 2016, 26, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Mott, H.R.; Owen, D. Structures of Ras superfamily effector complexes: What have we learnt in two decades? Crit. Rev. Biochem. Mol. Biol. 2015, 50, 85–133. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.; Chang, H.; Karim, F.; Laverty, T.; Michaud, N.; Morrison, D.; Rebay, I.; Tang, A.; Therrien, M.; Wassarman, D. Signal Transduction Downstream from Ras in Drosophila; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; pp. 347–352. [Google Scholar]
- Brand, A.H.; Perrimon, N. Raf acts downstream of the EGF receptor to determine dorsoventral polarity during Drosophila oogenesis. Genes Dev. 1994, 8, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Uhlirova, M.; Jasper, H.; Bohmann, D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc. Natl. Acad. Sci. USA 2005, 102, 13123–13128. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Nicolette, C.; Minden, A.; Polverino, A.; Van Aelst, L.; Karin, M.; Wigler, M.H. Multiple Ras functions can contribute to mammalian cell transformation. Cell 1995, 80, 533–541. [Google Scholar] [CrossRef]
- Kelso, R.J.; Hudson, A.M.; Cooley, L. Drosophila kelch regulates actin organization via Src64-dependent tyrosine phosphorylation. J. Cell Biol. 2002, 156, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Tominaga, T. mDia-interacting protein acts downstream of Rho-mDia and modifies Src activation and stress fiber formation. J. Biol. Chem. 2001, 276, 39290–39294. [Google Scholar] [CrossRef] [PubMed]
- Jasper, H.; Benes, V.; Schwager, C.; Sauer, S.; Clauder-Münster, S.; Ansorge, W.; Bohmann, D. The genomic response of the Drosophila embryo to JNK signaling. Dev. Cell 2001, 1, 579–586. [Google Scholar] [CrossRef]
- Benlali, A.; DRaskovic, I.; Hazelett, D.J.; Treisman, J.E. Act up controls actin polymerization to alter cell shape and restrict hedgehog signaling in the Drosophila eye disc. Cell 2000, 101, 271–281. [Google Scholar] [CrossRef]
- DeMali, K.A.; Wennerberg, K.; Burridge, K. Integrin signaling to the actin cytoskeleton. Curr. Opin. Cell Biol. 2003, 15, 572–582. [Google Scholar] [CrossRef]
- Homsy, J.G.; Jasper, H.; Peralta, X.G.; Wu, H.; Kiehart, D.P.; Bohmann, D. JNK signaling coordinates integrin and actin functions during Drosophila embryogenesis. Dev. Dyn. 2006, 235, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.; Holder, M.V.; Aerne, B.L.; Janody, F.; Tapon, N. Zyxin antagonizes the ferm protein expanded to couple F-actin and yorkie-dependent organ growth. Curr. Biol. 2015, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, W.; Yu, J.; Zheng, Y.; Qing, Y.; Pan, D. Spectrin regulates Hippo signaling by modulating cortical actoMyosin activity. eLife 2015, 4, e06567. [Google Scholar] [CrossRef] [PubMed]
- Dent, L.G.; Poon, C.L.C.; Zhang, X.; Degoutin, J.L.; Tipping, M.; Veraksa, A.; Harvey, K.F. The GTPase regulatory proteins pix and git control tissue growth via the Hippo pathway. Curr. Biol. 2015, 25, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.L.; Li, W.; An, Y.; Duan, Y.; Li, Z.; Kang, Y.; Yan, Y. Β-spectrin regulates the Hippo signaling pathway and modulates the basal actin network. J. Biol. Chem. 2015, 290, 6397–6407. [Google Scholar] [CrossRef] [PubMed]
- Sansores-Garcia, L.; Bossuyt, W.; Wada, K.-I.; Yonemura, S.; Tao, C.; Sasaki, H.; Halder, G. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011, 30, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Fernández, B.G.; Gaspar, P.; Brás-Pereira, C.; Jezowska, B.; Rebelo, S.R.; Janody, F. Actin-capping protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development 2011, 138, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Kizawa, D.; Ohsawa, S.; Igaki, T. JNK signaling is converted from anti- to pro-tumor pathway by Ras-mediated switch of warts activity. Dev. Biol. 2015, 403, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhuang, Y.; Han, M.; Xu, T.; Deng, K. Ras promotes cell survival by antagonizing both JNK and hid signals in the Drosophila eye. BMC Dev. Biol. 2009, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cagan, R.L. Drosophila models for cancer research. Curr. Opin. Genet. Dev. 2006, 16, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Bunker, B.D.; Nellimoottil, T.T.; Boileau, R.M.; Classen, A.K.; Bilder, D. The transcriptional response to tumorigenic polarity loss in Drosophila. eLife 2015, 4, e03189. [Google Scholar] [CrossRef] [PubMed]
- Colombani, J.; Andersen, D.S.; Leopold, P. Secreted peptide dilp8 coordinates Drosophila tissue growth with developmental timing. Science 2012, 336, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Garelli, A.; Gontijo, A.M.; Miguela, V.; Caparros, E.; Dominguez, M. Imaginal discs secrete insulin-like peptide 8 to mediate plasticity of growth and maturation. Science 2012, 336, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Boone, E.; Colombani, J.; Andersen, D.S.; Léopold, P. The Hippo signalling pathway coordinates organ growth and limits developmental variability by controlling dilp8 expression. Nat. Commun. 2016, 7, 13505. [Google Scholar] [CrossRef] [PubMed]
- Alter, J.; Rozentzweig, D.; Bengal, E. Inhibition of myoblast differentiation by tumor necrosis factor α is mediated by c-Jun N-terminal kinase 1 and leukemia inhibitory factor. J. Biol. Chem. 2008, 283, 23224–23234. [Google Scholar] [CrossRef] [PubMed]
- Gazel, A.; Banno, T.; Walsh, R.; Blumenberg, M. Inhibition of JNK promotes differentiation of epidermal keratinocytes. J. Biol. Chem. 2006, 281, 20530–20541. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Stepniak, E.; Hui, L.; Leibbrandt, A.; Katada, T.; Nishina, H.; Wagner, E.F.; Penninger, J.M. Antagonistic control of cell fates by JNK and p38-MAPK signaling. Cell. Death Differ. 2008, 15, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M. The transcription cofactor c-Jun mediates phenotype switching and bRaf inhibitor resistance in melanoma. Sci. Signal. 2015, 8, ra82. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, P.T. Taking out the JNK: A window of opportunity to improve cancer therapy. Mol. Cell. Oncol. 2016, 3, e1128515. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537. [Google Scholar] [CrossRef] [PubMed]
- Willecke, M.; Toggweiler, J.; Basler, K. Loss of PI3K blocks cell-cycle progression in a Drosophila tumor model. Oncogene 2011, 30, 4067–4074. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Chapter three—Autophagy in cell life and cell death. In Current Topics in Developmental Biology; Steller, H., Ed.; Academic Press: Cambridge, MA, USA, 2015; Volume 114, pp. 67–91. [Google Scholar]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhász, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.W.; Purkayastha, A.; Jones, K.T.; Thaker, S.K.; Banerjee, U. In vivo genetic dissection of tumor growth and the warburg effect. Elife 2016, 5, e18126. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Hay, B.A.; Maile, R.; Rubin, G.M. P element insertion-dependent gene activation in the Drosophila eye. Proc. Natl. Acad. Sci. USA 1997, 94, 5195–5200. [Google Scholar] [CrossRef] [PubMed]
- Ruberte, E.; Marty, T.; Nellen, D.; Affolter, M.; Basler, K. An absolute requirement for both the type II and type I receptors, punt and thick veins, for dpp signaling in vivo. Cell 1995, 80, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Luo, L. Mosaic analysis with a repressible cell marker (marcm) for Drosophila neural development. Trends Neurosci. 2001, 24, 251–254. [Google Scholar] [CrossRef]
- Toba, G.; Ohsako, T.; Miyata, N.; Ohtsuka, T.; Seong, K.H.; Aigaki, T. The gene search system. A method for efficient detection and rapid molecular identification of genes in Drosophila melanogaster. Genetics 1999, 151, 725–737. [Google Scholar] [PubMed]
- Gao, X.; Neufeld, T.P.; Pan, D. Drosophila PTEN regulates cell growth and proliferation through PI3K-dependent and -independent pathways. Dev. Biol. 2000, 221, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Leevers, S.J.; Weinkove, D.; MacDougall, L.K.; Hafen, E.; Waterfield, M.D. The Drosophila phosphoinositide 3-kinase dp110 promotes cell growth. EMBO J. 1996, 15, 6584–6594. [Google Scholar] [PubMed]
- Wodarz, A.; Ramrath, A.; Kuchinke, U.; Knust, E. Bazooka provides an apical cue for inscuteable localization in Drosophila neuroblasts. Nature 1999, 402, 544. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transgene | Control | Src42AGS | Src42A | Src42AACT | Src64B | |
|---|---|---|---|---|---|---|
| GAL4 driver | ||||||
| ey> | Wild-type | Wild-type | Wild-type | Range of rough eye phenotypes, from very small to reduced eye size | Range of rough eye phenotypes, from no eye to smaller reduced size | |
| ey > p35 | Wild-type | Wild-type | Wild-type | Rough, reduced eye size | Rough, reduced eye size | |
| ey > RasACT | Hyperplastic overgrowth | Enhanced hyperplastic overgrowth | No enhanced overgrowth | Enhanced growth in dorsal region, reduced in ventral region | Enhanced hyperplastic overgrowth | |
| Transgene | Src42AGS + | |||||
|---|---|---|---|---|---|---|
| Phenotype | Control | p35 | bskDN | RasACT | RasACT bskDN | |
| Clone size | Small clones; increased cell death | Small clones, with increased cell proliferation in adjacent wild-type clones | Increased clone size, and basal overgrowth | Large clones, wild type tissue present | Reduced clonal overgrowth; restored tissue morphology | |
| Differentiation | Normal | Normal but disrupted organisation | Normal but disrupted organisation | Reduced | Restored | |
| F actin | Normal, some clones show enriched F-actin | Accumulation | Cortical localisation | Accumulation | Cortical localisation | |
| Protrusive morphology | Yes | NA | NA | Enhanced | Suppressed | |
| JNK (Jun N-terminal kinase) pathway reporter | NA | Increased | NA | Increased | NA | |
| Adult phenotype | 1–2 day delay in adult eclosion, eye phenotype comparable to control | Overgrown eye tissue | Lethal at late larval/early pupal stage | Lethal at late L3, with melanotic masses | Lethal at late larval/early pupal stage | |
| Transgene | Src64B + | |||||
|---|---|---|---|---|---|---|
| Phenotype | Control | p35 | bskDN | RasACT | RasACT + bskDN | |
| Clone size | Small clones within epithelium; Discrete, rounded clones excluded from epithelia proper; increased cell death | Small clones, clones, with increased cell proliferation in adjacent wild-type clones | Increased clone size, basal over-growth | Large clones that out compete wild -type tissue | Reduced clone size; restored overall tissue morphology | |
| Differentiation | Normal differentiation in small clones; no differentiation in clones located apical to eye disc proper | Normal but disrupted organisation | Normal but disrupted organisation, with some differentiated cells localised basally | Reduced | Partially restored | |
| F-actin | Accumulation | Accumulation | Reduced cortically | Accumulation | Enriched cortically | |
| Protrusive morphology | NA | NA | NA | Increased | Decreased | |
| JNK pathway reporter | Increased | NA | NA | Increased | NA | |
| Adult phenotype | 1–2 day delay in adult eclosion, eye phenotype comparable to control | Larval lethal | Lethal at late larval/early pupal stage, with melanotic masses | Larval lethal | Lethal at late larval/early pupal stage | |
| Transgene | Src64B + | ||||
|---|---|---|---|---|---|
| Phenotype | RafGOF | RasACT S35 | RasACT + PTEN | RasACT + Dp110DN | |
| Clone size | No cooperative overgrowth | Large clones | Reduced clone size compared to Src + RasACT | Reduced clone size compared to Src + RasACT | |
| Differentiation | Suppressed ectopic RafGOF differentiation | Reduced | Reduced | Reduced | |
| F-actin | Enrichment at clone borders | Accumulation | Accumulation | Accumulation | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poon, C.L.C.; Brumby, A.M.; Richardson, H.E. Src Cooperates with Oncogenic Ras in Tumourigenesis via the JNK and PI3K Pathways in Drosophila epithelial Tissue. Int. J. Mol. Sci. 2018, 19, 1585. https://doi.org/10.3390/ijms19061585
Poon CLC, Brumby AM, Richardson HE. Src Cooperates with Oncogenic Ras in Tumourigenesis via the JNK and PI3K Pathways in Drosophila epithelial Tissue. International Journal of Molecular Sciences. 2018; 19(6):1585. https://doi.org/10.3390/ijms19061585
Chicago/Turabian StylePoon, Carole L.C., Anthony M. Brumby, and Helena E. Richardson. 2018. "Src Cooperates with Oncogenic Ras in Tumourigenesis via the JNK and PI3K Pathways in Drosophila epithelial Tissue" International Journal of Molecular Sciences 19, no. 6: 1585. https://doi.org/10.3390/ijms19061585