DNA Methylation Profiling of Breast Cancer Cell Lines along the Epithelial Mesenchymal Spectrum—Implications for the Choice of Circulating Tumour DNA Methylation Markers

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Ranking of Breast Cancer Cell Lines across the Epithelial–Mesenchymal Spectrum
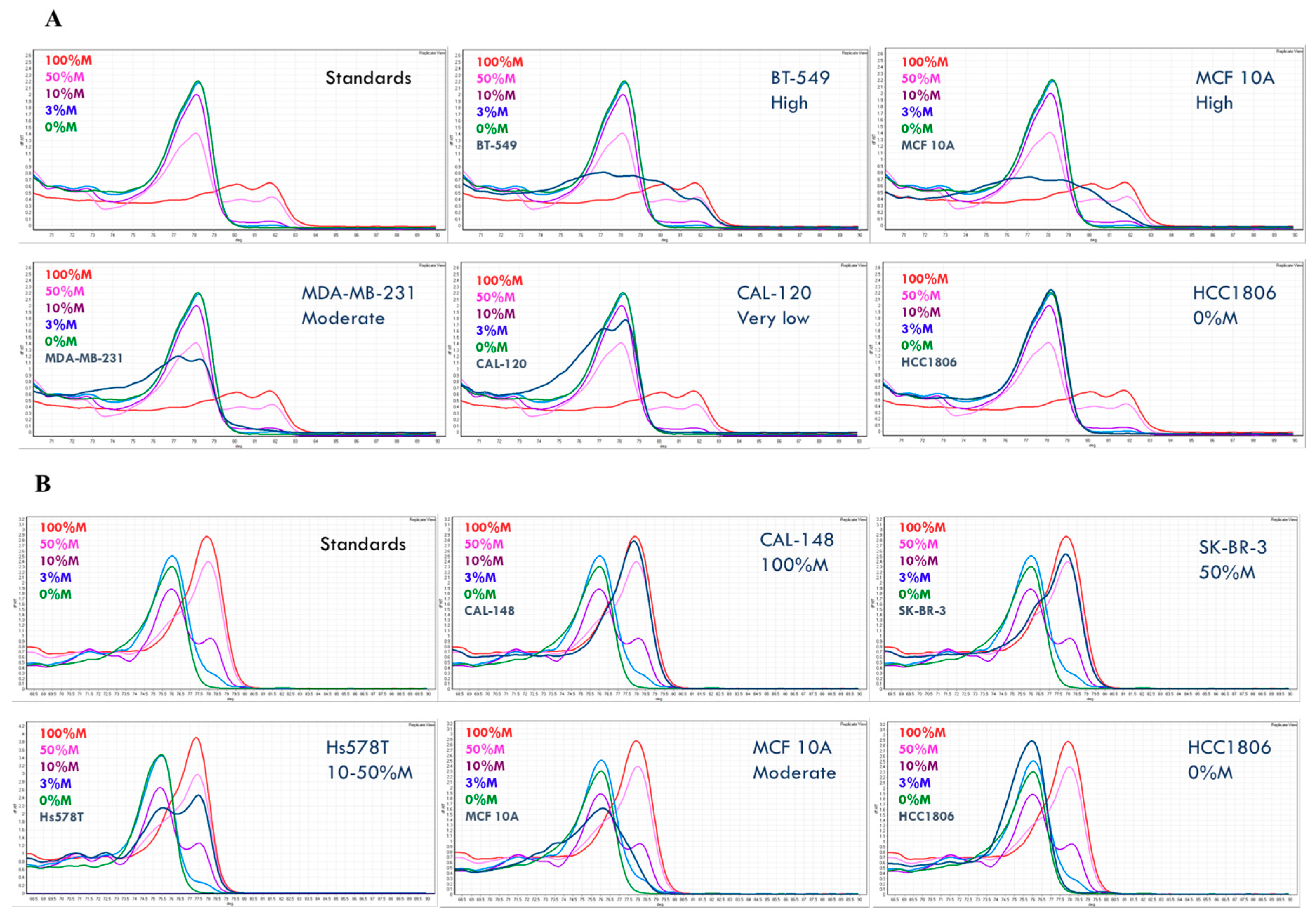
2.2. The DNA Methylation Status of the Breast Cancer Cell Lines
2.3. Heterogeneous Methylation Is Frequent for the Studied Markers
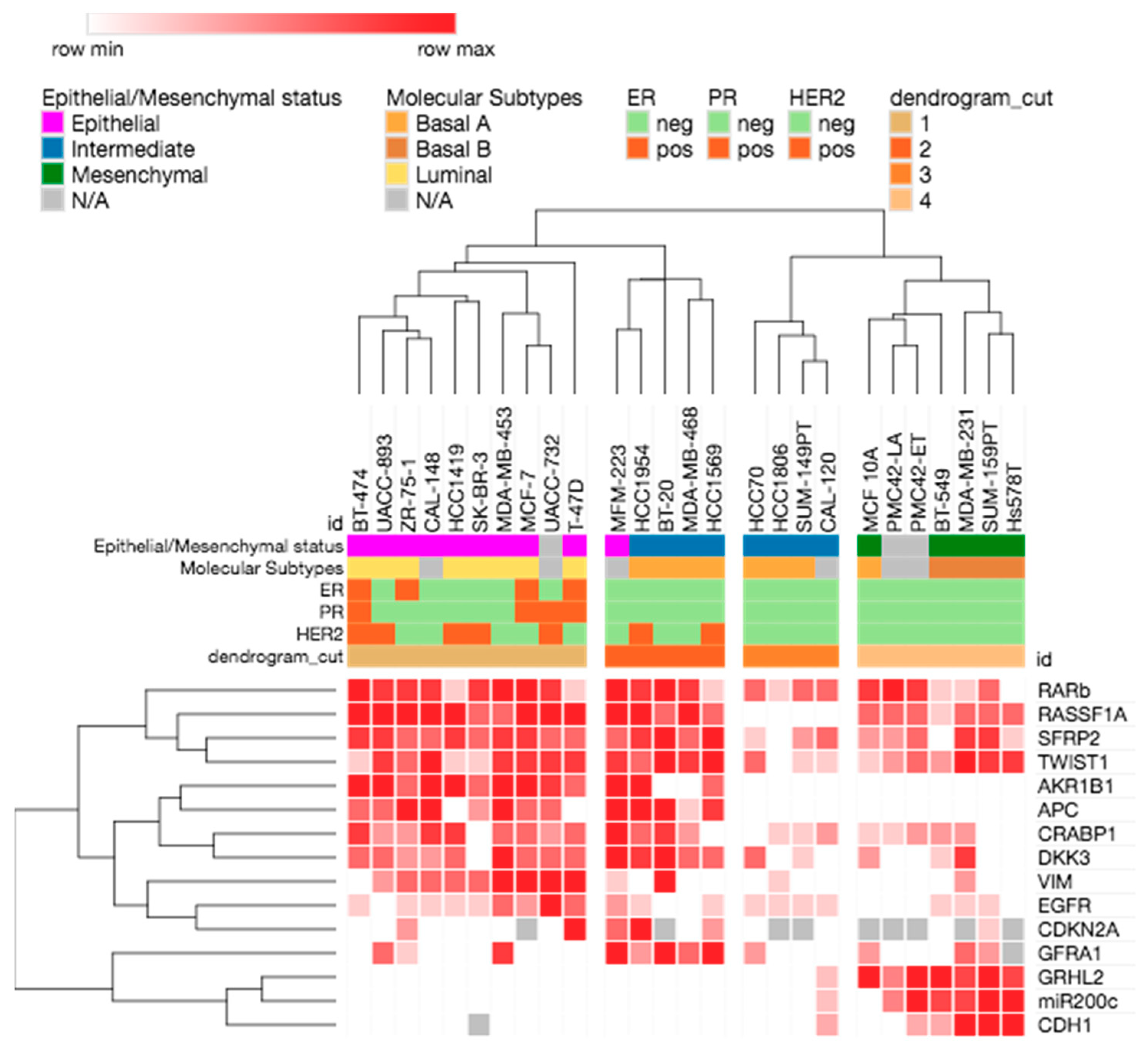
2.4. Methylation Patterns and EMP Status in Breast Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Breast Cancer Cell Lines
4.2. DNA Extraction from Cell Pellets
4.3. Bisulfite Conversion
4.4. Methylation-Sensitive High-Resolution Melting
4.5. Bisulfite Pyrosequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EMP | Epithelial–mesenchymal plasticity |
| EMT | Epithelial–mesenchymal transition |
| MET | Mesenchymal–epithelial transition |
| MS-HRM | Methylation-sensitive high-resolution melting |
| PCR | Polymerase chain reaction |
| MSP | Methylation-specific PCR |
| CpG | Cytosine–guanine dinucleotides |
| MRD | Minimal residual disease |
| CTCs | Circulating tumour cells |
| ctDNA | Circulating tumour DNA |
| ER | Estrogen receptor |
| PR | Progesterone receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| ATCC | American Type Culture Collection |
| AKR1B1 | Aldo-Keto Reductase Family 1 Member B1 |
| APC | Adenomatous Polyposis Coli |
| RASSF1A | Ras Association Domain Family Member 1A |
| RARß | Retinoic Acid Receptor Beta |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| BRCA1 | Breast Cancer 1 |
| TWIST1 | Twist Family BHLH Transcription Factor 1 |
| CRABP1 | Cellular Retinoic Acid Binding Protein 1 |
| DKK3 | Dickkopf WNT Signaling Pathway Inhibitor 3 |
| EGFR | Epidermal Growth Factor Receptor |
| VIM | Vimentin |
| GRHL2 | Grainyhead-Like Transcription Factor 2 |
| MIR200C | MicroRNA 200c |
| CDH1 | Cadherin 1 |
| SFRP2 | Secreted Frizzled Related Protein 2 |
| GFRA1 | GDNF Family Receptor Alpha 1 |
References
- Silva, J.M.; Dominguez, G.; Garcia, J.M.; Gonzalez, R.; Villanueva, M.J.; Navarro, F.; Bonilla, F. Presence of tumor DNA in plasma of breast cancer patients: Clinicopathological correlations. Cancer Res. 1999, 59, 3251–3256. [Google Scholar] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhou, Y.; Zhou, D.; Kan, M.; Niu, X.; Zhang, Z.; Liu, Y. Whole DNA methylome profiling in lung cancer cells before and after epithelial-to-mesenchymal transition. Diagn. Pathol. 2014, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genom. 2010, 11, 137. [Google Scholar] [CrossRef]
- Carmona, F.J.; Davalos, V.; Vidal, E.; Gomez, A.; Heyn, H.; Hashimoto, Y.; Sánchez-Mut, J.V. A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res. 2014, 74, 5608–5619. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metast. Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blick, T.; Widodo, E.; Hugo, H.; Waltham, M.; Lenburg, M.E.; Neve, R.M.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin. Exp. Metast. 2008, 25, 629–642. [Google Scholar] [CrossRef] [PubMed]
- America Type Culture Collection. Available online: https://www.atcc.org (accessed on 17 May 2016).
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.L.; Girard, L.; Gandhi, J.; Minna, J.D. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommers, C.L.; Byers, S.W.; Thompson, E.W.; Torri, J.A.; Gelmann, E.P. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res. Treat. 1994, 31, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.W.; Paik, S.; Brünner, N.; Sommers, C.L.; Zugmaier, G.; Clarke, R.; Martin, G.R. Association of increased basement membrane invasiveness with absence of estrogen receptor and expression of vimentin in human breast cancer cell lines. J. Cell. Physiol. 1992, 150, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Neves, P.L.; Morgado, E.; Faísca, M.; Carrasqueira, H.; Baptista, A.; Silva, A.P. Nutritional and inflammatory status influence darbepoetin dose in pre-dialysis elderly patients. Int. Urol. Nephrol. 2006, 38, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Zhou, W. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [PubMed]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Bayani, N. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Kalous, O.; Conklin, D.; Desai, A.J.; O’Brien, N.A.; Ginther, C.; Anderson, L.; Slamon, D.J. Dacomitinib (PF-00299804), an irreversible Pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol. Cancer Ther. 2012, 11, 1978–1987. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Dobrovic, A.; Hansen, L.L. Methylation-sensitive high-resolution melting. Nat. Protoc. 2008, 3, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Dobrovic, A. Methylation-sensitive high resolution melting (MS-HRM): A new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007, 35, e41. [Google Scholar] [CrossRef] [PubMed]
- Candiloro, I.L.; Mikeska, T.; Hokland, P.; Dobrovic, A. Rapid analysis of heterogeneously methylated DNA using digital methylation-sensitive high resolution melting: Application to the CDKN2B (p15) gene. Epigenet. Chromatin 2008, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Candiloro, I.L.; Mikeska, T.; Dobrovic, A. Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics 2011, 6, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Veeck, J.; Bektas, N.; Hartmann, A.; Kristiansen, G.; Heindrichs, U.; Knüchel, R.; Dahl, E. Wnt signalling in human breast cancer: Expression of the putative Wnt inhibitor Dickkopf-3 (DKK3) is frequently suppressed by promoter hypermethylation in mammary tumours. Breast Cancer Res. 2008, 10, R82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, A.J.; Díaz-Montero, C.M.; Mao, L.; Youssef, E.; Estecio, M.R.; Shen, L.; Issa, J.P. Epigenetic inactivation of EGFR by CpG island hypermethylation in cancer. Cancer Biol. Ther. 2006, 5, 1494–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, M.F.; Fraga, M.F.; Avila, S.; Guo, M.; Pollan, M.; Herman, J.G.; Esteller, M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003, 63, 1114–1121. [Google Scholar] [PubMed]
- Lombaerts, M.; Van Wezel, T.; Philippo, K.; Dierssen, J.W.F.; Zimmerman, R.M.E.; Oosting, J.; Cleton-Jansen, A.M. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer 2006, 94, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Deng, Z.; Zhuang, X.; Ju, S.; Mu, J.; Jiang, H.; Zhang, H.G. Grhl2 determines the epithelial phenotype of breast cancers and promotes tumor progression. PLoS ONE 2012, 7, e50781. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Riley, P.; Pifer, P.M.; Widmeyer, J.; Addison, J.B.; Ivanov, A.V.; Frisch, S.M. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012, 72, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Holcomb, T.; Januario, T.; Du, P.; Evangelista, M.; Kartha, N.; Modrusan, Z. DNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 2360–2373. [Google Scholar] [CrossRef] [PubMed]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2012, 31, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.; Scheel, C.; Weinhold, S.; Honisch, E.; Iwaniuk, K.M.; Trompeter, H.I.; Uhrberg, M. Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res. Notes 2010, 3, 219. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Wright, J.A.; Attema, J.L.; Gregory, P.A.; Bert, A.G.; Smith, E.; Goodall, G.J. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J. Cell Sci. 2013, 126, 2256–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, E.; Dubois-Marshall, S.; Sims, A.H.; Faratian, D.; Li, J.; Smith, E.S.; Langdon, S.P. A gene on the HER2 amplicon, C35, is an oncogene in breast cancer whose actions are prevented by inhibition of Syk. Br. J. Cancer 2010, 103, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahov, N.; Scrace, S.; Soto, M.S.; Grawenda, A.M.; Bradley, L.; Pankova, D.; Timpson, P. Alternate RASSF1 Transcripts Control SRC Activity, E-Cadherin Contacts, and YAP-Mediated Invasion. Curr. Biol. 2015, 25, 3019–3034. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozaki, M.; Hoon, D.S.; Giuliano, A.E.; Hansen, N.M.; Wang, H.J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Gort, E.H.; Suijkerbuijk, K.P.; Roothaan, S.M.; Raman, V.; Vooijs, M.; van der Wall, E.; van Diest, P.J. Methylation of the TWIST1 promoter, TWIST1 mRNA levels, and immunohistochemical expression of TWIST1 in breast cancer. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 3325–3330. [Google Scholar] [CrossRef] [PubMed]
- Veeck, J.; Noetzel, E.; Bektas, N.; Jost, E.; Hartmann, A.; Knüchel, R.; Dahl, E. Promoter hypermethylation of the SFRP2 gene is a high-frequent alteration and tumor-specific epigenetic marker in human breast cancer. Mol. Cancer 2008, 7, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.I.E.; Zhang, X.; Sun, B.; Lu, H.; Wang, D.; Yuan, X.; Huang, Z. Detection of aberrant promoter methylation of RNF180, DAPK1 and SFRP2 in plasma DNA of patients with gastric cancer. Oncol. Lett. 2014, 8, 1745–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, H.; Amatya, V.J.; Takeshima, Y.; Kushitani, K.; Hattori, N.; Kohno, N.; Inai, K. Aberrant promoter methylation of WIF-1 and SFRP1, 2, 4 genes in mesothelioma. Oncol. Rep. 2010, 24, 423–431. [Google Scholar] [PubMed]
- Nojima, M.; Suzuki, H.; Toyota, M.; Watanabe, Y.; Maruyama, R.; Sasaki, S.; Hirata, K. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007, 26, 4699–4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; Van Engeland, M.; Weijenberg, M.P.; Baylin, S.B. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002, 31, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhu, D.; Chen, F.; Qian, M.; Wei, H.; Chen, W.; Xu, J. SFRP2 augments WNT16B signaling to promote therapeutic resistance in the damaged tumor microenvironment. Oncogene 2016, 35, 4321–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, M.; Borg, Å.; Nilbert, M.; Andersson, T. Involvement of adenomatous polyposis coli (APC)/beta-catenin signalling in human breast cancer. Eur. J. Cancer 2000, 36, 242–248. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Zablocki, G.J.; Ruzycki, P.A.; Overturf, M.A.; Palla, S.; Reddy, G.B.; Petrash, J.M. Aldose reductase-mediated induction of epithelium-to-mesenchymal transition (EMT) in lens. Chem. Biol. Interact. 2011, 191, 351–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, X.; Fu, Q.; Cao, Q.; Chen, X.; Wang, M.; Wang, D. AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. J. Exp. Med. 2017, 214, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Dedeurwaerder, S.; Desmedt, C.; Calonne, E.; Singhal, S.K.; Haibe-Kains, B.; Defrance, M.; Lallemand, F. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol. Med. 2011, 3, 726–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fackler, M.J.; Umbricht, C.; Williams, D.; Argani, P.; Cruz, L.A.; Merino, V.F.; Marks, J. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res. 2011, 71, 6195–6207. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Fackler, M.J.; Lee, J.H.; Choi, C.; Park, M.H.; Yoon, J.H.; Sukumar, S. Basal-like breast cancer displays distinct patterns of promoter methylation. Cancer Biol. Ther. 2010, 9, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Subedee, A.; Bloushtain-Qimron, N.; Savova, V.; Krzystanek, M.; Li, L.; Li, M. Somatic Cell. Fusions Reveal Extensive Heterogeneity in Basal-like Breast Cancer. Cell Rep. 2015, 11, 1549–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, V.Y.; Tan, T.Z.; Tan, M.; Wong, M.K.; Kuay, K.T.; Yang, Z.; Thiery, J.P. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci. Rep. 2016, 6, 19943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malouf, G.G.; Taube, J.H.; Lu, Y.; Roysarkar, T.; Panjarian, S.; Estecio, M.R.; Tahara, T. Architecture of epigenetic reprogramming following Twist1-mediated epithelial-mesenchymal transition. Genome Biol. 2013, 14, R144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, L.M.; Fackler, M.J.; Lopez-Bujanda, Z.; Wolff, A.C.; Visvanathan, K.; Gray, J.W.; Umbricht, C.B. Do breast cancer cell lines provide a relevant model of the patient tumor methylome? PLoS ONE 2014, 9, e105545. [Google Scholar] [CrossRef] [PubMed]
- Smiraglia, D.J.; Rush, L.J.; Frühwald, M.C.; Dai, Z.; Held, W.A.; Costello, J.F.; Caligiuri, M.A. Excessive CpG island hypermethylation in cancer cell lines versus primary human malignancies. Hum. Mol. Genet. 2001, 10, 1413–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikeska, T.; Bock, C.; Do, H.; Dobrovic, A. DNA methylation biomarkers in cancer: Progress towards clinical implementation. Expert Rev. Mol. Diagn. 2012, 12, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Hansen, L.L.; Dobrovic, A. A new approach to primer design for the control of PCR bias in methylation studies. BMC Res. Notes 2008, 1, 54. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ranking | BC Cell Lines | ER Expression | PR Expression | HER2 Expression | VIM Status | Subtypes | Subtypes | Subtypes | HER2 Status | EMT Status |
|---|---|---|---|---|---|---|---|---|---|---|
| Tan 2014 | ATCC (1) Kao 2009 (2) Lehmann 2011 (3) | Thompson 1992 (4) Sommers 1994 (5) | Neve 2006 | Heiser 2012 (E-MTAB-181) | Hoeflich 2009 (GSE12790) | Tan 2014 | ||||
| 1 | MFM-223 | ER− (3) | PR− (3) | HER2− (3) | Luminal | Her2 amplified | Epithelial | |||
| 2 | MDA-MB-453 | ER− (1) | PR− (1) | HER2− (1) | VIM− (5) | Luminal | Luminal | Luminal | Her2 amplified | Epithelial |
| 3 | BT-474 | ER+ (2) | PR+ (2) | HER2+ (2) | VIM− (5) | Luminal | Luminal | Luminal | Her2 amplified | Epithelial |
| 4 | ZR-75-1 | ER+ (2) | PR− (2) | HER2− (2) | VIM− (4) | Luminal | Luminal | Luminal | Nonamplified | Epithelial |
| 5 | HCC1419 | ER− (1) | PR− (1) | HER2+ (1) | Luminal | Luminal | Her2 amplified | Epithelial | ||
| 6 | UACC-893 | ER− (1) | PR− (1) | HER2+ (1) | Luminal | Luminal | Her2 amplified | Epithelial | ||
| 7 | SK-BR-3 | ER− (2) | PR− (2) | HER2+ (2) | VIM− (4, 5) | Luminal | Luminal | Luminal | Her2 amplified | Epithelial |
| 8 | T-47D | ER+ (2) | PR+ (2) | HER− (2) | VIM− (4, 5) | Luminal | Luminal | Luminal | Nonamplified | Epithelial |
| 9 | CAL-148 | ER− (3) | PR− (3) | HER2− (3) | Basal | Nonamplified | Epithelial | |||
| 10 | MCF-7 | ER+ (2) | PR+ (2) | HER2− (2) | VIM− (4, 5) | Luminal | Luminal | Luminal | Nonamplified | Epithelial |
| 11 | UACC-732 | ER− (1) | PR+ (1) | HER2+ (1) | Epithelial | |||||
| 12 | MDA-MB-468 | ER− (1) | PR− (1) | HER2− (1) | VIM− (4, 5) | Basal A | Basal A | Basal | Nonamplified | Intermediate |
| 13 | HCC1954 | ER− (1) | PR− (1) | HER2+ (1) | Basal A | Basal A | Basal | Her2 amplified | Intermediate | |
| 14 | HCC70 | ER− (1) | PR− (1) | HER2− (1) | Basal A | Basal A | Basal | Nonamplified | Intermediate | |
| 15 | BT-20 | ER− (1) | PR− (1) | HER2− (1) | Basal A | Basal A | Basal | Nonamplified | Intermediate | |
| 16 | HCC1806 | ER− (1) | PR− (1) | HER2− (1) | Basal A | Basal | Nonamplified | Intermediate | ||
| 17 | HCC1569 | ER− (1) | PR− (1) | HER2+ (1) | Basal A | Basal A | Basal | Her2 amplified | Intermediate | |
| 18 | SUM-149PT | ER− (3) | PR− (3) | HER2− (3) | Basal B | Basal A | Intermediate | |||
| 19 | CAL-120 | ER− (3) | PR− (3) | HER2− (3) | Basal | Nonamplified | Intermediate | |||
| 20 | MCF 10A | ER− (2) | PR− (2) | HER− (2) | Basal B | Basal A | Mesenchymal | |||
| PMC42-LA | ER− | PR− | HER2− | VIM low | ||||||
| PMC42-ET | ER− | PR− | HER2− | VIM+ | ||||||
| 21 | MDA-MB-231 | ER− (1) | PR− (1) | HER2− (1) | VIM+ (4, 5) | Basal B | Basal B | Basal | Nonamplified | Mesenchymal |
| 22 | BT-549 | ER− (1) | PR− (1) | HER2− (1) | VIM+ (4, 5) | Basal B | Basal B | Basal | Nonamplified | Mesenchymal |
| 23 | SUM-159PT | ER− (3) | PR− (3) | HER2− (3) | Basal B | Basal B | Mesenchymal | |||
| 24 | Hs578T | ER− (1) | PR− (1) | HER2− (1) | VIM+ (4, 5) | Basal B | Basal B | Basal | Nonamplified | Mesenchymal |
| A | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| % of Methylated Allele | |||||||||
| CpG Positions Samples | Status by MS-HRM (Confirmed by Pyrosequencing) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 100%M Std | 90 | 88 | 89 | 86 | 90 | 83 | 96 | 94 | |
| 50%M Std | 41 | 44 | 40 | 41 | 44 | 39 | 45 | 47 | |
| 10%M Std | 12 | 16 | 15 | 14 | 16 | 16 | 18 | 19 | |
| 0%M Std | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | |
| BT-549 | high (yes) | 67 | 88 | 79 | 96 | 93 | 89 | 83 | 70 |
| MCF 10A | high (yes) | 74 | 77 | 72 | 88 | 89 | 76 | 79 | 53 |
| MDA-MB-231 | moderate (yes) | 17 | 27 | 15 | 25 | 20 | 23 | 21 | 13 |
| CAL-120 | very low (yes) | 6 | 12 | 7 | 7 | 9 | 8 | 8 | 5 |
| HCC1806 | 0%M (yes) | 2 | 2 | 1 | 1 | 2 | 1 | 1 | 1 |
| B | |||||||||
| % of Methylated Allele | |||||||||
| CpG Positions Samples | Status by MSHRM (Confirmed by Pyrosequencing) | 1 | 2 | 3 | 4 | ||||
| 100%M Std | 97 | 65 | 95 | 89 | |||||
| 50%M Std | 83 | 70 | 84 | 84 | |||||
| 10%M Std | 39 | 26 | 44 | 40 | |||||
| 0%M | 3 | 2 | 3 | 2 | |||||
| CAL-148 | 100%M (yes) | 89 | 92 | 96 | 96 | ||||
| SK-BR-3 | 50%M (yes) | 78 | 81 | 100 | 79 | ||||
| Hs578T | 10–50%M (yes) | 72 | 64 | 71 | 71 | ||||
| MCF 10A | moderate (yes) | 55 | 37 | 52 | 41 | ||||
| HCC1806 | 0%M (yes) | 0 | 0 | 0 | 0 | ||||
| 90–100 | 80–89 | 50–79 | 20–49 | 10–19 | 0–9 |
| Breast Cancer Cell Lines | EMP-Unassigned Group | Mesenchymal Group | Epithelial Group | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AKR1B1 | APC | RASSF1A | RARβ | SFRP2 | GFRA1 | CDKN2A | TWIST1 | CRABP1 | DKK3 | EGFR | VIM | GRHL2 | MIR200C | CDH1 | |
| MFM-223 | very high | 100% | 100% | 100% | 10–50% | 100% | moderate | high | 100% | 100% | low | very low | 0 | 0 | 0 |
| MDA-MB-453 | very high | 100% | moderate | 100% | 50–100% | high | 0 | high | moderate | 100% | 10–50% | 100% | 0 | 0 | 0 |
| BT-474 | 100% | 10–50% | 100% | 100% | 50–100% | 0 | 0 | very low | high | moderate | very low | 0 | 0 | 0 | 0 |
| ZR-75-1 | moderate | 100% | 100% | high | moderate | very low | low | moderate | low | low | very low | moderate | 0 | 0 | 0 |
| HCC1419 | very high | 0 | 100% | very low | high | 0 | 0 | very low | high | moderate | very low | moderate | 0 | 0 | 0 |
| UACC-893 | very high | 50% | 100% | high | high | moderate | 0 | high | low | moderate | 0 | low | 0 | 0 | 0 |
| SK-BR-3 | moderate | low | 50% | high | moderate | 0 | 0 | very low | 0 | 0 | very low | moderate | 0 | 0 | n.a. |
| T-47D | moderate | 0 | 100% | very low | moderate | 0 | 100% | high | moderate | moderate | moderate | 100% | 0 | 0 | 0 |
| CAL-148 | very high | 100% | 100% | high | moderate | 0 | 0 | 100% | high | low | very low | moderate | 0 | 0 | 0 |
| MCF-7 | very high | 10–50% | 100% | 100% | high | 0 | n.a. | high | moderate | moderate | low | 100% | 0 | 0 | 0 |
| UACC-732 | high | 50% | 100% | high | moderate | 0 | 0 | high | low | moderate | very high | 100% | 0 | 0 | 0 |
| MDA-MB-468 | 0 | 3% | 100% | 50–100% | moderate | moderate | 0 | high | low | moderate | 0 | 0 | 0 | 0 | 0 |
| HCC1954 | 100% | 100% | 100% | high | high | low | 100% | moderate | moderate | high | very low | 0 | 0 | 0 | 0 |
| HCC70 | 0 | 0 | 0 | 50% | very low | low | 0 | moderate | 0 | moderate | very low | 0 | 0 | 0 | 0 |
| BT-20 | 0 | 100% | 50% | 100% | 100% | 100% | n.a. | 100% | high | 100% | 0 | 100% | 0 | 0 | 0 |
| HCC1806 | 0 | 0 | 0 | very low | 0 | 0 | n.a. | 0 | very low | 0 | very low | very low | 0 | 0 | 0 |
| HCC1569 | 10–50% | 50–100% | 10–50% | very low | 100% | 100% | low | 100% | 0 | moderate | very low | 0 | 0 | 0 | 0 |
| SUM-149PT | 0 | 0 | 0 | moderate | low | 0 | n.a. | very low | very low | very low | very low | 0 | 0 | 0 | 0 |
| CAL-120 | 0 | 0 | 0 | 50% | moderate | 0 | 0 | very low | low | 0 | very low | 0 | very low | very low | very low |
| MCF 10A | 0 | 0 | moderate | high | low | low | n.a. | very low | very low | low | 0 | 0 | high | 0 | 0 |
| PMC42-LA | 0 | 0 | moderate | 100% | low | 0 | n.a. | low | very low | 0 | 0 | 0 | 10% | low | 0 |
| PMC42-ET | 0 | 0 | moderate | 50–100% | moderate | 0 | n.a. | moderate | low | 0 | 0 | 0 | 50–100% | high | very low |
| MDA-MB-231 | 0 | 0 | 50% | very low | high | moderate | n.a. | 100% | low | high | very low | 10% | moderate | moderate | moderate |
| BT-549 | 0 | 0 | very low | very low | 0 | 0 | 0 | low | low | very low | very low | 0 | high | moderate | very low |
| SUM-159PT | 0 | 0 | 50% | moderate | high | low | very low | high | 0 | 0 | very low | 0 | high | high | moderate |
| Hs578T | 0 | 0 | 10–50% | 0 | very low | n.a. | n.a. | high | 0 | 0 | 0 | 0 | moderate | high | moderate |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, A.V.-P.; Szaumkessel, M.; Tan, T.Z.; Thiery, J.-P.; Thompson, E.W.; Dobrovic, A. DNA Methylation Profiling of Breast Cancer Cell Lines along the Epithelial Mesenchymal Spectrum—Implications for the Choice of Circulating Tumour DNA Methylation Markers. Int. J. Mol. Sci. 2018, 19, 2553. https://doi.org/10.3390/ijms19092553
Le AV-P, Szaumkessel M, Tan TZ, Thiery J-P, Thompson EW, Dobrovic A. DNA Methylation Profiling of Breast Cancer Cell Lines along the Epithelial Mesenchymal Spectrum—Implications for the Choice of Circulating Tumour DNA Methylation Markers. International Journal of Molecular Sciences. 2018; 19(9):2553. https://doi.org/10.3390/ijms19092553
Chicago/Turabian StyleLe, Anh Viet-Phuong, Marcin Szaumkessel, Tuan Zea Tan, Jean-Paul Thiery, Erik W. Thompson, and Alexander Dobrovic. 2018. "DNA Methylation Profiling of Breast Cancer Cell Lines along the Epithelial Mesenchymal Spectrum—Implications for the Choice of Circulating Tumour DNA Methylation Markers" International Journal of Molecular Sciences 19, no. 9: 2553. https://doi.org/10.3390/ijms19092553