Interleukin-18 in Health and Disease


1
Department of Immunology, Hyogo College of Medicine, 1-1 Mukogawa-cho, Nishinomiya, Hyogo 663-8501, Japan
2
Department of Surgery, Hyogo College of Medicine, 1-1 Mukogawa-cho, Nishinomiya, Hyogo 663-8501, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(3), 649; https://doi.org/10.3390/ijms20030649
Submission received: 21 December 2018
/
Revised: 28 January 2019
/
Accepted: 29 January 2019
/
Published: 2 February 2019
(This article belongs to the Special Issue The Interleukins in Health and Disease)
Abstract
:Interleukin (IL)-18 was originally discovered as a factor that enhanced IFN-γ production from anti-CD3-stimulated Th1 cells, especially in the presence of IL-12. Upon stimulation with Ag plus IL-12, naïve T cells develop into IL-18 receptor (IL-18R) expressing Th1 cells, which increase IFN-γ production in response to IL-18 stimulation. Therefore, IL-12 is a commitment factor that induces the development of Th1 cells. In contrast, IL-18 is a proinflammatory cytokine that facilitates type 1 responses. However, IL-18 without IL-12 but with IL-2, stimulates NK cells, CD4+ NKT cells, and established Th1 cells, to produce IL-3, IL-9, and IL-13. Furthermore, together with IL-3, IL-18 stimulates mast cells and basophils to produce IL-4, IL-13, and chemical mediators such as histamine. Therefore, IL-18 is a cytokine that stimulates various cell types and has pleiotropic functions. IL-18 is a member of the IL-1 family of cytokines. IL-18 demonstrates a unique function by binding to a specific receptor expressed on various types of cells. In this review article, we will focus on the unique features of IL-18 in health and disease in experimental animals and humans.
1. Introduction
Th1 cells produce interferon (IFN)-γ upon stimulation with antigen (Ag) plus antigen presenting cells or anti-CD3 antibody in vitro and in vivo. Lipopolysaccharide (LPS)-stimulation of anti-CD3-stimulated Th1 cells does not induce the production of IFN-γ in vitro. However, the injection of LPS into Propionibacterium acnes-primed mice or Bacillus Calmette–Guerin (BCG)-infected mice, but not naïve mice, strongly induced IFN-γ production in vivo [1,2]. Furthermore, to our surprise, the addition of sera derived from P. acnes-primed and LPS-challenged mice strongly enhanced IFN-γ production by anti-CD3-stimulated Th1 cells in vitro, suggesting the presence of IFN-γ inducing factor(s) in the sera.
Because IL-12 is produced by LPS-stimulated macrophages and dendritic cells (DC), IL-12 from LPS-stimulated macrophages or DC in P. acnes-primed mice were initially thought to induce anti-CD3-stimulated Th1 cells to produce IFN-γ in vivo and in vitro. Indeed, the sera contained high levels of IL-12. However, only the addition of sera from P. acnes-primed and LPS-challenged mice, but not the addition of excess doses of IL-12, enhanced the production of IFN-γ from anti-CD3-stimulated Th1 cells, strongly suggesting the presence of IFN-γ inducing factors in the sera from P. acnes-primed and LPS-challenged mice.
Physicochemical studies and amino acid sequence analysis revealed that IFN-γ inducing factor (IGIF) is different from IL-12. The molecular cloning of IGIF was performed by Okamura in collaboration with Hayashibara Biochemical Laboratories. Soon after human IGIF was cloned [3], we and others found various functions of IGIF, including the induction of IL-2 production, IL-2 receptor (IL-2R) and Fas ligand (FasL) expression on Th1 cells, and the activation of natural killer (NK) cells. Based on these pleiotropic functions of IGIF, we named IGIF “IL-18” [2]. Although both IL-12 and IL-18 are major factors in IFN-γ production by Th1 cells, IL-12 is a differentiation factor that induces the development of Th1 cells—in contrast, IL-18 is a proinflammatory cytokine that facilitates IFN-γ production by Th1 cells particularly in conjunction with IL-12. Indeed, IL-12 and IL-18 from LPS-stimulated macrophages synergistically induced IFN-γ production from Th1 cells in P. acnes-primed and LPS-challenged mice.
Murine and human IL-18 proteins consist of 192 and 193 amino acids, respectively [1,3]. Based on the homology of its amino acid sequence compared with IL-1β, IL-18 is classified as a member of the IL-1 cytokine family. Human IL-18 and IL-1β share only 15% sequence homology although they contain a common β-pleated sheet structure. Furthermore, similar to IL-1β, IL-18 is produced as a biologically inactive precursor, pro-IL-18, which lacks a signal peptide and requires proteolytic processing to become active. The cleavage of pro-IL-18 or pro-IL-1β depends mainly on the action of the intracellular cysteine protease caspase-1 in the NLRP3 inflammasome [4,5].
The IL-18 receptor (IL-18R) consists of the inducible component IL-18Rα (IL-1 receptor-related protein [IL-1Rrp]) and the constitutively expressed component IL-18Rβ (IL-1R accessory protein-like [IL-1RAcPL]) [2]. Cytoplasmic domains of IL-18Rα and IL-18Rβ contain a common domain termed the Toll-like receptor (TLR)/IL-1R (TIR) domain, shared by other IL-1R family members and TLRs. Upon stimulation with IL-18, IL-18Rα forms a high-affinity heterodimeric complex with IL-18Rβ—which mediates intracellular signal transduction. Cytoplasmic TIR domains of the receptor complex interact with myeloid differentiation primary response 88 (MyD88), a signal adaptor containing a TIR domain [6], via TIR-TIR interactions. Then, MyD88-induced events result in the activation of nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK) via association with the signal adaptors IL-1R-associated kinase (IRAK) 1-4 and tumor necrosis factor (TNF) receptor-activated factor (TRAF) 6, respectively, which eventually leads to the appropriate gene expressions, such as Ifng, Tnfa, Cd40l, and FasL.
Although IL-18 was originally discovered as a factor that induces IFN-γ production from Th1 cells, it also acts on non-polarized T cells, NK cells, NKT cells, B cells, DC and macrophages to produce IFN-γ in the presence of IL-12. Moreover, IL-18 without IL-12 but with IL-2 induces Th2 cytokine production from CD4+ NKT cells, NK cells, and even established Th1 cells. Furthermore, IL-18 with IL-3 induces mast cells and basophils to produce IL-4 and IL-13. Therefore, IL-18 stimulates both innate immunity and acquired immunity [2,7].
The source of IL-18 was initially demonstrated to be from Kupffer cells, which constitutively express pro-IL-18. In addition, LPS binding to TLR4 induces the production of IL-18 via the activation of caspase-1. In contrast, upon stimulation with LPS, DC or macrophages increase their transcription of pro-IL-18 mRNA and subsequently their production of pro-IL-18, which is then processed by caspase-1 to be secreted as mature IL-18. In addition to these IL-18 producing cells, pro-IL-18 is produced by a wide variety of other cells, including keratinocytes, intestinal epithelial cells, and osteoblasts suggesting it has an important pathophysiological role in health and disease. Like other cytokines, IL-18 shows its pleiotropic action depending on its cytokine milieu (Figure 1).
2. Production of IL-18
Many cell types, both hematopoietic cells and non-hematopoietic cells, have the potential to produce IL-18. Originally, IL-18 production was recognized in Kupffer cells, liver-resident macrophages, even in the resting state without stimulation. However, subsequently, many investigators reported IL-18 production in non-hematopoietic cells, such as intestinal epithelial cells, keratinocytes, and endothelial cells, even in the steady state. In addition to its unique distribution and constitutive production in a wide variety of cell types and tissues, IL-18 is characterized by its unique process of cellular production. Usually, cytokines such as IFN-γ and IL-4 are secreted after the corresponding genes are expressed because their genes encompass a signal peptide that is necessary for their extracellular release through the endoplasmic reticulum to the Golgi. In contrast, the IL18 gene, similar to other IL-1 family members, lacks a signal peptide. It was reported that IL-18 is stored in the cytosol of IL-18 producing cells [1,2,8]. Furthermore, similar to IL-1β but unlike IL-1α or IL-33, IL-18 is produced as a biologically inactive precursor [1,2,8]. To become active and be released, precursor IL-18 (pro-IL-18) needs post-translational processing [2,4,9]. Therefore, the extracellular release of biologically active IL-18 is regulated by multiple processes, such as regular transcriptional gene regulation, post-transcriptional gene regulation, and post-translational regulation.
2.1. IL18 Gene Expression
The IL18 gene is located on chromosome 11 in humans and chromosome 9 in mice [2].
2.1.1. Transcriptional Gene Regulation
2.1.1.1. IL18 Gene Promoter
The IL18 gene contains 7 exons, where exons 1 and 2 are noncoding. An early study reported that promoter activity was detected upstream of exons 1 and 2 of the murine Il18 gene [10]. Furthermore, the promoter upstream of exon 1 (5′-flanking region) contains an interferon consensus sequence binding protein (ICSBP)-binding site and activator protein-1 (AP-1)-binding site [11], while another promoter upstream of exon 2 (intron 1) encompasses a PU.1-binding site [11]. Similar to the genomic sequence of murine Il18, human IL18 gene fragments were reported to contain a PU.1-binding site upstream of exon 2 and to have promoter activity [12].
A study on the detailed structure and sequence variations of the human IL18 promoter revealed five single nucleotide polymorphisms (SNPs) at the 5′-end of the IL18 gene: −656 G/T (rs1946519), −607 C/A (rs1946518), −137 G/C (rs187238), +113 T/G (rs360718), and +127 C/T (rs360717) [13]. The transcription activity of the IL18 gene promoter fragment demonstrated that −656 G/T (rs1946519), −607 C/A (rs1946518), and −137 G/C (rs187238) are in the promoter region and that the other two SNPs are in the 5′-untranslated region (Table 1). A change from C to A at position −607 disrupted a cAMP-responsive element binding protein (CREB) binding site [13]. A change from C to G at position −137 altered the histone H4 gene-specific transcription factor-1 (H4TF-1) nuclear factor binding site [13] (Table 1). A new putative IL18 gene variant was identified in systemic lupus erythematosus (SLE) patients [14]. These promoter variants were reported to reflect the protein levels of IL-18 produced by peripheral blood mononuclear cells (PBMCs) isolated from healthy individuals [15].
Intriguingly, many clinical study groups have extensively studied the association between these SNPs of IL18 gene promoters and various diseases. Table 1 shows a summary of representative meta-analyses and/or systematic reviews of individual diseases. Therefore, IL18 promoter variants are associated with diverse diseases such as chronic viral infection, chronic diseases, and cancer. Therefore, these promoter variants might influence pro-IL-18 production although they might not influence the release of biologically active IL-18. Therefore, how IL18 promoter variants are associated with the risk of individual diseases remains to be elucidated. Cytoplasmic IL-18 might exert unknown actions on cellular properties that might influence disease risk.
2.1.1.2. IL18 Gene Repressor
B cell lymphoma 6 protein (Bcl6) was demonstrated to repress the IL18 gene. Bcl6 was originally identified as a human proto-oncogene [16] and was recently demonstrated to be a master regulator of follicular helper CD4+ T cells [17]. A putative Bcl6-binding DNA located in the 5′-noncoding region at a site −2686 from exon 1 is a prerequisite for the Bcl6 repression of the expression of luciferase under control of the IL18 promoter. In response to LPS, bone marrow-derived macrophages from Bcl6−/− mice expressed higher levels of Il18 than those from control mice [18].
2.1.2. Post-Transcriptional Gene Regulation (miRNA)
MicroRNAs (miRNAs) are endogenous ~21 nucleotide-long noncoding RNAs that form a large family of post-transcriptional regulators of gene expression in metazoans and plants [19,20]. Humans have approximately 800 miRNAs, which participate in most cellular processes. However, changes in miRNA expression are involved in the pathogenesis of human disease. miRNAs interact with their mRNA targets by base pairing only using short sequences from these RNAs and mediate post-transcriptional gene regulation by translational repression or mRNA degradation. Multiple miRNAs in combination regulate their common target mRNA, whereas individual miRNAs regulate multiple different mRNAs. Therefore, individual miRNAs coordinate the expression of cellular proteins. The detailed mechanisms of post-translational regulation by miRNAs were extensively reviewed in recent articles [21,22,23].
Multiple miRNAs regulate IL18 gene expression, directly or indirectly, and might be associated with disease and/or disease severity [24] as discussed in the following examples.
Bruton’s tyrosine kinase (Btk) is a cytoplasmic non-receptor tyrosine kinase, and its loss-of-function mutation was verified to be responsible for a humoral immunodeficiency named X-linked agammaglobulinemia with impaired B cell development [25]. Recently, Btk was shown to be involved in the stabilization of various cytokine mRNAs including Tnfa mRNA [25]. Upon stimulation with LPS, BTK was similarly induced in human macrophages and human fibroblast-like synovial cells. However, IL18 was induced in the macrophages but not in the synovial cells. This was explained by the induction of miRNA-346 (miR-346) in synovial cells. Indeed, transfection with miR-346 antisense restored IL18 induction in synovial cells, which was Btk dependent. Therefore, miR-346 negatively regulates IL18 levels by reducing the induction of Btk expression, at least in synovial cells [26].
miRNA-197 (mi-R197) was shown to regulate IL18 mRNA expression. Base-pair sequences were identified in miR-197 and IL18 in THP-1 cells, a human macrophage cell line. THP-1 cells transfected with miR-197 showed lower IL18 expression and secreted lower levels of IL-18 than control cells. Intriguingly, there was a negative correlation between disease stage of hepatitis B virus (HBV)-infected hepatitis (asymptomatic HBV carrier, chronic hepatitis, and acute on chronic liver failure) and miR-197 expression levels in peripheral blood mononuclear cells (PBMCs), and a positive correlation between disease severity and IL18 expression levels in PBMCs. Therefore, miR-197 might be important for the reactivation of hepatitis by targeting IL18 [27].
2.2. Post-Translational Regulation of IL-18 (Processing of pro-IL-18)
Pro-IL-18 requires cleavage by appropriate enzymes to become active. Caspase-1, originally designated as IL-1β-converting enzyme and now a member of the cysteine protease (caspase) family, is a major IL-18-processing enzyme. Processing of IL-18 with caspase-1 occurs in cytoplasmic inflammasomes. Caspase-8, a pro-apoptotic caspase, was recently shown to also be involved in the activation of IL-18. Various other proteases produced by killer lymphocytes, neutrophils, and mast cells have the capacity to appropriately cleave pro-IL-18 into biologically active IL-18.
2.2.1. Caspases
Caspases (cysteine-aspartic proteases) are proteases responsible for important biological events including inflammation as well as programmed cell death such as apoptosis, necroptosis, and pyroptosis [28,29,30]. Caspases are produced as biologically inactive precursors (pro-), and require cleavage by other caspase members or itself to become an active caspase [29,30]. Caspase-1 is an essential enzyme for the conversion of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, respectively [4,9].
2.2.1.1. Caspase-1 (Inflammasome NLRP3, NLRC4, and AIM2)
Caspases are produced as biologically inactive precursors (pro-), and require cleavage by other caspase members or themselves to become active caspases [29]. The inflammasome is a large protein-complex generated in the cytoplasm after the activation of cells by appropriate stimuli [31]. Caspase-1 is activated within inflammasomes formed in the cytosol [31]. After stimulation with specific inflammasome activators, the corresponding monomeric pattern-recognition receptors (PRRs) gather together to assemble pro-caspase-1 with help from an adaptor protein, ASC (apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain; PYCARD), which forms a wheel-shaped inflammasome [32,33]. Pro-caspase-1 in the inflammasome undergoes autolysis to become active caspase-1, which converts pro-IL-18 into mature IL-18. Then, active caspase-1 cleaves gasdermin D to liberate a pore-forming domain, N-terminal, leading to the liberation of mature IL-1β and IL-18 and eventually pyroptosis [34,35,36]. Many PRRs participate in the inflammasome. In this review article, we briefly introduce NAIP-NLRC4, AIM2, and NLRP3 inflammasomes. Excellent reviews provide detailed features of these inflammasomes [37,38,39,40,41].
The nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 (NLRP3) inflammasome was originally demonstrated to be a cytoplasmic platform necessary for caspase-1 activation [42,43,44]. To activate the NLRP3 inflammasome in bone marrow-derived macrophages, two sequential signals are needed—the first signal, “priming”, is usually TLR-mediated, while the second signal, “activation”, is triggered by a wide variety of cellular responses. NLRP3 activators include highly diverse molecules such as inducers of K+ efflux, initiators of Ca2+ mobilization, microbial pore-forming toxins, endogenous or exogenous particulates, microcrystals, and endogenous metabolites [45,46,47]. However, how these biologically different signals activate the NLRP3 inflammasome is unclear. Recently, a common NLRP3 activation pathway was identified [48]. Zhong et al. demonstrated that macrophages began to synthesize mitochondrial DNA in response to TLR-mediated “priming”. Upon stimulation by subsequent NLRP3 activators including inducers of K+ efflux, Ca2+ influx, and particulates, these macrophages underwent mitochondrial insult and produced reactive oxygen species (ROS) in their mitochondria. During the activation process, newly synthesized mitochondrial DNA might be oxidized by ROS, and small fragmented oxidized mitochondrial DNA might translocate from the mitochondria to the cytosol through injured mitochondrial membranes. Indeed, transfection with oxidized mitochondrial DNA activated the NLRP3 inflammasome (Figure 2A).
The nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family of apoptosis inhibitory proteins (NAIPs), including human NAIP, and murine Naip1, Naip2, Naip5, and Naip6, are localized in the cytoplasm and sense bacterial components. Gram-negative bacteria exert pathological actions by injecting toxic molecules such as flagellin, a protein composed of flagella, via a syringe-like shaped type III secretion system consisting of a rod and needle [49]. Upon sensing these rods, needles, and flagellin, cytoplasmic NAIPs associate with NLR family CARD domain-containing protein 4 (NLRC4), initially designated as Ice protease-activating factor (IPAF), to generate the NAIP-NLRC4 inflammasome, which eventually leads to caspase-1-mediated IL-18 secretion (Figure 2B) [50,51,52]. Therefore, upon infection with Gram-negative bacteria such as Salmonella typhimurium, the resulting production of IL-18 might participate in host defense against certain bacterial infections [50,51].
Absence in melanoma 2 (AIM2) recognizes double-stranded (ds) DNA derived from microorganisms and host cells [53,54,55]. Infection with dsDNA viruses such as cytomegalovirus, induces free viral dsDNA in the cytosol of host cells. Certain intracellular bacterium, such as Francisella tularensis, exists in the phagosomes of macrophages and evade detection by entering the cytosol. These bacteria are regarded as healthy in the host cytosol. However, proteolytic enzymes activated in the cytosol damage the cytoplasmic bacterium cell wall, liberating free bacterial dsDNA, which activates the AIM2 inflammasome, leading to the release of biologically active IL-18. Host-derived dsDNA can activate caspase-1-mediated inflammation. Irradiation injured host cell DNA, and free host dsDNA activates the AIM2 inflammasome. In contrast to NLRC4, AIM2 directly senses dsDNA to activate the AIM2 inflammasome (Figure 2C).
In contrast to bone marrow-derived macrophages, Kupffer cells, resident macrophages in the liver, release IL-18 upon a single stimulation with LPS in the absence of priming [1]. To release mature IL-18, TLR4 [56], TRIF (TIR-domain-containing adapter-inducing interferon-β) [57], NLRP3 [57], ASC [58], and caspase-1 [4] are required [59]. We found that Kupffer cells produce ROS in response to LPS and that a ROS inhibitor completely inhibited LPS-induced-IL-18 release (our unpublished data). General irradiation to generate chimeric mice causes the deletion of hematopoietic cells including splenic macrophages but does not kill Kupffer cells [60,61]. Therefore, Kupffer cells might be different from bone marrow-derived macrophages in the context of their potential to generate ROS.
IL-18 maturation occurs by the activation of other types of inflammasomes, such as NLRP6 and NLRP9b inflammasomes, particularly in intestinal epithelial cells, which contribute to gut homeostasis and host defense, respectively (described below).
2.2.1.2. Caspase-8 (upon Fas Ligation)
Fas is an extracellularly expressed cell death receptor, and its ligation with FasL leads to apoptotic cell death, a programmed cell death without cell membrane insult. We and others found that upon Fas engagement, neutrophils and macrophages release biologically active mature IL-1β and IL-18, respectively that is caspase-1 independent [62,63]. Because pan-caspase inhibitors prevent Fas-mediated IL-1β and IL-18 release, we assumed that caspases other than caspase-1 might process these precursor proteins. Recently, we and others revealed that caspase-8, an apoptosis-initiating protease [29], was involved in converting pro-IL-18 into mature IL-18 after the stimulation of macrophages by FasL [64,65,66]. We found that the Fas-mediated pathway for IL-18 release was important for host defense against bacterial infection. Fas−/− mice were highly susceptible to Listeria monocytogenes, an intracellular Gram-positive bacterium that causes serious food-born infections in humans, with the impaired secretion of IL-18 and IL-1β [64]. Fas-mediated IL-18/IL-1β processing does not require NLRC4, NLRP3, or caspase-1, but does require ASC and caspase-8 [64,65]. Recently, we reported that IL-1β processed by Fas-mediated caspase-8 activation was profoundly involved in the development of Th17/Th1 cells, but not Th1 cells, during L. monocytogenes infection [67].
2.2.2. Other Proteases Involved in the Production of Biologically Active IL-18
Several proteases other than caspases can cleave pro-IL-18 to generate biologically active pro-IL-18 fragments.
Proteinase 3 is a 29 kDa serine protease [68], which is mainly produced by neutrophils and stored in their azurophilic granules [68]. Human oral epithelial cells that constitutively produce pro-IL-18 release biologically active IL-18 after stimulation with neutrophilic proteinase 3, even in the presence of caspase-1 or pan-caspase inhibitors [69]. The proteinase-3 cleavage site of human pro-IL-18 was later identified to be at I46, while the N-terminal amino acid residue of mature human IL-18 processed by caspase-1 is Y35 [70].
Mast cells predominantly accumulate in the dermis of atopic-dermatitis model mice, skin-specific caspase-1 transgenic mice [71], as well as human atopic dermatitis patients [72]. The incubation of human pro-IL-18 with human mast cell chymase produced biologically active IL-18, but its N-terminal amino acid residue was I56, which is different from mature IL-18 cleaved by caspase-1 or by proteinase 3 [70].
Granzyme B is a serine protease with aspartic protease activity, and is produced mainly by NK cells and cytotoxic T lymphocytes (CTLs). Upon the recognition of target cells, cytotoxic lymphocytes inject granzyme B through a target cell membrane pore generated by perforin polymerization, which results in the apoptotic death of the target cells [73,74]. Granzyme B cleaves human pro-IL-18 at the same site as caspase-1, and the granzyme B-cleaved pro-IL-18 fragment has the capacity to produce IFN-γ [75]. Human CD8+ T cells isolated from PBMCs express granzyme B and kill human keratinocytes that constitutively produce pro-IL-18, accompanied by the release of mature IL-18 [76].
2.3. Regulation of Circulating IL-18 by IL-18-Binding Protein (IL-18BP)
IL-18-binding protein (IL-18BP) binds to IL-18 with a higher affinity than IL-18R and inhibits IL-18 signaling [77,78,79]. This suggests that IL-18BP levels determine the free IL-18 concentration. Adult-onset Still’s disease is a multi-systemic inflammatory disease characterized by upregulated IL-18 levels and downregulated IL-18BP in the circulation, mediated by an unknown mechanism [80]. Recently, miRNA profiles in the plasma of adult-onset Still’s disease patients were investigated, and the plasma levels of miRNA-134 (miR-134) were positively correlated with disease activity scores and decreased after effective treatment [81]. IL18BP was identified as its target mRNA [81]. Therefore, elevated miR-134 in adult-onset Still’s disease might induce the upregulation of IL-18 by inhibiting IL18BP, and therefore, miR-134 might be a potential biomarker for this disease.
3. IL-18 Signaling
3.1. IL-18 Receptor
IL-18 mediates its effects by signaling through its receptor, belonging to the IL-1R family, composed of an IL-18Rα chain (IL-18R1, IL-1Rrp) and IL-18Rβ chain (IL-18R accessory protein, IL-1RAcPL) [2]. Following the binding of IL-18 to IL-18Rα, IL-18Rβ then binds to form a trimer. The intracellular region contains a TIR domain in common with TLR, and MyD88 binds to TIR to transmit a signal into the cell. Although IL-18Rα alone can bind to IL-18, its affinity is low [82]. The IL-18Rβ chain is required for high-affinity binding and cell signaling [83]. When IL-18Rα binds to IL-18Rβ, it causes a conformational change, resulting in a high-affinity receptor [84,85].
IL-18R expression, induced by stimulation with IL-12 and IFN-α (human) in T cells and NK cells or by signal transduction and transcriptional regulation via STAT4 (Signal transducer and activator of transcription 4), is essential for potent IFN-γ production [86,87,88,89]. However, IL-18R is also expressed in basophils, mast cells, and CD4+ NKT cells in the steady state, all of which produce Th2 cytokines such as IL-4 and IL-13 in response to IL-18 stimulation [2,7]. IL-18R is also expressed by non-immune cells such as epithelial cells and nerve cells and is involved in cell survival and differentiation. The regulatory mechanism of IL-18R expression in these cells is poorly understood.
IL-18Rα is important for inflammatory cytokine production following IL-18 stimulation. However, although contradictory, the inflammatory response was exacerbated in IL-18Rα-deficient mice because IL-18Rα also binds to the inhibitory cytokine, IL-37 (IL-1F7). When IL-37 binds to IL-18Rα, it prevents IL-18Rα binding to IL-18Rβ, which blocks the transmission of activation signals into the cell. Instead, the IL-37/IL-18Rα complex binds to IL-1R8 (TIR8, SIGIRR), which promotes anti-inflammatory effects by activating STAT3 and transmitting an inhibitory signal [90,91,92,93].
3.2. IL-18 Signaling Cascade
Following the trimerization of IL-18/IL-18Rα/IL-18Rβ, MyD88 binds to the Toll-IL-1 receptor (TIR) domain of IL-18Rα and IL-18Rβ [6]. IRAK1 and IRAK4 are combined via the death domain of MyD88 to form a Myddosome [94,95,96]. Furthermore, following binding to TRAF6, inhibitor of κB (IκB) is degraded, and phosphorylated p65/p50 NF-κB translocates into the nucleus [97]. The MAPK cascade of Extracellular Signal-regulated Kinase (ERK), c-jun N-terminal kinase (JNK), and p38 is also activated (Figure 3) [98]. These signals induce IFN-γ production in Th1 cells and promote cell proliferation. IL-18 stimulation also induces the phosphorylation and activation of phosphatidylinositol-3 kinase (PI3K)/Akt/S6 and mammalian target of rapamycin (mTOR), as well as autophagosome formation and the expressions of Bcl-xL and Bcl2 [99,100]. Although PI3K suppresses inflammatory cytokine production by TLR signaling in myeloid cells [101], this signal enhances the proliferation and survival of NK cells. The PI3K/Akt pathway is also important for the survival of non-immune system cells, such as keratinocytes and neurons following IL-18 stimulation [102,103].
IL-18R signaling is similar to that of IL-1R/TLR. On the basis of IL-1R signaling, the signaling pathway after TRAF6 is thought to be transmitted as follows. K63-polyubiquitin chain (K63-pUb) is formed by the E3 ubiquitin ligase activity of TRAF6 to recruit TAB2/TAB3 [104] and NF-κB essential modulator (NEMO) [105]. K63-pUb also activates TAK1 and the phosphorylation of TAB1, IκB kinase (IKK) α, IKKβ, and MAPK kinase (MKK) 3/6 by TAK1 promotes the subsequent activation and nuclear translocation of MAPKs and NF-κB, inducing the expressions of various genes [105,106]. Regarding the activation of PI3K, the direct binding of PI3K and MyD88, or the B-cell adapter for PI3K (BCAP) might be involved [100].
Regarding differences in signaling between IL-1 and IL-18, TRIF-related adaptor molecule (TRAM, TICAM2) is thought to be involved in IL-18R signaling. In TLR4 signaling, MyD88 and TRIF function downstream. Then, TIRAP (Mal) connects TLR4 and MyD88, and TRAM links TLR4 and TRIF. However, TIRAP does not participate in IL-18 signaling [107]. Instead, TRAM directly binds to IL-18Rα, IL-18Rβ and MyD88 and transduces IL-18 signals into the cell [108].
3.3. IL-18 Binding Protein
IL-18 binding protein (IL-18BP) is an endogenous soluble factor that specifically inhibits the action of IL-18. IL-1 receptor family proteins are generally characterized by an extracellular portion consisting of three immunoglobulin (Ig)-like domains; however, IL-18BP contains a single Ig domain and is similar to TIR8 (SIGIRR, IL-1R8). In addition to mammals, various viruses possess highly homologous genes. IL-18BP contains a signal peptide and its protein is secreted without a transmembrane domain [77]. When the extracellular region of IL-33 receptor (ST2) is secreted, as soluble ST2, competes for the binding of IL-33 and ST2. However, IL-18BP does not correspond to the extracellular ligand binding domain of the IL-18 receptor, which is encoded by another gene, and therefore it is different from classical soluble receptors.
The affinity of IL-18BP for IL-18 is about 400 pM, similar to that for IL-18Rα/IL-18Rβ, and much higher than for IL-18Rα alone (10–50 nM). IL-18BP inhibits the binding of IL-18 to IL-18R and neutralizes IL-18 activity, thereby suppressing IFN-γ production and limiting Th1 cell responses [77,109]. For example, the administration of IL-18BP substantially reduced the pathology in mouse models of experimental arthritis, colitis, endotoxin shock, and type 1 diabetes, disease models in which IL-18 is important for the pathology [110,111,112,113,114]. Furthermore, transgenic mice overexpressing IL-18BP were protected from acute renal injury induced by ischemia-reperfusion [115]. In a colitis model, IL-18 acted directly on intestinal epithelial cells to enhance inflammation, thereby exacerbating colitis via excessive IL-18 stimulation in IL-18BP-deficient mice. In this case, the inhibition of goblet cell maturation was observed in the intestinal tract [116].
In healthy humans, IL-18BP is present in the serum at a 20-fold molar excess compared with IL-18 [114], suggesting IL-18BP provides a threshold at which IL-18 does not mediate its effects until the concentration of IL-18 reaches a point where it does not induce systemic excessive Th1-type immune responses against general infection with low pathogenicity. However, because IL-18BP-deficient mice were reported to have markedly decreased levels of IL-18 in their blood, IL-18BP might function as a carrier to maintain a constant blood concentration of IL-18 [117].
In autoimmune inflammatory diseases where IFN-γ is involved in the pathology, the concentration of free IL-18 was more important for determining the severity of disease compared with IL-18 bound to IL-18BP [114]. In Wegener’s granulomatosis and systemic lupus erythematosus, the serum levels of IL-18BP and IL-18 were high [114,118], but the level of IL-18BP was insufficient to neutralize IL-18 and the level of free IL-18 was higher than that of healthy individuals. Macrophage activation syndrome is another disease where the clinical and hematologic abnormalities correlate with elevated free IL-18 levels [119]. These studies have shown that IL-18BP therapy may be of clinical value in situations where excessive IL-18 stimulation appears to cause disease or to enhance its severity. Indeed, clinical trials to investigate treatment with IL-18BP for adult-onset Still’s Disease and NLRC4-associated macrophage activation syndrome, inflammatory diseases associated with high plasma IL-18 levels, are ongoing [120,121,122] (ClinicalTrials.gov Identifier: NCT 02398435, NCT 03113760).
IL-37 is another endogenous factor that suppresses the action of IL-18. IL-37 has high homology with IL-18, and IL-18 BP also binds to IL-37 [90]. Binding to IL-37 enhances the ability of IL-18BP to inhibit IFN-γ induction stimulated by IL-18. Of note, IL-37 has not been found in mice and human IL-37 binds to IL-18R with very low affinity. However, mice expressing human IL-37 showed marked anti-inflammatory effects especially related to LPS-induced cytokine production and dendritic cell maturation [93]. Furthermore, human IL-37 expressing mice were also resistant to dextran sulphate sodium (DSS)-induced colitis [123].
The amount of IL-18BP is highly regulated at the level of gene expression. Because IFN-γ increases the gene expression and protein synthesis of IL-18BP [124,125], IFN-γ invokes a negative feedback loop for IL-18-mediated inflammation. This concept is supported by clinical data where patients treated with IFN-α for hepatitis show elevated levels of IL-18BP [126,127]. Moreover, in patients with familial hemophagia, the ability of IFN-γ to induce IL-18BP was decreased and inflammation might have been exacerbated by insufficient negative feedback [128]. IL-27 functions has both inflammatory and anti-inflammatory effects, but similar to IFN-γ, it utilizes STAT1 for signal transduction and increases the production of IL-18BP as a negative feedback loop against inflammation in skin keratinocytes, fibroblasts, ovarian epithelial cancer cells, and leukocytes [129,130].
4. Physiological Roles of IL-18
4.1. Cytokine and Immune Cell Milieu Determines the Biological Action of IL-18
4.1.1. IFN-γ Production
Naïve Th cells stimulated with Ag and IL-12 or IL-4 develop into IL-18R expressing Th1 or ST2 expressing Th2 cells, respectively. Therefore, the expressions of IL-18R and ST2 are convenient cell markers for Th1 cells and Th2 cells, respectively. IL-18, originally discovered as an IFN-γ-inducing factor, induces IFN-γ production from IL-18R expressing Th1 cells. IL-18 also induces IFN-γ production from NK cells and CD4+ NKT cells, which constitutively express IL-18R [2,7]. In general, IL-18-induced IFN-γ production by Th1 cells, NK cells and CD4+ NKT cells is strikingly enhanced by costimulation with IL-12. Moreover, IL-18 can synergize with IL-12 to induce IFN-γ production in DC, macrophages, and B cells [2,7]. It is well known that B cells develop into IgG1 and IgE-producing cells after stimulation with anti-CD40 antibodies and IL-4. We found that when B cells were stimulated with anti-CD40 antibodies and IL-4 in the presence of IL-12 and IL-18, these activated B cells produced IFN-γ that inhibited IL-4 dependent IgG1 and IgE production but enhanced IgG2a production [131]. T cells increase their expression of IL-18R in response to IL-12—similarly, IL-12-stimulated B cells also showed increased IL-18R expression and the production of IFN-γ in response to IL-18 and IL-12 [132].
How do IL-12 and IL-18 synergistically induce IFN-γ production? One major mechanism is the reciprocal induction of the other’s corresponding receptor expression [2]. Another mechanism is the synergistic induction of IFN-γ at the molecular level. The Ifng promoter contains a consensus sequence for NF-κB, AP-1 and the STAT4 binding site. IL-18 activates the IRAK/TRAF6 pathway resulting in the activation of NF-κB and AP-1, while IL-12 activates STAT4. In combination, these activate the Ifng promoter, resulting in the synergistic induction of IFN-γ production at the transcription level.
4.1.2. Innate-Type Basophil and Mast Cell Activation by IL-3 and IL-18
Following the cross-linkage of FcεR1 by the Ag/IgE complex, mast cells and basophils produce Th2 cytokines, including IL-4 and IL-13. We investigated whether mast cells or basophils also had the potential to produce IFN-γ when stimulated with a combination of IL-12 and IL-18. Unexpectedly, basophils and mast cells derived from bone marrow cell cultures supplemented with IL-3 for 10 days expressed the IL-18Rα chain [133]. Furthermore, basophils produced large amounts of IL-4 and IL-13 concurrently in response to stimulation with IL-3 and IL-18. In contrast, mast cells mainly produced IL-13 in response to IL-3 and IL-18. The cross-linkage of FcεR1 with the Ag/IgE complex modestly increased IL-4 and IL-13 production by basophils and mast cells stimulated with IL-3 and IL-18. Furthermore, mast cells and basophils did not produce IFN-γ in response to any combination of IL-3, IL-18 and IL-12. Because IL-18 in combination with IL-3 stimulated basophils and mast cells to produce chemical mediators and Th2 cytokines, we speculated that IL-18 might induce allergic inflammation without help from the Ag/IgE complex. Therefore, we described the IL-3 plus IL-18-dependent activation of basophils and mast cells as “innate-type basophil and mast cell activation”.
4.1.3. Innate-Type Allergic Inflammation by IL-18
We detected high levels of IgG1 and IgE in the sera of wild type mice and IFN-γ-deficient mice infected with Strongyloides venezuelensis at days 10 and 14 after infection. When a mixture of IL-12 and IL-18 was injected daily into these infected mice, IgG1 and IgE responses were only inhibited in wild type mice infected with S. venezuelensis. IL-12 and IL-18 stimulation induced T cells and B cells to produce IFN-γ, resulting in the inhibition of IL-4-dependent IgE production. To our surprise, the injection of IL-12 and IL-18 increased the serum levels of IgG1 and IgE in IFN-γ-deficient mice infected with S. venezuelensis. Most surprisingly, the daily administration of IL-18 or a combination of IL-2 and IL-18 induced a striking increase in the serum levels of IgE that was dependent on CD4+ T cells and IL-4/IL-4R/STAT6 [134]. Consistent with this result, transgenic mice overexpressing human caspase-1 in keratinocytes (KCasp1-Tg) produced IL-18 and IgE in the serum. Furthermore, they spontaneously developed atopic dermatitis (AD)-like skin lesions [71]. Therefore, we disrupted the gene encoding STAT6, which is required for the signal transduction of IL-4, or for IL-18 in KCasp1-Tg. Disruption of STAT6 resulted in no IgE production and did not affect the skin manifestations of KCasp1-Tg. In contrast, IL-18-deficient KCasp1-Tg mice had markedly diminished skin lesions. Therefore, the overproduction of IL-18 from keratinocytes induced AD-like skin lesions in the absence of IgE [71]. On the basis of these results, we described IL-18-induced allergic inflammation as “innate-type allergic inflammation” [71,135].
In collaboration with IL-2, but in the absence of IL-12, IL-18 stimulated NK cells, CD4+ NKT cells and splenic Th cells to produce IL-3, IL-9 and IL-13. Because IL-3 and IL-9 induce mucosal mastocytosis, we examined the capacity of mice injected with IL-2 and IL-18 to protect against infection with S. venezuelensis. C57BL/6 mice pretreated with IL-18 and IL-2 developed mucosal mastocytosis and had high levels of serum mMCP1 (mouse mast cell protease 1), an activation marker of mucosal mast cells. Furthermore, they could expel the intestinal nematode S. venezuelensis. Therefore, IL-18 is important for the expulsion of intestinal nematodes [136].
4.1.4. Th1 Cells Produce IFN-γ and IL-13 in Response to IL-18
Although IL-18 and IL-12 increased IFN-γ production from Ag-stimulated Th1 cells, the injection of a mixture of IL-12 and IL-18 into IFN-γ-deficient mice infected with S. venezuelensis increased the serum levels of IgG1 and IgE. These results suggested that IL-18 and IL-2 might stimulate even Th1 cells to produce Th2 cytokines. Therefore, we examined the capacity of IL-18 and Ovalbumine (OVA) peptide to stimulate OVA-specific Th1 cells to produce Th2 cytokines. We found that Th1 cells stimulated with OVA and IL-18 produced both Th1 cytokines (IFN-γ) and Th2 cytokines (IL-9, IL-13). Furthermore, additional IL-2 stimulation enhanced the production of Th2 cytokines, suggesting Th1 cells can alter their gene expression pattern in response to OVA, IL-2 and IL-18. We designated these Th2 cytokine-producing Th1 cells as “super Th1 cells”. Intriguingly, after several rounds of stimulation with Ag, IL-2 and IL-18, IL-18Rα-expressing and T-bet-expressing Ag-specific Th1 cells expressed GATA3, and started to produce both IFN-γ and IL-13 [137]. We verified that GATA3 was essential for the induction of IL-13 in Th1 cells after their stimulation with Ag, IL-2 and IL-18.
4.2. IL-18 in Host Defense
Microorganisms can be classified as intracellular or extracellular. Generally, intracellular microorganisms are killed by cellular immunity, whereas extracellular microorganisms are eliminated by humoral immunity. IL-18 plays an important role in host defense against various infectious microorganisms because it strongly enhances the induction of IFN-γ, nitric oxide (NO), and ROS in phagocytes. In addition, IL-18 directly activates CD8+ T cells, which play a central role in viral clearance. Furthermore, because IL-18 activates Th2 cytokine production and granulocytes in the absence of IL-12, it also acts defensively in helminth infection.
4.2.1. IL-18-Mediated Defense Against Extracellular Pathogens
4.2.1.1. Bacteria Infection
For defense against extracellular bacteria, it is important to activate macrophages by IFN-γ produced from NK cells and Th1 cells, opsonization by antibodies and activation of complement, and phagocytosis and ROS production by neutrophils and macrophages.
Patients undergoing severe surgical stress, i.e., trauma, burns, or major surgery, suffer a loss of immunity and physical barriers such as the skin and intestines, thereby increasing the risk of infection such as sepsis [138]. If the host is infected with bacteria where the host defense system is weakened, it cannot suppress bacterial proliferation, which might cause fatal multiorgan damage. In a mouse study, Kinoshita and colleagues showed that multiple doses of IL-18 restored a state of reduced immune function after injury suggesting the medical application of IL-18 for infections. Multiple injections of IL-18 in burned mice significantly increased IFN-γ production from mononuclear cells and improved bacterial clearance and mouse viability after Escherichia coli infection [139,140]. IL-18 also enhanced host defense against Pseudomonas aeruginosa infection by enhancing IgM natural antibody production from liver B1 cells [141]. Such antibodies can opsonize bacteria and promote their uptake by phagocytes before bacterial antigen-specific antibodies are produced. IL-18 may also be useful for preventing serious complications of pneumococcal respiratory infections in immunocompromised patients [138].
Burns activate neutrophils and cause tissue damage, whereas phagocytosis and pathogen killing are decreased. IL-18 restored the burn-related decrease in activity of neutrophils and enhanced phagocytosis and ROS production to prevent infection by methicillin-resistant Staphylococcus aureus [141,142,143].
Overall, these data suggest that IL-18 treatment might be an alternative and useful treatment for infection by extracellular pathogens, even in immunocompromised individuals.
4.2.1.2. Helminth Infection
Several types of intestinal parasitic nematodes are removed from the mammalian host Th2 responses [144]. S. venezuelensis is a rodent intestinal nematode that induces a Th2 type immune response in the host. It is removed mainly by the action of chondroitin sulphate and protease released from activated intestinal mucosal mast cells [145]. IL-18 induces mastocytosis by increasing the mast cell growth factor IL-3, thereby promoting the elimination of S. venezuelensis [136]. Trichuris muris is also expelled by mucosal mast cells activated by a Th2 response. However, in the case of T. muris, the immune response induced by infection varies by mouse strain. Mice that mount a Th1 type immune response following infection are susceptible, whereas mice that mount a Th2 type immune response are resistant. In C57BL/6 mice, IL-12 is produced by T. muris infection and IL-12 and IL-18 promote IFN-γ-producing Th1 cell differentiation. In IL-18-deficient mice that are resistant to chronic infection, Th2 cytokine-producing cells are increased in the lymph nodes in addition to Th1 cells. However, the administration of IL-18 inhibited Th2 cytokine production and prolonged infection [146,147]. A similar effect of IL-18 was also observed for Trichinella spiralis infection [148]. This indicates IL-18 inhibits the production of Th2 cytokines. Therefore, in a helminth infection in which a Th2 type immune response is important, IL-18 has a different role depending on the nematode type and may play an important role in host defense.
4.2.2. IL-18-Mediated Defense Against Intracellular Pathogens
4.2.2.1 Bacterial Infection
The ability of IFN-γ to increase the expression of inducible nitric oxide synthase (iNOS) is essential for killing intracellular organisms. Because NO production is critical for intracellular killing, IL-18, an IFN-γ inducer, plays an important role in controlling infection.
Mycobacterium avium is an intracellular microorganism that infects and proliferates in macrophages. In an M. avium infection mouse model, genetically susceptible BALB/c mice had decreased expressions of IL-12 and IL-18 and reduced IFN-γ/Th1 responses. In contrast, resistant DBA/2 mice showed the increased expressions of IL-12, IL-18 and IFN-γ [149] which correlated with the clearance of M. avium. Indeed, IL-18-deficient C57BL/6 mice had a severe infection and pathological changes that were significantly suppressed by treatment with exogenous IL-18 [150].
The role of IL-18 in host defense against M. tuberculosis infection has been demonstrated in studies using IL-18 knockout and IL-18 transgenic mice. IL-18-deficient mice are susceptible to M. tuberculosis infection, and IFN-γ levels in the serum, spleen, lung, and liver were lower than in wild type mice [151,152]. IFN-γ production by spleen cells stimulated with mycobacterial antigen was also impaired in IL-18 knockout mice. In contrast, IL-18 transgenic mice were more resistant to M. tuberculosis infection than control wild mice, and IFN-γ levels in the serum and production by mycobacterial antigen-stimulated splenocytes were increased. These data suggest the important contribution of IL-18 in the development of Th1 immunity [151]. In M. tuberculosis infected IL-18-deficient mice, IL-17 and MIP-1α were increased instead of decreasing blood IFN-γ and NO, and in the lung, the M2 macrophage markers Arginase-1 and Ym-1 were increase. Neutrophils are important for the control of M. tuberculosis in wild type mice, but Il18−/− mice had increased accumulation of neutrophils as assessed histologically. However, because Il18−/− mice cannot control M. tuberculosis, IL-18 might enhance the M. tuberculosis bactericidal activity of neutrophils [152].
In humans, the IL-18 promoter gene −137 G/C polymorphism is a risk factor for tuberculosis in the Chinese population, and PBMCs of the −137 GG type had lower IL-18 production compared with the GC and CC types [153]. Extensive case-control studies reported that a polymorphism of the IL-18Rα gene was associated with tuberculosis risk in people aged over 46 years old [154]. In addition, a SNP of the IL-18Rα promoter was associated with the methylation status of the gene and IL-18Rα expression, and the increased DNA methylation and decreased expression of IL-18Rα might be partially involved in the increased susceptibility to M. tuberculosis. In mice, even when IL-18Rα is deficient, there is no change in susceptibility to M. tuberculosis, indicating differences in susceptibility related to IL-18 between humans and mice. In humans, IL-37 uses IL-18Rα as a receptor to suppress immune responses. IL-37 levels were increased in the blood of M. tuberculosis patients, suggesting immunity is suppressed by a reduction in IFN-α production and the induction of M2 macrophage differentiation [155,156].
L. monocytogenes are Gram-positive intracellular bacteria that cause food listeriosis in humans. L. monocytogenes invade and multiply within the cytoplasm of various types of cells, including macrophages, epithelial cells, and hepatocytes. The role of IL-18 in resistance to L. monocytogenes infection is controversial. In a study using IL-18R antibodies, IL-18 potentiated the resistance of mice to L. monocytogenes infection [157]. Furthermore, IL-12- or IL-18-deficient mice lacked the ability to produce detectable levels of IFN-γ in the serum and showed significant and moderate L. monocytogenes sensitivity, respectively. Ifng−/− mice and Il12−/−Il18−/− mice had a higher sensitivity to L. monocytogenes infection than wild type mice [158]. Moreover, the administration of IL-18 decreased the number of bacterial cells in the liver and spleen of L. monocytogenes–infected mice [159]. However, Lockner et al. and Tsuchiya et al. reported that mice had increased resistance to L. monocytogenes infection because of the lack of IL-18 [160,161]. It is thought that IL-10 secreted from IL-18-stimulated NK cells was involved in the increase of L. monocytogenes susceptibility by IL-18 [162,163]. The influence of the presence or absence of IL-18 is thought to depend on the mouse strain and the infectious dose of L. monocytogenes used for infection.
Listeria infection induces IL-18 production by inflammasome-dependent and -independent mechanisms. Bacteria invade cells by listeriolysin O, which activates the caspase-1 inflammasome through NLRP3 and AIM2 and induces IL-18 production, which is also produced by Fas signaling independent of the caspase-1 pathway. Uchiyama et al. revealed the mechanism of the induction of IL-18/IL-1β production by L. monocytogenes infection via Fas. Upon L. monocytogenes infection, macrophages produce type I IFN, which induces the expression of the Il18 gene. NK cells express FasL in response to L. monocytogenes infection that stimulates Fas expressed by infected macrophages. ASC, ROS and caspase-8 are activated in Fas-stimulated macrophages, which then secrete IL-18/IL-1β in their active forms [64]. Ly6C+ monocytes produce IL-18 in L. monocytogenes–infected mice, which is important for the expression of IFN-γ, but not granzyme B, by CTLs [164].
Recently, a new mechanism for IL-18 production by Listeria infection was reported. Macrophages infected with L. monocytogenes recognize lipoteichoic acid in bacterial cells by NLRP6, which recruits caspase-11 and caspase-1 via ASC to form an inflammasome that processes IL-1β and IL-18 to their active forms. At this time, NLRP6 and caspase-11 are induced by type 1 IFN similar to IL-18. Mice deficient for NLRP6, caspase-11, or IL-18 are more resistant to L. monocytogenes than wild type mice, but this effect is reversed by the administration of IL-18 [165].
Caspase-1 and IL-18 are important for host defense against Shigella flexneri, the causative agent of Bacillus dysentery. IL-18-deficient mice nasally infected with S. flexneri developed more severe inflammation in the lungs compared with wild type mice, because they could not eliminate the bacterial infection. These studies indicate that IL-18 is important for the induction of inflammation and the effective elimination of bacteria [166].
In a mouse model of S. typhimurium infection, the protective role of IL-18 was demonstrated by the treatment of infected mice with anti-IL-18 Ab or the administration of IL-18. In mice susceptible to lethal S. typhimurium infection, the administration of IL-18 increased survival and reduced bacterial tissue load [167]. A beneficial effect of exogenous IL-18 was not observed in mice deficient for IFN-γ. Caspase-1-deficient mice with Salmonella infection died more rapidly than wild type mice. Both caspase-1 substrates, IL-18 and IL-1β, are involved in the control of S. typhimurium. Infection experiments with Il18−/− mice and the administration of recombinant IL-18 to caspase-1−/− mice showed that IL-18 was not important for resistance to the intestinal phase of infection, but rather to the systemic infection [168].
4.2.2.2. Protozoan Infection
Leishmania major is an intracellular parasitic protozoan that mainly infects monocytic cells such as macrophages. The healing of lesions caused by L. major infection requires the induction and expansion of Th1 cells and NO production, an important effector molecule involved in the clearance of L. major via iNOS induction by IFN-γ [171]. IL-12 or IL-18 was administered to L. major-sensitive mice did not induce wound healing, but combined IL-12 and IL-18 completely protected footpad swelling in a NO-dependent manner. Treatment with anti-IL-18 neutralizing Ab reduced NO production by the downregulation of IFN-γ induced by L. major infection and markedly reduced the tolerance to L. major infection. In addition, IL-18-deficient mice on a L. major-resistant background developed large lesions during the early stage of L. major infection compared with wild type littermates but these were eventually resolved [172,173]. However, IL-18 enhanced Th2 cytokine production and exacerbated footpad swelling in L. major susceptible BALB/c mice [174]. Neutralization of IL-18 by IL-18BP suppressed Th2 cytokine production by IL-18 and alleviated footpad swelling.
Trypanosoma cruzi is an intracellular protozoan parasite that causes Chagas disease, mainly in Latin America. Following infection with T. cruzi, early inflammation of the invasion site occurs followed by hepatoma, splenomegaly after lymphadenitis, and in some cases, acute myocarditis develops and the patient dies. Chronic infections cause chronic myocarditis, gigantic esophagus, and a giant colon. IFN-γ produced after infection is important for determining resistance or susceptibility. Infection with T. cruzi also induces the production of IL-12 and IL-18 [175]. IL-12 is the major factor important for defense because absence of IL-18 did not affect the resistance against the protozoa [176,177].
However, in humans there is a correlation between the rs360719 gene polymorphism and T. cruzi infection (seropositive). rs360719 is located in the promoter region of the IL18 gene and its polymorphism is thought to affect IL18 gene expression by creating a binding site for the transcription factor OCT-1 [178]. Therefore, IL-18 might be involved in the resistance to T. cruzi infection.
Infection with T. cruzi can cause myocarditis. Mice infected with a Colombian strain of T. cruzi had increased blood IFN-γ and IL-12 levels, and in IL-18-deficient mice, the trafficking of inflammatory cells to the myocardium and the number of protozoa in the tissues were reduced [179]. In addition, the myocardium of patients with chronic Chagas disease had a high expression of IL-18 [180], and in Brazilian patients a correlation with the rs2043055 SNP was shown [181]. Therefore, although IL-18 is not involved in the resistance to T. cruzi infection, it might affect the pathology of myocarditis.
The protozoan, Tritrichomonas musculis, a commensal parabasalid newly identified in 2016, activates the host epithelial inflammasome and induces IL-18 release. This epithelial-derived IL-18 promotes Th1 and Th17 immunity in dendritic cells and provides protection against mucosal bacterial infection. Colony formation by T. musculis exacerbated the development of T cell driven colitis and sporadic colorectal tumor formation [182].
Toxoplasma gondii is an intracellular parasitic protozoan that spreads by ingestion and vertical infection. Oral or intraperitoneal infection of low doses (<20 cysts) of T. gondii induced IFN-γ production from Th1 cells and NK cells, which mediated the resistance of mice against infection [183,184]. However, oral infection with a higher inoculum dose (50–100 cysts) resulted in a deleterious Th1 cell response characterized by the development of severe small intestinal necrosis caused by the overproduction of proinflammatory mediators [185]. In immunodeficient mice, the injection of IL-18 decreased the number of parasites isolated from the tissue and correlated with an increase in NK cell activity [186]. IL-18 contributed to the development of immunopathological findings in the small intestine after the oral high-dose infection of T. gondii via the induction of IFN-γ production [187]. By this mechanism, IL-18 acts on IL-15-dependent NKp46+ NK1.1+ cells to induce CCL3 production, which is involved in the accumulation of CCR1 positive inflammatory monocytes [188]. IL-22 is also an important cytokine for ileitis induced by T. gondii infection. IL-22 acts on intestinal epithelial cells to induce IL-18 expression, which induces IL-22 production from innate lymphoid cells (ILCs). This positive feedback causes excessive IFN-γ production and marked neutrophil infiltration in the ileum [189].
Malaria is a major cause of morbidity and mortality, especially in sub-Saharan Africa. When the malaria parasite invades the host body via mosquito blood sucking, it first infects hepatocytes and then red blood cells. The early production of IFN-γ and TNF-α is crucial for protective immune reactions [190]. However, the excessive and prolonged production of proinflammatory cytokines is involved in the etiology of diseases associated with symptoms characteristic of severe malaria. The role of IL-18 in host defense against blood stage mouse malaria infection was reported using the non-lethal strain Plasmodium yoelii 265 and the lethal strain P. berghei ANKA. Infection by P. yoelii 265 or P. berghei ANKA increased the production of IL-18, IL-12p40 and IFN-γ. The administration of IL-18 to infected mice increased the infiltration of inflammatory cells, consisting of mononuclear cells and Kupffer cells, into the liver and decreased necrosis and pigment hemozoin deposition. Furthermore, serum IFN-γ levels were elevated, the onset of parasitemia was delayed, and the survival rate of infected mice was increased. The administration of anti-IL-18 neutralizing Ab to mice exacerbated infection with P. berghei ANKA, impaired host resistance, and shortened the mean survival time. In addition, Il18−/− mice were more sensitive to P. berghei ANKA than wild type C57BL/6 mice. These data suggest that IL-18 has a protective role in host defense by enhancing IFN-γ production during blood stage infection by murine malaria [191,192]. IL-18 increased NK cell reactivity to IL-2 by increasing CD25 expression and inducing higher IFN-γ production [193].
Serum IL-18 levels are increased in human P. falciparum malaria patients [194]. When serum levels of IL-18 and IFN-γ were measured by dividing malaria patients into non-complicated, severe, and cerebral malaria, an increase in IL-18 levels was observed in all three groups. In cases with severe malaria, IL-18 tended to be high during the disease course. In addition, there was a significant correlation between the IL-18 level in severe malaria patients and the extent of parasitemia [195].
4.2.3. IL-18-Mediated Viral Clearance
In addition to the potent induction of IFN-γ, IL-18 activates CD8+ T cells, which play a central role in viral clearance, suggesting a role for IL-18 in viral infection. Infection with influenza A virus induced the production of IFN-α/β, TNF-α, IL-1β and IL-18 in human peripheral macrophages [196]. Virus infected macrophage-derived IL-18 acts synergistically with IFN-α/β, inducing rapid IFN-γ production by T cells, resulting in the induction of Th1 immune responses. In a mouse model of vaccinia virus infection, the intraperitoneal injection of IL-18 significantly inhibited tail pock formation in BALB/c mice. Ectromelia virus belonging to the orthopoxvirus family is used as a mouse model of human smallpox. In IL-18-deficient mice, resistance to E. virus infection was attenuated, CTL numbers were decreased, and Th2 cytokine producing cells and Treg cells were increased. Therefore, IL-18 contributes to antiviral immune responses by promoting IFN-γ production and the induction of CTLs [197]. IL-18 induces HIV replication and in vitro experiments using the monocyte cell line U1 showed an increase in HIV-1 production after stimulation with the proinflammatory cytokines IL-1 and TNF-α or after exposure to IL-6 [198]. IL-18 increases HIV-1 production from U1 cells [199] and treatment with TNF binding protein or neutralizing anti-IL-6 mAb decreased IL-18 stimulated HIV-1 production, suggesting a role for IL-18 in HIV-1 pathogenesis. IL-18 levels are elevated in the sera of HIV-infected patients 3-to-6 years after infection, and decrease thereafter. The serum of HIV patients contains a large amount of TGF-β and the ability of blood IL-18 to induce IFN-γ is low. Because TGF-β inhibits IL-18 production from peripheral blood monocytes, IL-18 and TGF-β concentrations are inversely correlated [200]. In a herpes simplex virus (HSV-1) infection mouse model, the administration of IL-18 prior to infection markedly improved survival through T and B cell independent enhancement of IFN-γ induced NO. This effect was also observed in athymic nude mice and SCID mice, suggesting that IL-18 potentiates innate immunity [201]. Resistance to primary infection with herpes simplex virus 2 (HSV-2) was attenuated in IL-18-deficient mice or anti-IL-18 antibody treated mice, and the survival rate was reduced after infection [202]. Inflammatory monocytes recruited to infected sites produce type I IFN and IL-18 induced by HSV-2 infection. IL-18 is important for IFN-γ production from NK cells [203]. Rotavirus (RV) infects small intestinal epithelial cells, causing severe dehydrating diarrhea in children and moderate intestinal distress in adults. Bacterial flagellin treatment prevents RV infection in mice through adaptive immunity, interferons (IFN, type I and type II), NLRC4, and TLR5, a receptor for the flagellin receptor, all of which combine to cure chronic RV infection. This effect is suppressed in IL-18BP transgenic mice and Il18−/− mice. The same protective effect was obtained by administering IL-18 and IL-22 instead of flagellin. The activation of TLR5 in dendritic cells by flagellin induced the production of IL-22 from ILCs via IL-23. Furthermore, flagellin also induced the production of IL-18, which was dependent on NLRC4 and independent of dendritic cells and ILCs. IL-22 induced protective gene expression in intestinal epithelial cells and IL-18 induced cell death by activating caspase-3 to eliminate RV-infected cells [204].
4.3. IL-18 in Metabolism
4.3.1. IL-18 in Metabolic Homeostasis
Dietary nutrients, such as carbohydrates, proteins, and lipids, are digested and absorbed through the alimentary tract. Glucose and amino acids generated from carbohydrates and proteins, respectively are absorbed in the small intestine and transported into the liver via the portal vein. Absorbed fatty acids from dietary lipids are synthesized into triglycerides, which are transported into the liver. In the liver, these dietary units are appropriately catabolized and/or anabolized into minimal substances, which are consumed in individual cells to produce cellular components, participate in cellular activities and biological functions. The residual energy in each cell is collected and stored as triglycerides in adipose tissues. However, excess energy initiates systemic diseases, termed “metabolic syndromes”, such as type 2 diabetes mellitus, atherosclerosis, and acute myocardial infarction.
Initially, PRRs were thought to distinguish between non-self patterns (e.g., bacterial lipopolysaccharide, viral single-stranded RNA) and self-derived molecules. However, soon after their discovery, PRRs were demonstrated to sense various self-derived molecules, such as monosodium urate crystals involved in gout [44], calcium pyrophosphate dehydrate crystals involved in pseudogout [44], amyloid b in Alzheimer’s disease [205], and islet amyloid polypeptide in type 2 diabetes mellitus [206]. Now we know that a wide variety of self-derived molecules can activate the NLRP3 inflammasome (as described above). Therefore, the activated NLRP3 inflammasome might be involved in metabolic syndrome. Furthermore, NLRP3 inflammasome-mediated IL-1β has been identified as a key pathogenic cytokine that triggers and/or promotes various metabolic disorders. In contrast, IL-18 is beneficial for metabolic homeostasis. In this section, we will discuss the beneficial roles of IL-18 in metabolic homeostasis and introduce topics regarding the unique pathogenic action of IL-18 in the development of metabolic syndrome.
IL-1β and IL-18 belong to the IL-1 superfamily, and are activated and secreted via a cytoplasmic, multimeric complex termed the inflammasome, to promote proinflammatory cytokine effects. In contrast to IL-1β, IL-18 is a prerequisite for metabolic homeostasis. Several cellular and molecular mechanisms for the maintenance of metabolic homeostasis by IL-18 have been reported.
4.3.1.1. IL-18 Regulation of Food Intake
IL-18-deficient mice spontaneously developed obesity and insulin resistance when fed a normal chow diet [207]. Il18−/− mice developed obese diabetes mellitus with hyperglycemia, hyperinsulinemia, impaired glucose- and insulin-tolerance tests, and ectopic lipid deposition in the aorta wall. This was also observed for transgenic mice systemically overexpressing IL-18BP, a functional serum decoy receptor for IL-18 [207]. Intriguingly, food intake in Il18−/− mice was significantly higher than that of control mice. Treatment with recombinant (r) IL-18 improved food intake in Il18−/− mice. Zorrilla et al. reported that Il18−/− mice fed a low-fat or high-fat diet (HFD) were hyperphagic as compared with control mice [208]. Furthermore, they found that the intracerebral ventricular administration of rIL-18 negatively regulated food intake in a dose dependent manner in HFD-fed wild type mice [209]. These reports suggest that IL-18 suppresses appetite and promotes energy expenditure even in people consuming an HFD.
4.3.1.2. IL-18 Regulation of Energy Expenditure by Activating Thermogenic Adipose Tissues
Il18−/− mice are characterized by impaired energy expenditure [208,209]. Recently, the mechanism for the regulation of energy expenditure by IL-18 was unveiled. Skeletal muscle cells are a prototype cell for energy expenditure [210]. Thermogenic adipose tissues, especially brown and beige adipocytes that are mitochondrion-rich cells, are secondary cell types that contribute to energy expenditure [211,212]. However, mitochondria in these cells cannot efficiently oxidize fatty acids, but dissipate energy as heat, thus contributing to energy expenditure [211,212]. Adipocytes in white adipose tissues, such as subcutaneous adipose tissue, have low numbers of mitochondria and are not thermogenic. Under certain conditions, however, white adipocytes can transdifferentiate into thermogenic beige cells [211]. Recently, it was reported that Il18−/− mice had an impaired activation of brown adipocytes, and the beiging of white adipocytes [213]. This impairment might account for the impaired energy expenditure in Il18−/− mice.
IL-33 is a pro-atopic innate immune cytokine and is a member of the IL-1 family. Of note, group 2 innate lymphoid cells (ILC2s) express the IL-33 receptor and IL-33 directly activated ILC2s to induce adipocyte beiging [214] via methionine enkephalin, a beiging-inducing molecule [215], as well as directly activating eosinophils to induce the beiging of adipocyte precursor cells [216]. Recently, certain populations of ILC2 were shown to express IL-18Rα but not the IL-33 receptor [217]. Skin-resident ILC2s dominantly express IL-18R, although some fat-resident ILC2s also express IL-18R [217]. IL-18 induction of adipocyte beiging might be explained by the activation of IL-18Rα-expressing ILC2s.
4.3.1.3. IL-18 Activation of AMPK and Lipid Oxidation in Skeletal Muscle
Adenosine monophosphate-activated protein kinase (AMPK) is a sensor of intracellular energy status and maintains energy stores by catabolic and anabolic pathways [218]. Skeletal muscles experience drastic energy changes during exercise and AMPK participates in cellular energy control [218]. Cytokines such as IL-6, were reported to activate AMPK in the skeletal muscle. Lindegaard et al. demonstrated that IL-18 activated AMPK in skeletal muscle cells and skeletal muscle strips in vitro and ex vivo [219]. Consistently, Il18ra−/− mice were reported to be highly susceptible to HFD, in terms of weight gain and insulin resistance [219]. Intriguingly, ectopic lipid deposition was induced in the skeletal muscle, but rarely in the liver, of HFD-fed Il18ra−/− mice accompanied by impaired activation of the AMPK pathway. Therefore, IL-18 might maintain metabolic homeostasis in part by activating the AMPK signal pathway in skeletal muscle.
4.3.1.4. IL-18 Processed by the NLRP1 Inflammasome Protects Against Metabolic Syndrome.
Recently, NLRP1 inflammasome-mediated IL-18 was reported to contribute to metabolic homeostasis in vivo [220]. It is well established that lethal toxins generated by Bacillus anthracis, a causative bacterium of anthrax, activate the NLRP1 inflammasome to release mature IL-1β and IL-18 [221]. Similar to Il18−/− mice, Nlrp1−/− mice spontaneously develop obesity (Figure 4A). Their metabolic alterations are characterized by ectopic lipid droplets in insulin-responsive organs, hepatic steatosis and adiposity in skeletal muscle, an increase in the size of adipose tissues, and systemically decreased lipolysis [220]. Nlrp1−/− mice suffer from metabolic syndrome with impaired glucose tolerance [220]. IL-18 levels in visceral adipose tissues were significantly decreased in HFD-fed Nlrp1−/− mice compared with control mice. Chimeric mice generated by bone marrow cell transfer revealed that the selective absence of NLRP1 in non-hematopoietic cells, but not hematopoietic cells, caused these alterations when fed an HFD (Figure 4B). In contrast, mice with a systemic gain-of-function mutation in Nlrp1, namely Nlrp1MUT mice, spontaneously developed lethal inflammation associated with the enhanced production of IL-1β. Furthermore, Il1r−/−Nlrp1MUT mice were resistant to the induction of inflammatory diseases [220]. Convincingly, plasma levels of IL-18 were still elevated in Il1r−/−Nlrp1MUT mice compared with Il1r−/− mice. Il1r−/−Nlrp1MUT mice showed a loss of fat in various organs associated with enhanced lipolysis and low glucose tolerance. When fed an HFD, Il1r−/−Nlrp1MUT mice exhibited cachexia-like morbidity and mortality with hepatic focal necrosis and highly elevated plasma levels of IL-18 (10-60 ng/mL) (Figure 4C) [220]. The depletion of Il18 rescued Il1r−/−Nlrp1MUT mice from the HFD-induced fatal changes and they exhibited a comparable phenotype to Il1r−/− mice (Figure 4C) [220]. These observations clearly demonstrate the importance of IL-18 for metabolic homeostasis, in particular by controlling lipolysis and insulin sensitivity. Currently, endogenous agonists of the NLRP1 inflammasome remain unclear.
4.3.2. Detrimental Role of the NLRP3 Inflammasome/IL-1β Axis in the Development of Metabolic Syndrome
It is well established that chronic inflammation, induced by proinflammatory cytokines including IL-1β processed by the activated NLRP3 inflammasome, is associated with obesity and resultant metabolic syndromes, such as type 2 diabetes mellitus, atherosclerosis, and cardiac vascular diseases, which are leading causes of death [222,223,224,225,226,227,228]. Many NLRP3 activators have been identified. First, oxidized low-density lipoprotein (LDL) was reported to activate macrophages through CD36, a scavenger receptor, to produce TNF-α and other proinflammatory cytokines, eventually leading to the development of atherosclerosis and cardiovascular diseases [229,230]. Under the conditions of oxidized LDL, cholesterol crystals that activate the NLRP3 inflammasome are generated in macrophages [231,232]. These events promote atherosclerosis. Second, saturated free fatty acids, such as palmitate, were identified as NLRP3 activators in addition to TLR4 agonists, which eventually cause type 2 diabetes [233,234]. Third, hyperglycemia activates the thioredoxin-interacting protein (TXNIP)-mediated NLRP3 inflammasome in adipose tissues and microvascular endothelial cells, which are relevant to type 2 diabetes and myocardial ischemic/reperfusion injury, respectively [235,236,237].
Patients with metabolic syndrome have elevated circulating levels of IL-18, a product of NLRP3 inflammasome activation [238,239]. Chronic inflammation in adipose tissues causes insulin resistance and type 2 diabetes mellitus. Notably, IL-1β, but not IL-18, participates in the development of metabolic syndromes by inhibiting adipocyte differentiation that is required for the maintenance of insulin sensitivity [240,241] and inducing inflammation [242]. IL-18 alone does not exert a proinflammatory effect when levels of co-activating cytokines, such as IL-12, are low [1,2,131,243,244]. Adipose tissue-associated macrophages of the M2 type produce anti-inflammatory and pro-fibrotic cytokines but low levels of proinflammatory cytokines [245,246]. This might explain the minor role of IL-18 in triggering obesity-associated metabolic diseases.
4.4. IL-18 in Intestinal Homeostasis
The gastrointestinal tract absorbs beneficial nutrients and paradoxically functions as a surface barrier to prevent harmful factors from entering the circulation. To transport dietary nutrients safely and efficiently, the gastrointestinal tract contains various control systems. Movement of the gastrointestinal tract is regulated by a unique nervous network termed the enteric nervous system as well as the central nervous system, under the control of sensing dietary contents, nutrients, and possible dietary toxins [247]. Once toxic foods enter the gastrointestinal tract, these nervous systems enhance gastrointestinal motility to rapidly exclude them via the production of diarrhea. In the gastrointestinal tract, especially the colon, various indigenous xenogeneic microorganisms, termed microbiomes, including viruses, bacteria, and fungi, reside symbiotically. Furthermore, the gastrointestinal tract transports food Ag and dietary nutrients into the tissues. Therefore, the gastrointestinal tract is always in contact with stimuli that activate innate and/or adaptive immunity. However, to accomplish their task of nutrient intake, the gastrointestinal tract negatively regulates immune responses directed to food Ags and microbiomes. Furthermore, symbiotic microbiome-derived metabolites, such as butyrate, are utilized to develop regulatory T cells that prevent pathological immune responses to food Ags and/or microbiomes in healthy individuals [248,249,250]. However, the gastrointestinal tract is a gate keeper, and should also detect and prevent the potential invasion of symbiotic microbes and food-borne pathogens [251,252]. Therefore, the gastrointestinal tract encompasses both a resistant barrier and a unique neural and immune system/network to prevent the entry on non-self factors.
Recently, IL-18 generated by intestinal epithelial cells was shown to be required for intestinal homeostasis [252,253]. In contrast, the uncontrolled release of IL-18 by immune cells infiltrating into lesions had an opposite detrimental effect in inflammatory bowel diseases (IBDs) [254,255]. Intriguingly, a recent article reviewed the importance of the NLRP3/IL-1β axis in IBDs [256]. Here, we introduce the beneficial roles of epithelial IL-18 in gut homeostasis.
4.4.1. Intestinal Epithelial Cells Constitutively Produce Constituents of the Inflammasome for IL-18 Maturation
As described above, pro-IL-18 is produced and stored in a wide variety of cells. In contrast to pro-IL-18, pro-IL-1β is rarely expressed in the intestinal epithelium under normal conditions. Intestinal mucosal epithelial cells were reported to store pro-IL-18 in the cytoplasm [257,258,259]. These cells produce pro-caspase-1 and the caspase-1 activation adaptor molecule, ASC under a steady state [260,261,262]. Furthermore, several sensors or regulators of inflammasomes are also expressed constitutively, such as NLRP3 [263], NAIP-NLRC4 [264,265,266], and NLRP6 [267,268]. Therefore, intestinal epithelial cells have pre-formed components for inflammasomes. Of note, pro-IL-18, but not pro-IL-1β, is constitutively produced in tissue parenchymal cells in the steady state. Therefore, upon activation of the gut epithelial inflammasome, only mature IL-18 might be selectively activated and secreted. Therefore, the constitutive distribution of pro-IL-18 in specific cell types might determine the pro-homeostatic action of IL-18 on parenchymal cells.
4.4.2. Importance of the Gut Microbiome for Homeostatic IL-18 Release
Gut microbiota are necessary for the appropriate development of innate and adaptive immunity. Indeed, intestinal T cells fail to develop into Th17 and Treg cells, under germ-free conditions [250,269,270,271,272]. However, supplementation with commensal bacteria or their products corrected this effect leading to their healthy development [250,269,270,271,272]. Recently, a microbiome-derived metabolite was demonstrated to be a prerequisite for the intestinal epithelial cell secretion of IL-18, which is necessary for intestinal homeostasis [273]. Mice fed a high-fiber diet were resistant to DSS-induced colitis. The microbiome catabolizes food into short chain fatty acids, such as acetate, in the mammalian gut. Mice deficient for G protein-coupled receptors that recognize short chain fatty acids, are highly susceptible to DSS challenge even when fed a high-fiber diet. Consistently, supplementation with acetate restored their resistance to DSS challenge. Intriguingly, the induction of IL-18 by acetate was dependent on NLRP3 but not NLRP6 [273]. These results suggest that metabolites generated by the microbiome participate in the constitutive secretion of IL-18 for gut homeostasis.
4.4.3. Roles of NLRP6 Inflammasome-Mediated Epithelial IL-18 in Gut Homeostasis
Nlrp6, a member of the NLR family, is preferentially expressed in the kidney, liver, lung, and intestines, but rarely in the thymus or spleen [274]. In the colon, Nlrp6 is expressed dominantly in epithelial cells, whereas Asc and Caspase1 are similarly expressed in epithelial cells and CD45+ hematopoietic cells [267]. Similar to the NLRP3 inflammasome, the NLRP6 inflammasome is thought to consist of NLRP6, ASC, and pro-caspase-1 [261,275]. A recent report clearly demonstrated the requirement of caspase-11 for the optimal processing of caspase-1 by the NLRP6 inflammasome in macrophages upon transfection with lipoteichoic acids derived from a Gram-positive bacterium [276]. The loss of NLRP6 was reported to cause dysbiosis [261,267,275] and to impair the mucin layer in the colon of mice [277]. However, which stimuli initiate the sustaining activation of the NLRP6 inflammasome in intestinal epithelial cells remain to be elucidated. We need to identify whether lipoteichoic acid, presumably derived from the microbiome, contributes to the sustaining activation of the NLRP6 inflammasome.
4.4.3.1. The NLRP6 Inflammasome is Indispensable for the Healthy Microbiota
Both Nlrp6−/− and Asc−/− mice are highly susceptible to DSS-induced colitis, strongly suggesting the importance of the NLRP6 inflammasome in this disorder [267]. Wild type mice co-housed with Nlrp6−/− mice have a similar susceptibility to DSS challenge as observed for wild type mice co-housed with Asc−/−, caspase1−/− or Il18−/− mice [267], indicating colonic IL-18 is important for maintenance of the healthy microbiome (Figure 5). Indeed, the fecal bacterial phylogenetic architecture in these mice was clearly different from that in wild type mice [267]. Of note, wild type mice co-housed with Il1r−/− mice show comparable clinical course after DSS challenge as wild type mice co-housed with wild type mice [267]. This clearly suggests that IL-1β is not generated by the NLRP6 inflammasome in colonic epithelial cells possibly because of the absence of pro-IL-1β. Alternatively, IL-1β generated through the NLRP6 inflammasome is not involved in the microbiome. Furthermore, wild type mice co-housed with Aim2−/− mice or Nlrc4−/− mice have an intact microbiome [267], suggesting that the AIM2 inflammasome or NLRC4 inflammasome are not activated in colon epithelial cells in the steady state. Therefore, the NLRP6 inflammasome is activated in the murine colon, leading to IL-18 release which maintains gut homeostasis by preventing dysbiosis.
Colon tissues from wild type mice under specific pathogen-free (SPF) conditions, but not under germ-free conditions, constitutively produce mature IL-18 [261]. Consistent with this, levels of IL-18 spontaneously produced by the colon of SPF mice are very low at birth, but are markedly increased at around 3 weeks, with continuous high levels of IL-18 thereafter [261]. Therefore, the microbiome might initiate IL-18 production (Figure 5). Upon stimulation with recombinant IL-18, colon tissues from germ-free mice produced antimicrobial peptides, which were beneficial for host defense on the barrier surfaces [278,279]. Colon tissues from wild type mice, but not Asc−/−, Il18−/−, caspase1/11−/− or Nlrp6−/− mice, constitutively produced antimicrobial peptides (Figure 5). Exogenous IL-18 restored the antimicrobial peptide production by these inflammasome-deficient mice concomitant with the restoration of dysbiosis [261]. Metagenomic sequencing of germ-free wild type mice co-housed with wild type mice or Asc−/− mice revealed decreased taurine and increased histamine and spermidine in the metabolites of feces from the wild type mice co-housed with Asc−/− mice compared with the wild type mice co-housed with wild type mice. The administration of spermidine or histamine in the drinking water impaired the spontaneous activation of caspase-1 and spontaneous production of IL-18 in the colon of wild type mice. Those observations suggest that NLRP6 inflammasome-mediated IL-18 sustains a healthy microbiome via the induction of antimicrobial peptides in the murine colon [275]. Endogenous ligands that activate the NLRP6 inflammasome for the continuous release of IL-18 remain to be elucidated.
4.4.3.2. Involvement of the NLRP6 Inflammasome in the Formation of the Colonic Mucin Layer
The mucin layer is essential for intestinal host defense [280,281]. Nlrp6−/− mice have colonic goblet cells numerically comparable to wild type mice. However, a detailed histological analysis showed that the width of the colonic inner mucus layer had almost disappeared in Nlrp6−/− mice [277]. This was also observed in Asc−/− mice and caspase1/11−/− mice. In contrast, the inner mucin layer was intact in Il1r−/− mice and Il18−/− mice, indicating other biological events downstream of the NLRP6 inflammasome pathway might be involved in mucin formation (Figure 5). Nlrp6−/−, Asc−/−, and caspase1/11−/− mice are highly susceptible to enteric infection with Citrobacter rodentium. Mice deficient in the NLRP6 inflammasome have impaired autophagosome formation [277] suggesting the NLRP6 inflammasome might contribute to healthy mucin layer formation by modulating the autophagosome pathway because the autophagy-mediated pathway contributes to other secretory pathways [282], and mice deficient in the molecule responsible for autophagosome generation have an abnormal inner mucin layer in the intestine [277]. The mechanism underlying the NLRP6 inflammasome induction of autophagy remains to be elucidated.
4.4.4. Importance of Pyrin Inflammasome-Mediated Mucosal IL-18 for Tight Junction Formation
It is well established that mutations in PYRIN, alternatively named MEFV, are associated with a hereditary autoinflammatory disease termed Mediterranean fever as well as severe IBDs [283,284,285,286,287]. Pyrin is a member of the innate sensor family and forms part of the inflammasome after cells are stimulated by bacterial Rho GTPase [288,289]. Pyrin−/− mice are susceptible to infection with pathogens that produce Rho GTPase as an exotoxin, including Burkholderia cenocepacia [288,289]. Intriguingly, ligands for TLRs, such as LPS, and TNF-α/TNFR1-mediated signaling induced Pyrin expression [290]. Recently, it was demonstrated that the pyrin inflammasome contributes to tight junction integrity to alleviate colitis and colitis-associated colon cancer in mice [290] (Figure 5). Pyrin−/− mice are highly predisposed to colitis-associated colon cancer [290], which is induced by periodical treatment with AOM-DSS [291]. Pyrin−/− mice had a larger burden of colon cancer with more severe colitis compared with wild type mice. Furthermore, colon tissues from Pyrin−/− mice spontaneously produced higher amounts of proinflammatory cytokines/chemokines, but lower amounts of IL-18 despite comparable Il18 expression, when compared with control mice. Pyrin−/− mice exhibited a loss of epithelial barrier integrity with histologically and biologically impaired tight junctions, although the production of antimicrobial peptides and mucin were normal. Furthermore, the colons of DSS-treated Pyrin−/− mice expressed higher levels of stem cell markers compared with DSS-treated control mice. Intriguingly, treatment with exogenous recombinant IL-18 restored epithelial permeability, colitis, and the tumor burden [291]. Therefore, in colon epithelial cells, the pyrin inflammasome might be constitutively activated to release IL-18, which sustains epithelial barrier integrity and prevents tumorigenesis. Intriguingly, IL18 and IL18RB expressions were significantly lower in colorectal cancer patients than colon biopsies from healthy donors [291]. These observations suggest that the supplementation of IL-18 might be beneficial for certain types of colitis to protect against colorectal cancer.
4.4.5. Newly Identified NLRP9b-Mediated IL-18 Release is Involved in Rotavirus Clearance
Very recently, Zhu et al. demonstrated that NLRP9b formed a new inflammasome comprised of ASC and pro-caspase-1, and that upon rotavirus infection it released IL-18, but not IL-1β, to induce pyroptotic cell death [41,292]. Pyroptotic cell death, but not IL-18, is important for rotavirus eradication. NLRP9b, similar to NLRP6, is constitutively expressed in intestinal epithelial cells. Nlrp9b−/− mice, and mice selectively deficient in caspase-1 in epithelial cells, but not Il18−/− mice, were highly susceptible to rotavirus infection, in terms of exaggerated diarrhea [292]. Furthermore, the NLRP9b inflammasome recognizes rotavirus RNA pathogen associated molecular patterns through DHX9, an RNA helicase [293]. Therefore, the gut mucosal epithelium is equipped with several inflammasomes to protect against various pathogens. Currently, whether NLRP9b is involved in the homeostatic production of IL-18 for intestinal homeostasis is unknown.
5. IL-18 in Disease
5.1. Endotoxin-Induced Systemic and Tissue Diseases
5.1.1. Induction of Endotoxin Shock in P. acnes-Primed Mice.
Sepsis is still a common, life-threatening disorder, in which endotoxin is a key player. Paradoxically, patients with high serum levels of endotoxin do not necessarily develop lethal shock, whereas some patients die of septic shock even when their serum endotoxin levels are low. To understand this paradox, we measured serum IL-6 levels of patients, because LPS induces IL-6 production in vivo. The simultaneous measurement of serum levels of LPS and IL-6 indicated that there were at least two groups: the high IL-6 group was endotoxin shock susceptible and the low IL-6 group was endotoxin shock resistant [294]. These results suggested that limiting factors determine the sensitivity of patients to endotoxin shock.
Rodents are genetically resistant to LPS. Therefore, naïve BALB/c mice are resistant to challenge with high doses of LPS (100 μg/mouse). However, BALB/c mice primed with heat-killed P. acnes, a Gram-positive skin habituating bacterium, or BCG, become highly susceptible to the lethal shock-inducing effect of LPS. Furthermore, upon LPS (1 μg/mouse) challenge, they rapidly produced IL-1, TNF-α and IL-6 and died of endotoxin shock or, if they survived, they suffered from acute liver injury through apoptosis-mediated hepatocytotoxicity [294,295]. Moreover, P. acnes-primed mice became highly susceptible to the lethal shock-inducing effects of IL-1 and TNF-α, producing high levels of IL-6 and dying after challenge with IL-1 and TNF-α. Therefore, priming with P. acnes or BCG induced lethal endotoxin shock in mice highly susceptible to LPS by the enhanced production of IL-1, TNF-α, and IL-6.
After publishing these results [294], we observed that P. acnes-primed BALB/c nu/nu mice were resistant to LPS-induced lethal shock, but died of fulminant hepatitis [1]. However, nu/nu mice reconstituted with splenic T cells died of lethal shock before the development of fulminant hepatitis after sequential treatment with P. acnes and LPS [296]. Therefore, P. acnes pretreatment rendered mice highly susceptible to the lethal shock-inducing effect of LPS by the induction of Th1 cells. Indeed, IL-12p40-deficient mice or IFN-γ-deficient mice were highly resistant to P. acnes-primed and LPS-challenged endotoxin shock, revealing the importance of IFN-γ as a limiting factor to determine the sensitivity to LPS shock.
5.1.2. LPS-Induced Liver Injury in P. acnes-Primed Mice
As noted above, P. acnes-primed and LPS-challenged nu/nu mice eventually died of fulminant hepatitis. However, the administration of anti-IL-18 Ab prevented LPS-induced liver injury in P. acnes-primed nu/nu mice [1]. We found that IL-18 induced FasL expression on Th1 cells, NK cells and unique liver T cells. Therefore, IL-18 is a key player in LPS-induced liver injury and induced fulminant hepatitis through Fas-mediated hepatocytotoxicity [295]. Indeed, P. acnes-primed IL-18-deficient mice were resistant to liver injury after LPS challenge. However, the administration of IL-18 induced liver injury in P. acnes-primed IL-18-deficient mice via the induction of FasL and TNF-α [63]. Therefore, we are very interested in how IL-18 is released after LPS challenge in P. acnes-primed mice [295].
Wild type mice primed with P. acnes developed dense granulomas in the liver, and developed acute liver injury when subsequently challenged with a sublethal dose of LPS [295]. These mice had elevated serum IL-18 levels after LPS challenge. Furthermore, P. acnes-primed IL-18-deficient mice exhibited granulomas in the liver comparable with P. acnes-primed WT mice, but were resistant to acute hepatitis induced by LPS. In contrast, MyD88-deficient mice, which lack signaling common to many TLRs as well as IL-18/IL-1β signaling, primed with P. acnes had low hepatic granuloma formation and undetectable levels of IL-18 after LPS challenge [57], although MyD88-deficient Kupffer cells secreted IL-18 in response to LPS in vitro [56,295]. Therefore, we examined the contribution of TRIF for P. acnes-induced hepatic granuloma formation and LPS-induced IL-18 secretion [57]. Unlike MyD88-deficient mice, P. acnes-primed TRIF-deficient mice normally develop hepatic dense granulomas, but do not release IL-18 or develop liver injury. Therefore, we concluded that P. acnes treatment induced hepatic granuloma formation that was dependent on MyD88. Subsequent LPS challenge activated caspase-1 via the NLRP3 inflammasome and induced IL-18 release, which was dependent on TRIF, eventually leading to liver injury [57].
5.2. IL-18 in Allergy
5.2.1. Induction of IgE Production by IL-18
The daily administration of IL-18, especially with IL-2, markedly increased serum levels of IgE in naïve wild type mice [132]. An in vitro study revealed the increased expression of CD40L and production of IL-4 in CD4+NK1.1+ T cells stimulated with IL-2 and IL-18. These IL-18-stimulated NKT cells induced the development of naïve B cells into IgG1 and IgE-producing cells by the simultaneous stimulation of B cells with CD40L and IL-4 [134].
5.2.2. Innate-Type Allergic Inflammation Induced by IL-18
We established transgenic mice overexpressing human caspase-1 in keratinocytes (KCasp1-Tg). These mice spontaneously produced IL-18 and IgE, and developed atopic dermatitis (AD)-like skin lesions. Stat6-deficient KCasp1 Tg mice did not produce IgE, but still developed similar skin lesions. Therefore, the overproduction of IL-18 from keratinocytes induces skin lesion even in the absence of IgE [71]. We described this inflammation as “innate-type allergic inflammation” [135].
5.2.3. The Induction of IFN-γ and IL-13 Producing Super Th1 Cells by IL-2 and IL-18
Th1 cells produce both Th1 cytokines (IFN-γ) and Th2 cytokines (IL-9 and IL-13) in response to IL-18 plus IL-2. Furthermore, the intranasal administration of Ag, IL-2 and IL-18 to naïve mice bearing resting Th1 memory cells induced the development of airway inflammation and hyperresponsiveness [297]. We found that upon challenge with Ag, IL-2 and IL-18, resting memory Th1 cells produced both Th1 cytokines (IFN-γ) and Th2 cytokines (IL-9 and IL-13), which induced severe bronchial asthma. The administration of Ag and LPS also induced bronchial asthma by the induction of endogenous IL-18 from LPS-stimulated bronchial epithelial cells [298]. Therefore, Th1 cells, after stimulation with Ag and IL-18, become harmful cells that produce IFN-γ and IL-13, which induced difficult to control bronchial asthma [297,298]. We termed pathological Th1 cells as “super Th1 cells”, because they induced difficult to control asthma or AD-like skin lesions. This prominent feature of IL-18 might explain the mechanism for infection-associated allergic diseases.
5.2.4. Bronchial Asthma Induced by the Intranasal Administration of IL-2 and IL-18
The nasal administration of IL-2 and IL-18 induced airway hyperresponsiveness, pulmonary eosinophilia, and goblet cell hyperplasia in wild type mice, but not in Rag2-deficient mice [299]. However, the nasal administration of IL-33 induced similar changes in wild type mice and Rag2-deficient mice [300]. Therefore, IL-2 plus IL-18 induced pulmonary changes in a T cell-dependent manner, while IL-33 treatment induced the same changes in a T cell-independent and innate cell-dependent manner.
5.3. IL-18 in Kidney Diseases
IL-18 is well documented as being involved in various types of kidney diseases. For example, mice deficient in Il18 or those administered neutralizing anti-IL-18 Ab are resistant to acute kidney disease induced by ischemia/reperfusion [301] or by cisplatin treatment [302]. IL-18 blockade was also shown to protect against chronic kidney disease in mice induced by unilateral ureteric obstruction [303]. Recent excellent review articles have addressed this issue, in particular, focusing on its role in inflammasomes [304,305,306,307]. Here, we describe two topics of IL-18: its role in human IgA nephropathy and IL-18 as a clinical biomarker of acute kidney injury (AKI) that influences long-term outcomes of cardiac surgery.
5.3.1. Association between Serum IL-18 Levels and Renal Prognosis in IgA Nephropathy
IgA nephropathy is a primary mesangial proliferative glomerulonephritis with the prevalent deposition of IgA in mesangial cells in the glomerulus. IgA nephropathy is regarded as a benign kidney disease. However, recent clinical studies revealed that IgA nephropathy had an extremely variable clinical course and that it led to end-stage renal disease with slow progression [308,309]. It was reported that serum IL-18 levels were a potent prognostic factor for IgA nephropathy [310]. Notably, serum concentrations of IL-18 in IgA nephropathy patients were significantly elevated compared with healthy controls [310]. Patients sensitive to corticosteroid therapy showed a significant reduction in serum levels of IL-18 after therapy, while patients resistant to therapy exhibited no reduction. Moreover, the renal survival of IgA nephropathy patients with higher than median serum IL-18 levels at baseline was approximately 20% at the end of the follow-up period (four years), and approximately 80% for total IgA nephropathy patients [310]. Furthermore, immunohistochemical analyses revealed that the intensity of IL-18 and NLRP3 proteins in renal biopsy samples from patients with IgA nephropathy correlated with the severity of proteinuria [311]. Therefore, serum IL-18 concentration might be a predictor for renal prognosis in this disease.
5.3.2. Urinary IL-18 as A Biomarker of AKI after Cardiac Surgery
Because murine tubular epithelial cells secrete IL-18 and contain the components required for inflammasome activation [305,312,313], urine IL-18 levels might be elevated after acute tubular injury in human [314,315]. Urinary IL-18 is now recognized as a biomarker for AKI [316]. AKI often occurs in adults and children undergoing cardiac surgery and is a risk factor for morbidity and mortality [317,318]. Levels of serum creatinine, a biomarker for the diagnosis of AKI, increase late in the course of the disease, delaying timely treatment. Many studies have investigated new AKI biomarkers and several urinary proteins including IL-18 have been identified as early AKI biomarkers [319]. Recently, urinary biomarkers of AKI, particularly IL-18, were reported to be an additional prognostic factor for long-term postoperative mortality. Urinary IL-18 levels on post cardiac surgery days 1-3 were well correlated with the mortality rate at three-year follow-up [320]. AKI contributes to multiple organ failures [321,322], suggesting that long-term postoperative mortality might be directly evoked by AKI. However, a recent study revealed that postoperative AKI might be indicative of cardiac vascular stress, rather than an independent renal pathway for adverse cardiovascular death [323].
5.4. IL-18 in Metabolic Disorders
Early clinical studies revealed that IL-18 levels were elevated in the circulation and atherosclerotic plaques of patients with atherosclerosis [324,325]. In a prospective study of 1229 patients with coronary artery disease, at the 4-year follow-up, serum IL-18 levels were significantly higher in patients with fatal cardiovascular events than in those who did not die [326]. A community-based prospective cohort study showed that plasma IL-18 levels were a predictor of coronary evens in healthy European men [327]. Recently, a meta-analysis of the association of IL-18 with coronary heart disease identified circulating IL-18 as a possible risk factor of cardiovascular disease [328]. These reports suggest IL-18 is involved in metabolic syndrome. However, as described above (4.3.2), during metabolic disorders caused by excess energy, the NLRP3 inflammasome is likely to be activated by aberrant lipid metabolites and/or high glucose levels, which subsequently results in the secretion of IL-18 as well as IL-1β, which can induce inflammatory responses [242]. In contrast, IL-18 has a neutral or beneficial role in triggering obesity-associated metabolic diseases as described above (4.3.2). Therefore, an increase in circulating IL-18 concentrations might be an indicator of the activation levels of the NLRP3 inflammasome in the early phase of disease. IL-18, together with IL-12 and/or IL-15, exerts proinflammatory effects such as the activation of Th1 cells and the induction of IFN-γ by various immune cells including NK cells. During the progress of metabolic syndrome, abnormal metabolites, such as oxidized LDL and hyperglycemia activate the TLR4 and/or TLR2-mediated pathways [230,329], potentially leading to the production of IL-12 and/or IL-15. Under these conditions, IL-18 might activate NK cells and/or Th1 cells to produce large amounts of IFN-γ and/or TNF-α [1,2,132,243,244].
5.5. IL-18 in Cancer
As initially reported, IL-18 activates NK cells to produce IFN-γ and enhance cytotoxicity against tumor cells in synergy with IL-12 [1,243,244,330]. Because NK cells, and recently identified innate lymphoid cells, are well-established tumor-killing cells [331], many researchers have addressed whether IL-18 therapy rescues cancer expansion [332,333,334]. Here, we describe two recent theories on the beneficial roles of IL-18 in protecting against cancer. One is the establishment of cancer therapy by IL-18-activated human γδT cells. The other topic is the importance of fungi in microbiota for protection against colitis-associated colorectal cancer by inducing IL-18.
5.5.1. IL-18 Robustly Expands Human γδT Cells
γδT cells have several innate cell-like properties [335,336]. For example, similar to αβT cells, γδT cells are activated upon T cell receptor (TCR) engagement. Whereas the TCR-mediated activation of αβT cells occurs in an MHC-restricted manner, the TCR engagement of γδT cells is independent of the MHC. To exert their biological function, naïve αβT cells require the appropriate differentiation into effector T cells, whereas γδT cells, including NK cells, can rapidly produce large amounts of cytokines and kill tumor cells. Indeed, γδT cells are well documented to exert tumoricidal activity [335,336,337]. However, low numbers of γδT ells are present in the peripheral blood of humans. Therefore, the bottleneck for the development of γδT cell-mediated cancer therapy has been the lack of an established method suitable for the efficient and safe expansion of γδT cells. Recently, Okamura’s group reported a protocol to obtain high numbers of γδT cells using IL-18 [338,339,340,341]. The incubation of human PBMCs including 1%–2% γδT cells with γδT cell Ag and IL-2 and IL-18, induced the proliferation of γδT cells, but not αβT cells, by approximately several thousand-fold in a 2-week culture [338,339,340,341] (Figure 6A). They used zoledronate as an activator of endogenous γδT cell Ag, which induces the accumulation of intermediate isopentenyl pyrophosphate, an endogenous γδT cell Ag, by blocking farnesyl pyrophosphate synthase in human monocytes [337,342,343]. The depletion of monocytes from PBMCs prevented the expansion of γδT cells [339]. Intriguingly, the IL-18-mediated expansion of human γδT cells requires CD56+CD11c+cells [338,339], initially termed NK-like dendritic cells (NKDCs) [344,345]. Indeed, CD56intCD11c+ cells in PBMCs co-cultured with monocytes in the presence of IL-12 and IL-18 robustly expanded and differentiated into CD56brightCD11c+ cells [338]. However, how CD56brightCD11c+ cells contribute to the expansion of γδT cells remains unknown. They also demonstrated that combination therapy with IL-18 and immune-checkpoint therapy with anti-PD-L1 and/or anti-CTLA4 mAb, synergistically prevented the mortality of mice harboring various tumor cell lines [346]. Combination therapy with IL-18 induced the expansion of precursor mature NK cells (counter cells of human CD56+CD11c+ cells) but did not affect regulatory T cells. The in vivo depletion of precursor mature NK cells or CD8+ T cells abrogated these therapeutic effects [346]. Therefore, IL-18 in combination with immune-checkpoint therapy might be a potential treatment for the early stages of cancer in humans.
5.5.2. Mycobiome-Mediated IL-18 Protects Against Colitis-Associated Colorectal Cancer
A recent report confirmed the anti-cancer effect of IL-18 released from macrophages in response to commensal fungi on colitis-associated cancer [347]. The microbiome contains fungi as well as bacteria and other microorganisms [348]. C-type lectin receptors including Dectin-1, Dectin-2, Dectin-3, and Mincle are expressed on host cells and recognize β-glucan and α-mannans expressed by fungi [349,350,351,352]. Upon ligation with their ligands, C-type lectin receptors recruit Syk kinase, followed by NF-κB and MAPK signaling by assembling the CARD9/MALT/BCL10 complex [353]. Malik et al. reported that the recognition of commensal fungi by C-type lectin receptor induced Syk-dependent CARD9 inflammasome activation induced the release of mature IL-18 [347] (Figure 6B). Card9−/− mice and mice selectively deficient for Syk in myeloid cells were predisposed to azoxymethane (AOM)/DSS-induced colitis-associated colorectal cancer, concomitant with reduced mature IL-18 in colon explants and an impaired accumulation of anti-tumorigenic T cells in the colon. Exogenous IL-18 prevented these mutant mice from colorectal cancer and restored the migration of anti-tumorigenic T cells. Of note, the administration of antifungal drugs rendered wild type mice highly susceptible to AOM/DSS-induced colitis-associated colorectal cancer, and supplementation with IL-18 rescued their predisposition to colorectal cancer [347]. These observations suggest the careful per os treatment of IBD patients with antifungal drugs might be of benefit. The depletion of mycobiota by antifungal drugs might initiate and/or promote colorectal cancer in IBD patients.
6. Similarities and Differences between IL-18 and IL-33
IL-33 is an IL-1 cytokine family member that uses ST2 and IL-1RacP (IL-1 receptor accessory protein) as a receptor to transduce signals via MyD88 [354], similar to IL-18. In contrast to IL-18R expressed on Th1 cells, which enhances IFN-γ production, ST2 is expressed on Th2 cells and enhances Th2 cytokine production. Furthermore, ST2 is expressed on various cells including ILC2s, mast cells, eosinophils, and basophils, and IL-33 stimulates these cells to induce Th2-type cytokines including IL-4, IL-5 and IL-13. By the action of these cytokines, IL-33 is involved in allergic inflammation and anthelminthic immunity by enhancing eosinophilic inflammation and mucus secretion [355].
Both IL-33 and IL-18 are released from epithelial cells or macrophages, but the individual production mechanisms are different. IL-33 exists as an active full-length form in the nucleus, which is released when cells undergo necrosis or are stressed [356]. In contrast, IL-18 stored in the cytoplasm as an inactive precursor and is released in response to stimulation by proteases such as caspase-1 [357]. When IL-18 is produced from macrophages by TLR stimulation, IL-12 may be also produced, to markedly induce the production of IFN-γ in vivo. Caspase-1 inactivates IL-33, and therefore, IL-33 is not produced when cells are activated to form an inflammasome by infection with bacteria harboring TLR ligands [358]. However, granule proteases in neutrophils and mast cells cleave IL-18 and IL-33 to enhance their activity [69,70,359,360]. IL-18 and IL-33 can stimulate basophils and mast cells to produce IL-4 and IL-13 and might be involved in Th2 type inflammation [300].
Mice overexpressing IL-18 or IL-33 in the skin developed spontaneous dermatitis, although their pathogenic phenotypes are different. Eosinophil infiltration was frequently observed in the lesion areas of skin-specific IL-33 transgenic mice [361], while neutrophil infiltration was mainly observed in skin-specific IL-18 transgenic mice [71]. The nasal administration of IL-2 and IL-18 induced airway hyperresponsiveness, pulmonary eosinophilia and goblet cell hyperplasia in wild type mice, but not in Rag2-deficient mice [299]. However, the administration of IL-33 induced these changes in wild type and Rag2-deficient mice [300]. IL-33 increased the number of ILC2s and promoted the production of IL-5 and IL-13 independent of T cells, thereby inducing lung eosinophilia and goblet cell hyperplasia. Therefore, IL-33 plays an important role in inducing Th2 cell-dependent and ILC2-dependent allergic diseases. ILC2s are activated by IL-33 during nematode infection. Infection of the lung by the intestinal nematode S. venezuelensis increases the number of IL-33-producing alveolar epithelium type II (ATII) cells in wild type and Rag2-deficient mice, which develop eosinophilic inflammation and goblet cell hyperplasia (Loeffler syndrome) [362]. Furthermore, ILC2s induced by S. venezuelensis became memory-like ILC2s, with a higher reactivity than ILC2s from naïve mice, and protected against new parasite infections by a non-specific mechanism [363]. As described above, lung ILC2s have strong reactivity to IL-33, but recently, differences in ILC2s isolated from specific organs were reported. ILC2s in the skin express IL-18Rα [364], and the effect of IL-18 on skin ILC2s is expected to be clarified in future studies.
7. IL-18 as A Therapeutic Target
Because of the strong proinflammatory activity of IL-18, many researchers are investigating IL-18 as a therapeutic target for the treatment of inflammatory diseases. To neutralize IL-18, IL-18 BP or anti-IL-18 Ab formulations were devised and clinical trials have been conducted to verify its safety and efficacy [365,366]. Clinical trials are underway to investigate the treatment of adult-onset Still’s disease and NLRC4-related macrophage activation syndrome (inflammatory diseases associated with high plasma IL-18 levels) using IL-18BP [120,121,122] (ClinicalTrials.gov Identifier: NCT 02398435, NCT 03113760).
In addition, the immunostimulatory effects of IL-18 have been investigated for treatments. The first attempts to administer IL-18 to cancer patients showed that its toxicity was generally mild-to-moderate [367]. For optimal cancer therapy, combination with other therapies is being considered. Anti-CD20 Ab is used to treat CD20 positive B cell lymphoma. Clinical studies using IL-18 with an anti-CD20 Ab are underway, and it was reported that the effect of anti-CD20 Ab was enhanced by the administration of IL-18 [368].
Although it is still at the stage of animal experiments, a new treatment method using IL-18 for cancer treatment has been studied. Recently, immune-checkpoint therapy by the neutralization of PD-1 or CTLA4 has dramatically improved cancer treatment. Combination therapy with IL-18 and an immune-checkpoint inhibitor synergistically reduced mortality in mice harboring various tumor cell lines. Therefore, IL-18 in combination with immune-checkpoint therapy might be a potential treatment for the early stages of cancer in humans [346]. Furthermore, chimeric antigen receptor (CAR) T cells artificially expressing a cancer antigen-specific TCR were effective treatments for B cell lymphoma and leukemia [369,370]. Studies on the effect on tumors of expressing IL-18 in CAR T cells in mice, demonstrated that IL-18 enhanced the antitumor effect [371,372]. These new therapy methods are expected to be applied to humans in the future and to save those suffering from cancer.
Funding
This research received no external funding.
Acknowledgments
We express our sincere gratitude to William Paul and Tadamitsu Kishimoto for their great help in all stages of our research. We also thank all members of the Department of Immunology and Medical Zoology, Hyogo College of Medicine, and all of our collaborators inside and outside of Japan. We also thank J. Ludovic Croxford, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Ag | Antigen |
| AIM2 | Absence in melanoma 2 |
| AKI | Acute kidney injury |
| AMPK | Adenosine monophosphate-activated protein kinase |
| AP-1 | Activator protein-1 |
| ASC | Apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain |
| BCG | Bacillus Calmette–Guerin |
| Bcl6 | B cell lymphoma 6 |
| Btk | Bruton’s tyrosine kinase |
| CTL | Cytotoxic T lymphocyte |
| ds | Double-stranded |
| DSS | Dextran sulphate sodium |
| FasL | Fas ligand |
| HFD | High-fat diet |
| IBD | Inflammatory bowel disease |
| IFN | Interferon |
| IGIF | IFN-γ inducing factor |
| IL-18BP | IL-18 binding protein |
| IL-18R | IL-18 receptor |
| ILC | Innate lymphoid cell |
| iNOS | Inducible nitric oxide synthase |
| IRAK | IL-1R-associated kinase |
| LDL | Low-density lipoprotein |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| miRNA | MicroRNA |
| MyD88 | Myeloid differentiation primary response 88 |
| NAIP | NLR family of apoptosis inhibitory protein |
| NF-kB | Nuclear factor (NF)-κB |
| NK | Natural killer |
| NLR | Nucleotide-binding oligomerization domain (NOD)-like receptor |
| NLRC | NLR family CARD domain-containing protein |
| NLRP | Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing |
| NO | Nitric oxide |
| OVA | Ovalbumin |
| PBMC | Peripheral blood mononuclear cell |
| ROS | Reactive oxygen species |
| SNP | Single nucleotide polymorphism |
| SPF | Specific pathogen-free |
| STAT | Signaltransducer and activator of transcription |
| TCR | T cell receptor |
| TIR | TLR/IL-1R |
| TLR | Toll-like receptors |
| TNF | Tumor necrosis factor |
| TRAF6 | TNF receptor-activated factor 6 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
References
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef] [PubMed]
- Ushio, S.; Namba, M.; Okura, T.; Hattori, K.; Nukada, Y.; Akita, K.; Tanabe, F.; Konishi, K.; Micallef, M.; Fujii, M.; et al. Cloning of the cDNA for human IFN-g-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J. Immunol. 1996, 156, 4274–4279. [Google Scholar] [PubMed]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-g inducing factor mediated by interleukin-1b converting enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar] [CrossRef]
- Nakanishi, K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front. Immunol. 2018, 9, 763. [Google Scholar] [CrossRef]
- Bazan, J.F.; Timans, J.C.; Kastelein, R.A. A newly defined interleukin-1? Nature 1996, 379, 591. [Google Scholar] [CrossRef]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 processes IFN-g-inducing factor and regulates LPS-induced IFN-g production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef]
- Tone, M.; Thompson, S.A.; Tone, Y.; Fairchild, P.J.; Waldmann, H. Regulation of IL-18 (IFN-g-inducing factor) gene expression. J. Immunol. 1997, 159, 6156–6163. [Google Scholar]
- Kim, Y.-M.; Kang, H.-S.; Paik, S.-G.; Pyun, K.-H.; Anderson, K.L.; Torbett, B.E.; Choi, I. Roles of IFN consensus sequence binding protein and PU.1 in regulating IL-18 gene expression. J. Immunl. 1999, 163, 2000–2007. [Google Scholar]
- Takeuchi, M.; Okura, T.; Mori, T.; Akita, K.; Ohta, T.; Ikeda, M.; Ikegami, H.; Kurimoto, M. Intracellular production of interleukin-18 in human epithelial-like cell lines is enhanced by hyperosmotic stress in vitro. Cell Tissue Res. 1999, 297, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Giedraitis, V.; He, B.; Huang, W.-X.; Hillert, J. Cloning and mutation analysis of the human IL-18 promoter: A possible role of polymorphisms in expression regulation. J. Neuroimmunol. 2001, 112, 146–152. [Google Scholar] [CrossRef]
- Sánchez, E.; Palomino-Morales, R.J.; Ortega-Centeno, N.; Jiménez-Alonso, J.; González-Gay, M.A.; López-Nevot, M.A.; Sánchez-Román, J.; de Ramón, E.; Francisca González-Escribano, M.; Pons-Estel, B.A.; et al. Identification of a new putative functional IL18 gene variant through an association study in systemic lupus erythematosus. Hum. Mol. Genet. 2009, 18, 3739–3748. [Google Scholar] [CrossRef] [PubMed]
- Khripko, O.P.; Sennikova, N.S.; Lopatnikova, J.A.; Khripko, J.I.; Filipenko, M.L.; Khrapov, E.A.; Gelfgat, E.L.; Yakushenko, E.V.; Kozlov, V.A.; Sennikov, S.V. Association of single nucleotide polymorphisms in the IL-18 gene with production of IL-18 protein by mononuclear cells from healthy donors. Mediat. Inflamm. 2008, 2008, 309721. [Google Scholar] [CrossRef]
- Ye, B.H.; Lista, F.; Lo Coco, F.; Knowles, D.M.; Offit, K.; Chaganti, R.S.K.; Dalla-Favera, R. Alternations of a zink finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 1993, 262, 747–750. [Google Scholar] [CrossRef]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef]
- Takeda, N.; Arima, M.; Tsuruoka, N.; Okada, S.; Hatano, M.; Sakamoto, A.; Kohno, Y.; Tokuhisa, T. Bcl6 is a transcriptional repressor for the IL-18 gene. J. Immunol. 2003, 171, 426–431. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Robert Fabian, M.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Luiz Marques-Rocha, J.; Samblas, M.; Milagro, F.I.; Bressan, J.; Alfredo Marínez, J.; Marti, A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015, 29, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Sideras, P.; Smith, C.I.; Vorechovsky, I.; Chapman, V.; Paul, W.E. Colocalization of X-Linked Agammaglobulinemia and X-Linked Immunodeficiency Genes. Science 1993, 261, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, G.; Suffert, G.; Semaan, N.; Juncker, T.; Frenzel, L.; Gottenberg, J.E.; Sibilia, J.; Pfeffer, S.; Wachsmann, D. Bruton’s tyrosine kinase is involved in miR-346-related regulation of IL-18 release by lipopolysaccharide-activated rheumatoid fibroblast-like synoviocytes. J. Immunol. 2009, 182, 5088–5097. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, C.; Peng, Z.; Zhao, J.; Gong, G.; Tan, D. miR-197 Expression in Peripheral Blood Mononuclear Cells from Hepatitis B Virus-Infected Patients. Gut Liver 2013, 7, 335–342. [Google Scholar] [CrossRef]
- Alnemri, E.S.; Livingston, D.J.; Nicholson, D.W.; Salvesen, G.; Thornberry, N.A.; Wong, W.W.; Yuan, J. Human ICE/CED-3 protease nomenclature. Cell 1996, 87, 171. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2665–2672. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawer, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Tschopp, J.; Martinon, F.; Burns, K. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 2003, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermine-mediated programmed necrotic cell death. Trend Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Feng, S.; Fox, D.; Ming Man, S. Mechanisms of Gasdermin familly members in inflammasome signaling and cell death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef] [PubMed]
- Ming Man, S.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar]
- Lamkanfi, M.; Vande Walle, L.; Kanneganti, T.-D. Deregulated inflammasome signaling in disease. Immunol. Rev. 2011, 243, 163–173. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trend Biochem. Sci. 2016, 41, 1012–1020. [Google Scholar] [CrossRef]
- Mathur, A.; Hayward, J.A.; Ming Man, S. Molecular mechanisms of inflammasome signaling. J. Leukoc Biol 2018, 103, 233–257. [Google Scholar] [CrossRef]
- Place, D.E.; Kanneganti, T.-D. Recent advances in infllasome biology. Curr. Opin. Immunol. 2018, 50, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trend Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Scjnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Ratner, D.; Orning, M.P.A.; Lien, E. Bacterial secretion systems and regulation of inflammasome activation. J. Leukoc. Biol. 2017, 101, 165–181. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Duncan, J.A.; Canna, S.W. The NLRC4 inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liou, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Chrrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and fors a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.M.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. LPS-induced IL-18 secretion from murine Kupffer cells independently of MyD88 that is critically involved in induction of production of IL-12 and IL-1β. J. Immunol. 2001, 166, 2651–2657. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Tsutsui, H.; Yasuda, K.; Uchiyama, R.; Yumikura-Futatsugi, S.; Mitani, K.; Hayashi, S.; Akira, S.; Taniguchi, S.; Van Rooijen, N.; et al. Contribution of TIR domain-containing adapter inducing IFN-β-mediated IL-18 release to LPS-induced liver injury in mice. J. Hepatol. 2009, 51, 333–341. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yaginuma, K.; Tsutsui, H.; Sagara, J.; Guan, X.; Seki, E.; Yasuda, K.; Yamamoto, M.; Akira, S.; Nakanishi, K.; et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adapter molecules. Gene Cell 2004, 9, 1055–1067. [Google Scholar] [CrossRef]
- Tsutsui, H.; Imamura, M.; Fujimoto, J.; Nakanishi, K. The TLR4/TRIF-mediated activation of NLRP3 inflammasome underlies endotoxin-induced liver injury in mice. Gastroenterol. Res. Pract. 2010, 2010, 641865. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Asano, M.; Horai, R.; Iwakura, Y.; Nagata, S.; Suda, T. Caspase-1-independent IL-1b release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 1998, 4, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kayagaki, N.; Kuida, K.; Nakano, H.; Hayashi, N.; Takeda, K.; Matsui, K.; Kashiwamura, S.-I.; Hada, T.; Akira, S.; et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999, 11, 359–367. [Google Scholar] [CrossRef]
- Uchiyama, R.; Yonehara, S.; Tsutsui, H. Fas-mediated inflammatory response in Listeria monocytogenes infection. J. Immunol. 2013, 190, 4245–4254. [Google Scholar] [CrossRef] [PubMed]
- Bassaller, L.; Chiang, P.-I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.K.; Mocarski, E.S.; Subramanan, D.; Green, D.R.; Silverman, N.; et al. Fas (CD95) mediates noncanonical IL-1b and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef]
- Uchiyama, R.; Tsutsui, H. Caspases as the key effectors of inflammatory responses against bacterial infection. Arch. Immunol. Ther. Exp. 2015, 63, 1–13. [Google Scholar] [CrossRef]
- Uchiyama, R.; Yonehara, S.; Taniguchi, S.; Ishido, S.; Ishii, K.J.; Tsutsui, H. Inflammasome and Fas-Mediated IL-1beta Contributes to Th17/Th1 Cell Induction in Pathogenic Bacterial Infection In Vivo. J. Immunol. 2017, 199, 1122–1130. [Google Scholar] [CrossRef]
- Kao, R.C.; Wehner, N.G.; Skubitz, K.M.; Gray, B.H.; Hoidal, J.R. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J. Clin. Investig. 1988, 1963–1973. [Google Scholar] [CrossRef]
- Sugawara, S.; Uehara, A.; Nochi, T.; Yamaguchi, T.; Ueda, H.; Sugiyama, A.; Hanzawa, K.; Kumagai, K.; Okamura, H.; Takada, H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J. Immunol. 2001, 167, 6568–6575. [Google Scholar] [CrossRef]
- Omoto, Y.; Tokime, K.; Yamanaka, K.; Habe, K.; Morioka, T.; Kurokawa, I.; Tsutsui, H.; Yamanishi, K.; Nakanishi, K.; Mizutani, H. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J. Immunol. 2006, 177, 8315–8319. [Google Scholar] [CrossRef]
- Konishi, H.; Tsutsui, H.; Murakami, T.; Yumikura-Futatsugi, S.; Yamanaka, K.; Tanaka, M.; Iwakura, Y.; Suzuki, N.; Takeda, K.; Akira, S.; et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 11340–11345. [Google Scholar] [CrossRef] [PubMed]
- Irani, A.-M.; Bradford, T.R.; Kepley, C.L.; Schechter, N.M.; Schwartz, L.B. Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti-tryptase and anti-chymase antibodies. J. Histochem. Cytochem. 1989, 37, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Catalfamo, M.; Henkart, P.A. Perforin and the granule exocytosis cytotoxicity pathway. Curr. Opin. Immunol. 2003, 15, 522–527. [Google Scholar] [CrossRef]
- Chowdhury, D.; Lieberman, J. Death by a Thousand Cuts: Granzyme Pathways of Programmed Cell Death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef] [PubMed]
- Omoto, Y.; Yamanaka, K.; Tokime, K.; Kitano, S.; Kakeda, M.; Akeda, T.; Kurokawa, I.; Gabazza, E.C.; Tsutsui, H.; Katayama, N.; et al. Granzyme B is a novel interleukin-18 converting enzyme. J. Dermatol. Sci. 2010, 59, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Akeda, T.; Yamanaka, K.; Tsuda, K.; Omoto, Y.; Gabazza, E.C.; Mizutani, H. CD8+T cell granzyme B activates keratinocyte endogenous IL-18. Arch. Dermatol. Res. 2014, 306, 125–130. [Google Scholar] [CrossRef]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 1999, 10, 127–136. [Google Scholar] [CrossRef]
- Aizawa, Y.; Akita, K.; Taniai, M.; Torigoe, K.; Mori, T.; Nishida, Y.; Ushio, S.; Nukada, Y.; Tanimoto, T.; Ikegami, H.; et al. Cloning and expression of interleukin-18 binding protein. FEBS Lett. 1999, 445, 338–342. [Google Scholar] [CrossRef]
- Novick, D.; Rubinstein, M. The tale of soluble receptors and binding proteins: From bench to bedside. Cytokine & Growth Factor Rev. 2007, 18, 525–533. [Google Scholar]
- Kawashima, M.; Yamamura, M.; Taniai, M.; Yamauchi, H.; Tanimoto, T.; Kurimoto, M.; Amano, T.; Takeuchi, T.; Makino, H. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001, 44, 550–560. [Google Scholar] [CrossRef]
- Liao, T.L.; Chen, Y.M.; Hsieh, C.W.; Chen, H.H.; Lee, H.C.; Hung, W.T.; Tang, K.T.; Chen, D.Y. Upregulation of circulating microRNA-134 in adult-onset Still’s disease and its use as potential biomarker. Sci. Rep. 2017, 7, 4214. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Tsutsui, H.; Kawai, T.; Takeda, K.; Nakanishi, K.; Takeda, Y.; Akira, S. Generation of IL-18 receptor-deficient mice: Evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J. Immunol. 1999, 162, 5041–5044. [Google Scholar] [PubMed]
- Kim, S.H.; Reznikov, L.L.; Stuyt, R.J.L.; Selzman, C.H.; Fantuzzi, G.; Hoshino, T.; Young, H.A.; Dinarello, C.A. Functional reconstitution and regulation of IL-18 activity by the IL-18Rb chain. J. Immunol. 2001, 166, 148–154. [Google Scholar] [PubMed]
- Wu, C.; Sakorafas, P.; Miller, R.; McCarthy, D.; Scesney, S.; Dixon, R.; Ghayur, T. IL-18 receptor beta-induced changes in the presentation of IL-18 binding sites affect ligand binding and signal transduction. J. Immunol. 2003, 170, 5571–5577. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, N.; Kimura, T.; Arita, K.; Ariyoshi, M.; Ohnishi, H.; Yamamoto, T.; Zuo, X.; Maenaka, K.; Park, E.Y.; Kondo, N.; et al. The structural basis for receptor recognition of human interleukin-18. Nat. Commun. 2014, 5, 5340. [Google Scholar] [CrossRef] [PubMed]
- Tomura, M.; Maruo, S.; Mu, J.; Zhou, X.Y.; Ahn, H.J.; Hamaoka, T.; Okamura, H.; Nakanishi, K.; Clark, S.; Kurimoto, M.; et al. Differential capacities of CD4+, CD8+, and CD4-CD8- T cell subsets to express IL-18 receptor and produce IFN-gamma in response to IL-18. J. Immunol. 1998, 160, 3759–3765. [Google Scholar] [PubMed]
- Nakahira, M.; Tomura, M.; Iwasaki, M.; Ahn, H.-J.; Bian, Y.; Hamaoka, T.; Ohta, T.; Kurimoto, M.; Fujiwara, H. An absolute requirement for STAT4 and a role for IFN-g as an amplifying factor in IL-12 induction fo the functional IL-18 receptor complex. J. Immunol. 2001, 167, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, M.A.; Merlin, T.; Kalis, C.; Chvatchko, Y.; Stübig, H.; Galanos, C. A murine, IL-12-independent pathway of IFN-g induction by gram-negative bacteria based on STAT4 activation by type 1 IFN and IL-18 signaling. J. Immunol. 2002, 169, 1665–1668. [Google Scholar] [CrossRef]
- Matikainen, S.; Paananen, A.; Miettinen, M.; Kurimoto, M.; Timonen, T.; Julkunen, I.; Sareneva, T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: Differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur. J. Immunol. 2001, 31, 2236–2245. [Google Scholar] [CrossRef]
- Bufler, P.; Azam, T.; Gamboni-Robertson, F.; Reznikov, L.L.; Kumar, S.; Dinarello, C.A.; Kim, S.H. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc. Natl. Acad. Sci. USA 2002, 99, 13723–13728. [Google Scholar] [CrossRef]
- Kumar, S.; Hanning, C.R.; Brigham-Burke, M.R.; Rieman, D.J.; Lehr, R.; Khandekar, S.; Kirkpatrick, R.B.; Scott, G.F.; Lee, J.C.; Lynch, F.J.; et al. Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 2002, 18, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Allen, J.L.; Tsen, M.; Dubnicoff, T.; Danao, J.; Liao, X.C.; Cao, Z.; Wasserman, S.A. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J. Immunol. 1999, 163, 978–984. [Google Scholar] [PubMed]
- Kanakaraj, P.; Ngo, K.; Wu, Y.; Angulo, A.; Ghazal, P.; Harris, C.A.; Siekierka, J.J.; Peterson, P.A.; Fung-Leung, W.-P. Defective interleukin (IL)-18-mediated natural killer and T helper cell type 1 responses in IL-1 receptor-associated kinase (IRAK)-deficient mice. J. Exp. Med. 1999, 189, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Chen, N.-J.; Miller, D.G.; Suzuki, S.; Horacek, T.; Hara, H.; Bouchard, D.; Nakanishi, K.; Penninger, J.M.; Ohashi, P.S.; et al. IRAK-4 is essential for interleukin-18-mediated natural killer and T helper cell type 1 response. J. Immunol. 2003, 170, 4031–4035. [Google Scholar] [CrossRef]
- Robinson, D.; Shibuya, K.; Mui, A.; Zonin, F.; Murphy, E.; Sana, T.; Hartley, S.B.; Menon, S.; Kastelein, R.; Bazan, F.; et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NF-kB. Immunity 1997, 7, 571–581. [Google Scholar] [CrossRef]
- Kalina, U.; Kauschat, D.; Koyama, N.; Nuernberger, H.; Ballas, K.; Koschmieder, S.; Bug, G.; Hofmann, W.K.; Hoelzer, D.; Ottmann, O.G. IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN-gamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk-21. J. Immunol. 2000, 165, 1307–1313. [Google Scholar] [CrossRef]
- El-Darawish, Y.; Li, W.; Yamanishi, K.; Pencheva, M.; Oka, N.; Yamanishi, H.; Matsuyama, T.; Tanaka, Y.; Minato, N.; Okamura, H. Frontline Science: IL-18 primes murine NK cells for proliferation by promoting protein synthesis, survival, and autophagy. J. Leukoc. Biol. 2018, 104, 253–264. [Google Scholar] [CrossRef]
- Deason, K.; Troutman, T.D.; Jain, A.; Challa, D.K.; Mandraju, R.; Brewer, T.; Ward, E.S.; Pasare, C. BCAP links IL-1R to the PI3K-mTOR pathway and regulates pathogenic Th17 cell differentiation. J. Exp. Med. 2018, 215, 2413–2428. [Google Scholar] [CrossRef]
- Fukao, T.; Tanabe, M.; Terauchi, Y.; Ota, T.; Matsuda, S.; Asano, T.; Kadowaki, T.; Takeuchi, T.; Koyasu, S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 2002, 3, 875–881. [Google Scholar] [CrossRef]
- Hosotani, Y.; Kashiwamura, S.; Kimura-Shimmyo, A.; Sekiyama, A.; Ueda, H.; Ikeda, T.; Mimura, O.; Okamura, H. Interleukin-18 prevents apoptosis via PI3K/Akt pathway in normal human keratinocytes. J. Dermatol. 2008, 35, 514–524. [Google Scholar] [CrossRef]
- Zhou, J.; Ping, F.F.; Lv, W.T.; Feng, J.Y.; Shang, J. Interleukin-18 directly protects cortical neurons by activating PI3K/AKT/NF-kappaB/CREB pathways. Cytokine 2014, 69, 29–38. [Google Scholar] [CrossRef]
- Kanayama, A.; Seth, R.B.; Sun, L.; Ea, C.K.; Hong, M.; Shaito, A.; Chiu, Y.H.; Deng, L.; Chen, Z.J. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell 2004, 15, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Horng, T.; Barton, G.M.; Flavell, R.A.; Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002, 420, 329–333. [Google Scholar] [CrossRef]
- Ohnishi, H.; Tochio, H.; Kato, Z.; Kawamoto, N.; Kimura, T.; Kubota, K.; Yamamoto, T.; Funasaka, T.; Nakano, H.; Wong, R.W.; et al. TRAM is involved in IL-18 signaling and functions as a sorting adaptor for MyD88. PLoS ONE 2012, 7, e38423. [Google Scholar] [CrossRef]
- Kim, S.H.; Eisenstein, M.; Reznikov, L.; Fantuzzi, G.; Novick, D.; Rubinstein, M.; Dinarello, C.A. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc. Natl. Acad. Sci. USA 2000, 97, 1190–1195. [Google Scholar] [CrossRef]
- Faggioni, R.; Cattley, R.C.; Guo, J.; Flores, S.; Brown, H.; Qi, M.; Yin, S.; Hill, D.; Scully, S.; Chen, C.; et al. IL-18-binding protein protects against lipopolysaccharide-induced lethality and prevents the development of Fas/Fas ligand-mediated models of liver disease in mice. J. Immunol. 2001, 167, 5913–5920. [Google Scholar] [CrossRef]
- Banda, N.K.; Vodracek, A.; Kraus, D.; Dinarello, C.A.; Kim, S.-H.; Bendele, A.; Senaldi, G.; Arend, W.P. Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J. Immunol. 2003, 170, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, B.; Sennello, J.A.; Lehr, H.A.; Senaldi, G.; Dinarello, C.A.; Fantuzzi, G. Frontline: Interferon regulatory factor-1 as a protective gene in intestinal inflammation: Role of TCR gamma delta T cells and interleukin-18-binding protein. Eur. J. Immunol. 2004, 34, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, P.; Phillips, J.; Conget, I.; Cooke, A.; Nicoletti, F. IL-18 binding protein fusion construct delays the development of diabetes in adoptive transfer and cyclophosphamide-induced diabetes in NOD mouse. Clin. Immunol. 2005, 115, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Elbirt, D.; Miller, G.; Dinarello, C.A.; Rubinstein, M.; Sthoeger, Z.M. High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J. Autoimmun. 2010, 34, 121–126. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Lu, L.; Altmann, C.; Hoke, T.S.; Ljubanovic, D.; Jani, A.; Dinarello, C.A.; Faubel, S.; Edelstein, C.L. Interleukin-18 binding protein transgenic mice are protected against ischemic acute kidney injury. Am. J. Physiol. Renal Physiol. 2008, 295, F1414–F1421. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Siong Low, J.; Harman, C.C.D.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Immunity 2015, 163, 1444–1456. [Google Scholar] [CrossRef]
- Harms, R.Z.; Creer, A.J.; Lorenzo-Arteaga, K.M.; Ostlund, K.R.; Sarvetnick, N.E. Interleukin (IL)-18 Binding Protein Deficiency Disrupts Natural Killer Cell Maturation and Diminishes Circulating IL-18. Front. Immunol. 2017, 8, 1020. [Google Scholar] [CrossRef]
- Novick, D.; Elbirt, D.; Dinarello, C.A.; Rubinstein, M.; Sthoeger, Z.M. Interleukin-18 binding protein in the sera of patients with Wegener’s granulomatosis. J. Clin. Immunol. 2009, 29, 38–45. [Google Scholar] [CrossRef]
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Priori, R.; Alessandri, C.; Perricone, C.; Pendolino, M.; Picarelli, G.; Valesini, G. IL-18 Serum Level in Adult Onset Still’s Disease: A Marker of Disease Activity. Int. J. Inflamm. 2012, 2012, 156890. [Google Scholar] [CrossRef]
- Gabay, C.; Fautrel, B.; Rech, J.; Spertini, F.; Feist, E.; Kotter, I.; Hachulla, E.; Morel, J.; Schaeverbeke, T.; Hamidou, M.A.; et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis. 2018, 77, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; Girard, C.; Malle, L.; de Jesus, A.; Romberg, N.; Kelsen, J.; Surrey, L.F.; Russo, P.; Sleight, A.; Schiffrin, E.; et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 2017, 139, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; McManus, M.; Grenz, A.; Collins, C.B.; Nold, M.F.; Nold-Petry, C.; Bufler, P.; Dinarello, C.A.; et al. Interleukin 37 expression protects mice from colitis. Proc. Natl. Acad. Sci. USA 2011, 108, 16711–16716. [Google Scholar] [CrossRef] [PubMed]
- Muhl, H.; Kampfer, H.; Bosmann, M.; Frank, S.; Radeke, H.; Pfeilschifter, J. Interferon-g mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem. Biophys. Res. Commun. 2000, 267, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Hurgin, V.; Novick, D.; Rubinstein, M. The promoter of IL-18 binding protein: Activation by an IFN-gamma -induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc. Natl. Acad. Sci. USA 2002, 99, 16957–16962. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Novick, D.; Rubinstein, M.; Siegmund, B.; Enrich, B.; Koch, R.O.; Vogel, W.; Kim, S.H.; Dinarello, C.A.; Tilg, H. Interferon-alpha induces interleukin-18 binding protein in chronic hepatitis C patients. Clin. Exp. Immunol. 2002, 129, 332–338. [Google Scholar] [CrossRef]
- Ludwiczek, O.; Kaser, A.; Novick, D.; Dinarello, C.A.; Rubinstein, M.; Vogel, W.; Tilg, H. Plasma levels of interleukin-18 and interleukin-18 binding protein are elevated in patients with chronic liver disease. J. Clin. Immunol. 2002, 22, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Nold-Petry, C.A.; Lehrnbecher, T.; Jarisch, A.; Schwabe, D.; Pfeilschifter, J.M.; Muhl, H.; Nold, M.F. Failure of interferon gamma to induce the anti-inflammatory interleukin 18 binding protein in familial hemophagocytosis. PLoS ONE 2010, 5, e8663. [Google Scholar] [CrossRef]
- Wittmann, M.; Doble, R.; Bachmann, M.; Pfeilschifter, J.; Werfel, T.; Muhl, H. IL-27 Regulates IL-18 binding protein in skin resident cells. PLoS ONE 2012, 7, e38751. [Google Scholar] [CrossRef]
- Carbotti, G.; Barisione, G.; Orengo, A.M.; Brizzolara, A.; Airoldi, I.; Bagnoli, M.; Pinciroli, P.; Mezzanzanica, D.; Centurioni, M.G.; Fabbi, M.; et al. The IL-18 antagonist IL-18-binding protein is produced in the human ovarian cancer microenvironment. Clin. Cancer Res. 2013, 19, 4611–4620. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Okamura, H.; Tagawa, Y.; Iwakura, Y.; Nakanishi, K. Interleukin 18 together with interleukin 12 inhibits IgE production by induction of interferon-g production from activated B cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3948–3953. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-g production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar] [PubMed]
- Yoshimoto, T.; Tsutsui, H.; Tominaga, K.; Hoshino, K.; Okamura, H.; Akira, S.; Paul, W.E.; Nakanishi, K. IL-18, although anti-allergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc. Natl. Acad. Sci. USA 1999, 96, 13962–13966. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Mizutani, H.; Tsutsui, H.; Noben-Trauth, N.; Yamanaka, K.; Tanaka, M.; Izumi, S.; Okamura, H.; Paul, W.E.; Nakanishi, K. IL-18 induction of IgE: Dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 2000, 1, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Yoshimoto, T.; Hayashi, N.; Mizutani, H.; Nakanishi, K. Induction of allergic inflammation by interleukin-18 in experimental animal models. Immunol. Rev. 2004, 202, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Yoshimoto, T.; Maruyama, H.; Tegoshi, T.; Ohta, N.; Arizono, N.; Nakanishi, K. IL-18 with IL-2 protects against Strongyloides venezuelensis infection by activating mucosal mast cell-dependent type 2 innate immunity. J. Exp. Med. 2005, 202, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, M.; Nakanishi, K. Requirement of GATA-binding protein 3 for II13 gene expression in IL-18-stimulated Th1 cells. Int. Immunol. 2011, 23, 761–772. [Google Scholar] [CrossRef]
- Kinoshita, M.; Miyazaki, H.; Ono, S.; Seki, S. Immunoenhancing therapy with interleukin-18 against bacterial infection in immunocompromised hosts after severe surgical stress. J. Leukoc. Biol. 2013, 93, 689–698. [Google Scholar] [CrossRef]
- Kinoshita, M.; Seki, S.; Ono, S.; Shinomiya, N.; Hiraide, H. Paradoxical effect of IL-18 therapy on the severe and mild Escherichia coli infections in burn-injured mice. Ann. Surg. 2004, 240, 313–320. [Google Scholar] [CrossRef]
- Kinoshita, M.; Kuranaga, N.; Matsumoto, A.; Ono, S.; Shinomiya, N.; Hiraide, H.; Seki, S. Multiple interleukin-18 injections promote both mouse Th1 and Th2 responses after sublethal Escherichia coli infection. Clin. Exp. Immunol. 2006, 143, 41–49. [Google Scholar] [CrossRef]
- Kinoshita, M.; Shinomiya, N.; Ono, S.; Tsujimoto, H.; Kawabata, T.; Matsumoto, A.; Hiraide, H.; Seki, S. Restoration of natural IgM production from liver B cells by exogenous IL-18 improves the survival of burn-injured mice infected with Pseudomonas aeruginosa. J. Immunol. 2006, 177, 4627–4635. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Yamagata, T.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M.; Kanamaru, A. Blockade of IL-18 receptor signaling delays the onset of autoimmune disease in MRL-Faslpr mice. J. Immunol. 2004, 173, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Miyazaki, H.; Ono, S.; Inatsu, A.; Nakashima, H.; Tsujimoto, H.; Shinomiya, N.; Saitoh, D.; Seki, S. Enhancement of neutrophil function by interleukin-18 therapy protects burn-injured mice from methicillin-resistant Staphylococcus aureus. Infect. Immun. 2011, 79, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, F.D.; Wynn, T.A.; Donaldson, D.D.; Urban, J.F. The role of IL-13 in helminth-induced inflammation and protective immunity against nematode infections. Curr. Opin. Immunol. 1999, 11, 420–426. [Google Scholar] [CrossRef]
- Grencis, R.K. Th2-mediated host protective immunity to intestinal nematode infections. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Helmby, H.; Takeda, K.; Akira, S.; Grencis, R.K. Interleukin (IL)-18 promotes the development of chronic gastrointestinal helminth infection by downregulating IL-13. J. Exp. Med. 2001, 194, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Hardman, M.J.; Grencis, R.K. The role of sex hormones in the development of Th2 immunity in a gender-biased model of Trichuris muris infection. Eur. J. Immunol. 2010, 40, 406–416. [Google Scholar] [CrossRef]
- Helmby, H.; Grencis, R.K. IL-18 regulates intestinal mastocytosis and Th2 cytokine production independently of IFN-g during Trichinella spiralis infection. J. Immunol. 2002, 169, 2553–2560. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nakata, N.; Kai, M.; Kasama, T.; Hanyuda, Y.; Hatano, Y. Decreased expression of cytokines that induce type 1 helper T cell/interferon-g responses in genetically susceptible mice infected with Mycobacterium avium. Clin. Immunol. Immunopathol. 1997, 85, 112–116. [Google Scholar] [CrossRef]
- Sugawara, I.; Yamada, H.; Kaneko, H.; Mizuno, S.; Takeda, K.; Akira, S. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 1999, 67, 2585–2589. [Google Scholar]
- Kinjo, Y.; Kawakami, K.; Uezu, K.; Yara, S.; Miyagi, K.; Koguchi, Y.; Hoshino, T.; Okamoto, M.; Kawase, Y.; Yokota, K.; et al. Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: A comparative study with IL-12p40. J. Immunol. 2002, 169, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.E.; Korbel, D.; Hagens, K.; Koch, M.; Raupach, B.; Enders, J.; Kaufmann, S.H.; Mittrucker, H.W.; Schaible, U.E. A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur. J. Immunol. 2010, 40, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ouyang, N.; Li, Q.H.; Luo, S.X.; He, Q.; Lei, H.; Liu, Q. The -137G/C single nucleotide polymorphism in IL-18 gene promoter contributes to tuberculosis susceptibility in Chinese Han population. Infect. Genet. Evol. 2015, 36, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, L.; Zhu, D.; An, H.; Yang, Y.; Liang, Y.; Zhao, W.; Ding, W.; Wu, X. Polymorphisms in the interleukin 18 receptor 1 gene and tuberculosis susceptibility among Chinese. PLoS ONE 2014, 9, e110734. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gao, C.; Chi, X.; Hu, Y.W.; Zheng, L.; Zeng, T.; Wang, Q. IL-37 Expression is Upregulated in Patients with Tuberculosis and Induces Macrophages Towards an M2-like Phenotype. Scand. J. Immunol. 2015, 82, 370–379. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, R.; Wang, P.; Yang, H.; He, X.; Ji, Q.; Bai, W.; Chen, H.; Chen, J.; Peng, W.; et al. IL-37 Confers Protection against Mycobacterial Infection Involving Suppressing Inflammation and Modulating T Cell Activation. PLoS ONE 2017, 12, e0169922. [Google Scholar] [CrossRef] [PubMed]
- Neighbors, M.; Xu, X.; Barrat, F.J.; Ruuls, S.R.; Churakova, T.; Debets, R.; Bazan, J.F.; Kastelein, R.A.; Abrams, J.S.; O’Garra, A. A critical role for interleukin 18 in primary and memory effector responses to Listeria monocytogenes that extends beyond its effects on Interferon gamma production. J. Exp. Med. 2001, 194, 343–354. [Google Scholar] [CrossRef]
- Seki, E.; Tsutsui, H.; Tsuji, N.M.; Hayashi, N.; Adachi, K.; Nakano, H.; Futatsugi-Yumikura, S.; Takeuchi, O.; Hoshino, K.; Akira, S.; et al. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 2002, 169, 3863–3868. [Google Scholar] [CrossRef]
- Maltez, V.I.; Tubbs, A.L.; Cook, K.D.; Aachoui, Y.; Falcone, E.L.; Holland, S.M.; Whitmire, J.K.; Miao, E.A. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 2015, 43, 987–997. [Google Scholar] [CrossRef]
- Lochner, M.; Kastenmuller, K.; Neuenhahn, M.; Weighardt, H.; Busch, D.H.; Reindl, W.; Forster, I. Decreased susceptibility of mice to infection with Listeria monocytogenes in the absence of interleukin-18. Infect. Immun. 2008, 76, 3881–3890. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Hara, H.; Fang, R.; Hernandez-Cuellar, E.; Sakai, S.; Daim, S.; Chen, X.; Dewamitta, S.R.; Qu, H.; Mitsuyama, M.; et al. The adaptor ASC exacerbates lethal Listeria monocytogenes infection by mediating IL-18 production in an inflammasome-dependent and -independent manner. Eur. J. Immunol. 2014, 44, 3696–3707. [Google Scholar] [CrossRef]
- Clark, S.E.; Filak, H.C.; Guthrie, B.S.; Schmidt, R.L.; Jamieson, A.; Merkel, P.; Knight, V.; Cole, C.M.; Raulet, D.H.; Lenz, L.L. Bacterial Manipulation of NK Cell Regulatory Activity Increases Susceptibility to Listeria monocytogenes Infection. PLoS Pathog. 2016, 12, e1005708. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Schmidt, R.L.; McDermott, D.S.; Lenz, L.L. A Batf3/Nlrp3/IL-18 Axis Promotes Natural Killer Cell IL-10 Production during Listeria monocytogenes Infection. Cell. Rep. 2018, 23, 2582–2594. [Google Scholar] [CrossRef] [PubMed]
- Soudja, S.M.; Ruiz, A.L.; Marie, J.C.; Lauvau, G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 2012, 37, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Nunez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Phalipon, A.; Arondel, J.; Thirumalai, K.; Banerjee, S.; Akira, S.; Takeda, K.; Zychlinsky, A. Caspase-1 activation of IL-1b and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 2000, 12, 581–590. [Google Scholar] [CrossRef]
- Mastroeni, P.; Clare, S.; Khan, S.; Harrison, J.A.; Hormaeche, C.E.; Okamura, H.; Kurimoto, M.; Dougan, G. Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulent Salmonella typhimurium. Infect. Immun. 1999, 67, 478–483. [Google Scholar]
- Raupach, B.; Peuschel, S.K.; Monack, D.M.; Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006, 74, 4922–4926. [Google Scholar] [CrossRef]
- Bohn, E.; Sing, A.; Zumbihl, R.; Bielfeldt, C.; Okamura, H.; Kurimoto, M.; Heesemann, J.; Autenrieth, I.B. IL-18 (IFN-g-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J. Immunol. 1998, 160, 299–307. [Google Scholar]
- Hein, J.; Sing, A.; Di Genaro, M.S.; Autenrieth, I.B. Interleukin-12 and interleukin-18 are indispensable for protective immunity against enteropathogenic Yersinia. Microb. Pathog. 2001, 31, 195–199. [Google Scholar] [CrossRef]
- Diefenbach, A.; Schindler, H.; Donhause, N.; Lorenz, E.; Laskay, T.; MacMicking, J.; Röllinghoff, M.; Gresser, I.; Bogdan, C. Type 1 interferon (IFNa/b) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 1998, 8, 77–87. [Google Scholar] [CrossRef]
- Ohkusu, K.; Yoshimoto, T.; Takeda, K.; Ogura, T.; Kashiwamura, S.; Iwakura, Y.; Akira, S.; Okamura, H.; Nakanishi, K. Potentiality of interleukin-18 as a useful reagent for treatment and prevention of Leishmania major infection. Infect. Immun. 2000, 68, 2449–2456. [Google Scholar] [CrossRef]
- Monteforte, G.M.; Takeda, K.; Rodriguez-Sosa, M.; Akira, S.; David, J.R.; Satoskar, A.R. Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. J. Immunol. 2000, 164, 5890–5893. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Karki, R.; Vogel, P.; Watanabe, M.; Bix, M.; Lamkanfi, M.; Kanneganti, T.D. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J. Clin. Investig. 2015, 125, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Basso, B.; Cervetta, L.; Moretti, E.; Carlier, Y.; Truyens, C. Acute Trypanosoma cruzi infection: IL-12, IL-18, TNF, sTNFR and NO in T. rangeli-vaccinated mice. Vaccine 2004, 22, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Graefe, S.E.; Jacobs, T.; Gaworski, I.; Klauenberg, U.; Steeg, C.; Fleischer, B. Interleukin-12 but not interleukin-18 is required for immunity to Trypanosoma cruzi in mice. Microbes Infect. 2003, 5, 833–839. [Google Scholar] [CrossRef]
- Rodrigues, A.A.; Saosa, J.S.; da Silva, G.K.; Martins, F.A.; da Silva, A.A.; Souza Neto, C.P.; Horta, C.V.; Zamboni, D.S.; da Silva, J.S.; Ferro, E.A.; et al. IFN-gamma plays a unique role in protection against low virulent Trypanosoma cruzi strain. PLoS Negl. Trop. Dis. 2012, 6, e1598. [Google Scholar] [CrossRef] [PubMed]
- Leon Rodriguez, D.A.; Carmona, F.D.; Echeverria, L.E.; Gonzalez, C.I.; Martin, J. IL18 Gene Variants Influence the Susceptibility to Chagas Disease. PLoS Negl. Trop. Dis. 2016, 10, e0004583. [Google Scholar] [CrossRef]
- Esper, L.; Utsch, L.; Soriani, F.M.; Brant, F.; Esteves Arantes, R.M.; Campos, C.F.; Pinho, V.; Souza, D.G.; Teixeira, M.M.; Tanowitz, H.B.; et al. Regulatory effects of IL-18 on cytokine profiles and development of myocarditis during Trypanosoma cruzi infection. Microbes Infect. 2014, 16, 481–490. [Google Scholar] [CrossRef]
- Nogueira, L.G.; Santos, R.H.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; Bocchi, E.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Myocardial gene expression of T-bet, GATA-3, Ror-gammat, FoxP3, and hallmark cytokines in chronic Chagas disease cardiomyopathy: An essentially unopposed TH1-type response. Mediat. Inflamm. 2014, 2014, 914326. [Google Scholar] [CrossRef]
- Nogueira, L.G.; Frade, A.F.; Ianni, B.M.; Laugier, L.; Pissetti, C.W.; Cabantous, S.; Baron, M.; Peixoto Gde, L.; Borges Ade, M.; Donadi, E.; et al. Functional IL18 polymorphism and susceptibility to Chronic Chagas Disease. Cytokine 2015, 73, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Liesenfeld, O.; Heimesaat, M.M. Immunology of Toxoplasma gondii. Immunol. Rev. 2011, 240, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-gamma: The major mediator of resistance against Toxoplasma gondii. Science 1988, 240, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Liesenfeld, O.; Kosek, J.; Remington, J.S.; Suzuki, Y. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J. Exp. Med. 1996, 184, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Kastelein, R.; Hunter, C.A. Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect. Immun. 2000, 68, 6932–6938. [Google Scholar] [CrossRef]
- Vossenkamper, A.; Struck, D.; Alvarado-Esquivel, C.; Went, T.; Takeda, K.; Akira, S.; Pfeffer, K.; Alber, G.; Lochner, M.; Forster, I.; et al. Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur. J. Immunol. 2004, 34, 3197–3207. [Google Scholar] [CrossRef]
- Schulthess, J.; Meresse, B.; Ramiro-Puig, E.; Montcuquet, N.; Darche, S.; Begue, B.; Ruemmele, F.; Combadiere, C.; Di Santo, J.P.; Buzoni-Gatel, D.; et al. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 2012, 37, 108–121. [Google Scholar] [CrossRef]
- Munoz, M.; Eidenschenk, C.; Ota, N.; Wong, K.; Lohmann, U.; Kuhl, A.A.; Wang, X.; Manzanillo, P.; Li, Y.; Rutz, S.; et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity 2015, 42, 321–331. [Google Scholar] [CrossRef]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Singh, R.P.; Kashiwamura, S.; Rao, P.; Okamura, H.; Mukherjee, A.; Chauhan, V.S. The role of IL-18 in blood-stage immunity against murine malaria Plasmodium yoelii 265 and Plasmodium berghei ANKA. J. Immunol. 2002, 168, 4674–4681. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.P.; Lepenies, B.; Kamena, F.; Holscher, C.; Freudenberg, M.A.; Burchard, G.D.; Wagner, H.; Kirschning, C.J.; Liu, X.; Seeberger, P.H.; et al. MyD88/IL-18-dependent pathways rather than TLRs control early parasitaemia in non-lethal Plasmodium yoelii infection. Microbes Infect. 2008, 10, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, K.A.; De Souza, J.B.; Riley, E.M. IL-18-induced expression of high-affinity IL-2R on murine NK cells is essential for NK-cell IFN-gamma production during murine Plasmodium yoelii infection. Eur. J. Immunol. 2015, 45, 3431–3440. [Google Scholar] [CrossRef] [PubMed]
- Torre, D.; Giola, M.; Speranza, F.; Matteelli, A.; Basilico, C.; Biondi, G. Serum levels of interleukin-18 in patients with uncomplicated Plasmodium falciparum malaria. Eur. Cytokine Netw. 2001, 12, 361–364. [Google Scholar] [PubMed]
- Nagamine, Y.; Hayano, M.; Kashiwamura, S.; Okamura, H.; Nakanishi, K.; Krudsod, S.; Wilairatana, P.; Looareesuwan, S.; Kojima, S. Involvement of interleukin-18 in severe Plasmodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 236–241. [Google Scholar] [CrossRef]
- Sareneva, T.; Matikainen, S.; Kurimoto, M.; Julkunen, I. Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J. Immunol. 1998, 160, 6032–6038. [Google Scholar] [PubMed]
- Wang, Y.; Chaudhri, G.; Jackson, R.J.; Karupiah, G. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J. Immunol. 2009, 183, 3324–3331. [Google Scholar] [CrossRef]
- Poli, G.; Fauci, A.S. The effect of cytokines and pharmacologic agents on chronic HIV infection. AIDS Res. Hum. Retrovir. 1992, 8, 191–197. [Google Scholar] [CrossRef]
- Shapiro, L.; Puren, A.J.; Barton, H.A.; Novick, D.; Peskind, R.L.; Shenkar, R.; Gu, Y.; Su, M.S.; Dinarello, C.A. Interleukin 18 stimulates HIV type 1 in monocytic cells. Proc. Natl. Acad. Sci. USA 1998, 95, 12550–12555. [Google Scholar] [CrossRef]
- Ahmad, R.; Sindhu, S.T.; Toma, E.; Morisset, R.; Ahmad, A. Elevated levels of circulating interleukin-18 in human immunodeficiency virus-infected individuals: Role of peripheral blood mononuclear cells and implications for AIDS pathogenesis. J. Virol. 2002, 76, 12448–12456. [Google Scholar] [CrossRef]
- Fujioka, N.; Akazawa, R.; Ohashi, K.; Fujii, M.; Ikeda, M.; Kurimoto, M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J. Virol. 1999, 73, 2401–2409. [Google Scholar] [PubMed]
- Harandi, A.M.; Svennerholm, B.; Holmgren, J.; Eriksson, K. Interleukin-12 (IL-12) and IL-18 are important in innate defense against genital herpes simplex virus type 2 infection in mice but are not required for the development of acquired gamma interferon-mediated protective immunity. J. Virol. 2001, 75, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chassaing, B.; Shi, Z.; Uchiyama, R.; Zhang, Z.; Denning, T.L.; Crawford, S.E.; Pruijssers, A.J.; Iskarpatyoti, J.A.; Estes, M.K.; et al. Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science 2014, 346, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.; Latz, E.; Moore, K.J.; Golenbock, D.T. The Nalp3 inflammsome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1b in type 2 diabetes. Nat. Immunol. 2010, 11, 897. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.B.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Jan Kullberg, B.; Tack, C.J.; van Krieken, H.; Kim, S.-H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Sanchez-Alavez, M.; Sugama, S.; Brennan, M.; Fernandez, R.; Bartfai, T.; Conti, B. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 11097–11102. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Conti, B. Interleukin-18 null mutation increases weight and food intake and reduces energy expenditure and lipid substrate utilization in high-fat diet fed mice. Brain Behav. Immun. 2014, 37, 45–53. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscle, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2015, 17, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. Assessmentofbrownadiposetissuefunction. Front. Physiol. 2013, 4, Article128. [Google Scholar]
- Pazos, P.; Lima, L.; Tovar, S.; Gonzalez-Touceda, D.; Dieguez, C.; Garcia, M.C. Divergent responses to thermogenic stimuli in BAT and subcutaneous adipose tissue from interleukin 18 and interleukin 18 receptor 1-deficient mice. Sci. Rep. 2015, 5, 17977. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P.; et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef]
- Lee, M.-W.; Odegaard, J.I.; Mukundan, L.; Qin, Y.; Molofsky, A.B.; Nussbaum, J.C.; Yun, K.; Locksley, R.M.; CHawla, A. Activated type 2 innate lyphoid cells regulate beige fat biogenesis. Cell 2015, 160, 74–87. [Google Scholar] [CrossRef]
- Ricardo-Gonzalez, R.R.; Van Dyken, S.J.; Shcneider, C.; Lee, J.; Nussbaum, J.C.; Liang, H.-E.; Vaka, D.; Eckalbar, W.L.; Molofsky, A.B.; Erle, D.J.; et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018, 19, 1093–1099. [Google Scholar] [CrossRef]
- Kjøbsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.-N.; Pehmøller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef]
- Lindegaard, B.; Matthews, V.B.; Brandt, C.; Hojman, P.; Allen, T.L.; Estevez, E.; Watt, M.J.; Bruce, C.R.; Mortensen, O.H.; Syberg, S.; et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes 2013, 62, 3064–3074. [Google Scholar] [CrossRef]
- Murphy, A.J.; Kraakman, M.J.; Kammoun, H.L.; Dragoljevic, D.; Lee, M.K.; Lawlor, K.E.; Wentworth, J.M.; Vasanthakumar, A.; Gerlic, M.; Whitehead, L.W.; et al. IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab. 2016, 23, 155–164. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trend Immunol. 2011, 32, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; sepahvand, F.; Rashidi, B.; Sahebkar, A.; Mosoudifar, A.; Mizaei, H. NLRP3 inflammasome: Its regulation and involvment in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018, 419, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephenes, J.M.; Deep Dixit, V. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Steinstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, H.; van de Veerdonk, F.L.; Perera, D.; Neeale, G.A.; Hooiveld, G.J.; Hijimans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Stewart, C.R.; stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.C.; Halle, A.; Ryaner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Klinkner, A.M.; Robbie Waites, C.; Kerns, W.D.; Bugelski, P.J. Evidence of form cell and cholesterol crystal foration in macrophages incubated with oxidized LDL by fluorescence and electron microscopy. J. Histochem. Cytochem. 1995, 43, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauemfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Tze-Han Huang, M.; June Brickey, W.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC nflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Koenen, T.B.; Stienstra, R.; can Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.H.; Joosten, L.A.B.; Tack, C.J.; Netea, M.G. Hyperglycemia Activates Caspase-1 and TXNIP-Mediated IL-1b Transcription in Human Adipose Tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef]
- Lee, H.-M.; Kim, J.-J.; Shong, M.; Jeong Ku, B.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Kursawe, R.; Dixit, V.D.; Scherer, P.E.; santoro, N.; Narayan, D.; Gordillo, R.; Giannini, C.; Lopez, X.; Pierpont, B.; Nouws, J.; et al. A role of the inflammasome in the low strage capacity of the abdominal subcutaneous adipose tissue in obese addolescents. Diabetes 2016, 65, 610–618. [Google Scholar] [CrossRef]
- Steinstra, R.; Joosten, L.A.B.; Koenen, T.B.; van Tits, B.; van Diepen, J.A.; van den Berg, S.; Rensen, P.C.N.; van Rooijen, N.; Wabitsch, M.; Kullber, B.-J.; et al. The Inflammasome-Mediated Caspase-1 Activation Controls Adipocyte Differentiation and Insulin Sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef]
- Netae, M.G.; Joosten, L.A.B. The NLRP1-IL18 Connection: A Stab in the Back of Obesity-Induced Inflammation. Cell Metab. 2016, 23, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [PubMed]
- Hyodo, Y.; Matsui, K.; Hayashi, N.; Tsutsui, H.; Kashiwamura, S.; Yamauchi, H.; Hiroishi, K.; Takeda, K.; Tagawa, Y.; Iwakura, Y.; et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999, 162, 1662–1668. [Google Scholar]
- Takeda, K.; Tsutsui, H.; Yoshimoto, T.; Adachi, O.; Yoshida, N.; Kishimoto, T.; Okamura, H.; Nakanishi, K.; Akira, S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 1998, 8, 383–390. [Google Scholar] [CrossRef]
- Castordi, A.; Naffah de Souza, C.; Olsen Saraiva Cåmara, N.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2016, 6, 637. [Google Scholar]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell 2016, 165, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Ka-Ming Chan, F. Inflammasome, Inflammation, and Tissue Homeostasis. Trend Mol. Med. 2018, 24, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Lei-Leston, A.C.; Murphy, A.G.; Maloy, K.J. Epithelial cell inflammasomes in intestinal immunity and inflammation. Front. Immunol. 2017, 8, 1168. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.; Franchi, L. Role of inflammasomes in intestinal inflammation and Crohn’s disease. Inflamm. Bowel. Dis. 2015, 21, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamet, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 65, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Kitani, A.; Strober, W.; Fuss, I.J. The Role of NLRP3 and IL-1beta in the Pathogenesis of Inflammatory Bowel Disease. Front. Immunol 2018, 9, 2566. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Nishizaki, Y.; Sano, O.; Ohta, T.; Ikeda, M.; Kurimoto, M. Immunohistochemical and immuno-electron-microscopic detection of interferon-g-inducing factor (interelukin-18) in mouse intestinal epithelial cells. Cell Tissue Res. 1997, 289, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, T.T.; Michie, M.H.; Bents, M.; Woraratanadharm, J.; Smith, M.F., Jr.; Foley, E.; Moskaluk, C.A.; Bickston, S.J.; Cominelli, F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: Expression and localization in intestinal mucosal cells. J. Immunol. 1999, 162, 6829–6835. [Google Scholar]
- Kempster, S.; Belteki, G.; Forhead, A.J.; Fowden, A.L.; Catalano, R.D.; Lam, B.Y.; McFarlane, I.; Stephen Charnock-Junes, D.; Smith, G.C. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G253–G263. [Google Scholar] [CrossRef]
- Knodler, L.A.; Crowley, S.M.; Pan Sham, H.; Yang, H.; Wrande, M.; Ma, C.; Emst, R.K.; Steele-Mortimer, O.; Celli, J.; Callance, B.A. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell Host Microbe 2014, 16, 249–256. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilbeman-Schapira, G.; Ali Mahdi, J.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Song-Zhao, G.X.; Srinivasan, N.; Pott, J.; Baban, D.; Frankel, G.; Maloy, K.J. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 2014, 7, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Deets, K.A.; Ji, D.X.; von Moltke, J.; Tenthorey, J.L.; Lee, A.Y.; Philip, N.H.; Ayres, J.S.; Brodsky, I.E.; Gronert, K.; et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity 2017, 46, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Nordiander, S.; Pott, J.; Moloy, K.J. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 2015, 7, 775–785. [Google Scholar] [CrossRef] [PubMed]
- sellin, M.E.; Müller, A.A.; Dolowschiak, T.; Diard, M.; Tardivel, A.; Maslowski, K.M.; Hardt, W.-D. Epithelium-Intrinsic NAIP/NLRC4 Inflammasome Drives Infected Enterocyte Expulsion to Restrict Salmonella Replication in the Intestinal Mucosa. Cell Host Microbe 2014, 16, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; HGrandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 2011, 108, 9601–9606. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, C.G.; Santee, C.A.; Lych, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kumahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of Colonic Regulatory T Cellsby Indigenous Clostridium Species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut Immune Maturation Depends on Colonization with a Host-Specific Microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Staley, D.; Luong, S.; Maruya, M.; McKenzie, C.I.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trend Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Somanna, N.K.; Yariswamy, M.; Garagliano, J.M.; Siebenlist, U.; Mummidi, S.; Valete, A.J.; Chadrasekar, B. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell. Signal. 2015, 27, 1928–1938. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.-P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 Inflammasome Orchestrates the Colonic Host-Microbial Interface by Regulating Goblet Cell Mucus Secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Wang, M.L.; Keilbaugh, S.A.; He, W.; Brenes, M.; Swain, G.P.; Knight, P.A.; Donaldson, D.D.; Lazar, M.A.; Miller, H.R.; et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2004, 101, 13596–13600. [Google Scholar] [CrossRef]
- Tsuji, S.; Uehori, J.; Matsumoto, M.; Suzuki, Y.; Matsuhisa, A.; Toyoshima, K.; Seya, T. Human intelectin is a novel soluble lectin that recognizes galactofuranose in carbohydrate chains of bacterial cell wall. J. Biol. Chem. 2001, 276, 23456–23463. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.E.; De Bruun, A.C.J.M.; Velcich, A.; Meuerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That Muc2 Is Critical for Colonic Protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodriguez-Plñlro, A.M.; Nystrøm, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Ushio, H.; Ueno, T.; Kojima, Y.; Komatsu, M.; Tanaka, S.; Yamamoto, A.; Ichimura, Y.; Ezki, J.; Nishida, K.; Komazawa-Sakon, S.; et al. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 2011, 127, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- consortium, T.i.F. Ancient Missense Mutations in a New Member of the RoRet Gene Family Are Likely to Cause Familial Mediterranean Fever. Cell 1997, 90, 797–807. [Google Scholar] [CrossRef]
- Consortium, T.F.F. A candidate gene for familial Mediterranean fever. Nat. Genet. 1997, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cattan, D.; Notarnicola, C.; Molinari, N.; Touitou, I. Inflammatory bowel disease in non-Ashkenazi Jews with familial Mediterranean fever. Lancet 2000, 355, 378–379. [Google Scholar] [CrossRef]
- Giaglis, S.; Mimidis, K.; Papadopoulos, V.; Thomopoulos, K.; Sidiropoulos, P.; Rafail, S.; Nikolopoulos, V.; Fragouli, E.; Kartalis, G.; Tzioufas, A.; et al. Increased Frequency of Mutations in the Gene Responsible for Familial Mediterranean Fever (MEFV) in a Cohort of Patients with Ulcerative Colitis: Evidence for a Potential Disease-Modifying Effect? Dig. Dis. Sci. 2006, 51, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Ming Man, S.; Kanneganti, T.-D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gavrillin, M.A.; Abdelaziz, D.H.A.; Mostafa, M.; Abdulrahman, B.A.; Grandhi, J.; Akhter, A.; Abu Khweek, A.; Aubert, D.F.; Valvano, M.A.; Wewers, M.D.; et al. Activation of the Pyrin Inflammasome by Intracellular Burkholderia cenocepacia. J. Immunol. 2012, 188, 3469–3477. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.-N.; Peng, X.; Jeff Xi, J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Sharma, D.; Malik, A.; Gui, C.S.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology 2018, 154, 948–964. [Google Scholar] [CrossRef]
- Hasan Zaki, M.; Vogel, P.; Body-Malapel, M.; Lamkafi, M.; Kanneganti, T.-D. IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. J. Immunol 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.-B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Pelletier, J. The biology of DHX9 and its potential as a therapeutic target. Oncotarget 2016, 7, 42716–42739. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Nakanishi, K.; Hirose, K.; Hiroishi, K.; Okamura, H.; Takemoto, Y.; Kanamaru, A.; Hada, T.; Tamura, T.; Kakishita, E.; et al. High serum IL-6 level reflects susceptible status of the host to endotoxin and IL-1/tumor necrosis factor. J. Immunol. 1992, 148, 3596–3603. [Google Scholar] [PubMed]
- Tsutsui, H.; Matsui, K.; Okamura, H.; Nakanishi, K. Pathophysiological roles of interleukin-18 for inflammatory liver diseases. Immunol. Rev. 2000, 174, 192–209. [Google Scholar] [CrossRef] [PubMed]
- Kawa, K.; Tsutsui, H.; Uchiyama, R.; Kato, J.; Matsui, K.; Iwakura, Y.; Matsumoto, T.; Nakanishi, K. IFN-g is a master regulator of endotoxin shock syndrome in mice primed with heat-killed Propionibacterium acnes. Int. Immunol. 2010, 22, 157–166. [Google Scholar] [CrossRef]
- Sugimoto, T.; Ishikawa, Y.; Yoshimoto, T.; Hayashi, B.; Fujimoto, J.; Nakanishi, K. IL-18 acts on memory Th1 cells to induce airway inflammatin and hyperresponsiveness in a naive host mouse. J. Exp. Med. 2004, 199, 535–545. [Google Scholar] [CrossRef]
- Hayashi, N.; Yoshimoto, T.; Izuhara, K.; Matsui, K.; Tanaka, T.; Nakanishi, K. T helper 1 cells stimulated with ovalbunmin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-g and IL-13 production. Proc. Natl. Acad. Sci. USA 2007, 104, 14765–14770. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Yoshimoto, T.; Nakanishi, K. Contribution of IL-18-induced innate T cell activation to airway inflammation with mucus hypersecretion and airway hyperresponsiveness. Int. Immunol. 2006, 18, 847–855. [Google Scholar] [CrossRef]
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-Yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lung in the absence of adaptive immune system. Int. Immunol. 2008, 20, 791–800. [Google Scholar] [CrossRef]
- Wu, H.; Craft, M.L.; Wang, P.; Wyburn, K.R.; Chen, G.; Ma, J.; Hambly, B.; Chadban, S.J. IL-18 contributes to renal damage after ischemia-reperfusion. J. Am. Soc. Nephrol. 2008, 19, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Ljubanovic, D.; Reznikov, L.; Somerset, H.; Dinarello, C.A.; Edelstein, C.L. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004, 66, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Masood, H.; Che, R.; Zhang, A. Inflammasomes in the Pathophysiology of Kidney Diseases. Kidney Dis. (Basel) 2015, 1, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Purves, J.T.; Hughes, F.M., Jr. Inflammasomes in the urinary tract: A disease-based review. Am. J. Physiol. Renal Physiol. 2016, 311, F653–F662. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.; Tan, L.; Miethke, T.; Anand, P.K. Immunity to uropathogens: The emerging roles of inflammasomes. Nat. Rev. Urol. 2017, 14, 284–295. [Google Scholar] [CrossRef]
- D’Amico, G. Natural history of idiopathic IgA nephropathy: Role of clinical and histological prognostic factors. Am. J. Kidney Dis. 2000, 36, 227–237. [Google Scholar] [CrossRef]
- Koyama, A.; Igarashi, M.; Kobayashi, M. Natural history and risk factors for immunoglobulin A nephropathy in Japan. Research Group on Progressive Renal Diseases. Am. J. Kidney Dis. 1997, 29, 526–532. [Google Scholar] [CrossRef]
- Shi, B.; Ni, Z.; Cao, L.; Zhou, M.; Mou, S.; Wang, Q.; Zhang, M.; Fang, W.; Yan, Y.; Qian, J. Serum IL-18 is closely associated with renal tubulointerstitial injury and predicts renal prognosis in IgA nephropathy. Mediat. Inflamm. 2012, 2012, 728417. [Google Scholar] [CrossRef]
- Liu, D.; Xu, M.; Ding, L.H.; Lv, L.L.; Liu, H.; Ma, K.L.; Zhang, A.H.; Crowley, S.D.; Liu, B.C. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, V.Y.; Ecder, T.; Fantuzzi, G.; Siegmund, B.; Lucia, M.S.; Dinarello, C.A.; Schrier, R.W.; Edelstein, C.L. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J. Clin. Investig. 2001, 107, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, V.Y.; Faubel, S.; Siegmund, B.; Lucia, M.S.; Ljubanovic, D.; Edelstein, C.L. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J. Clin. Investig. 2002, 110, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Jani, A.; Melnikov, V.Y.; Faubel, S.; Edelstein, C.L. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am. J. Kidney Dis. 2004, 43, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Mishra, J.; Thiessen-Philbrook, H.; Dursun, B.; Ma, Q.; Kelly, C.; Dent, C.; Devarajan, P.; Edelstein, C.L. Urinary IL-18 is an early predictive biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2006, 70, 199–203. [Google Scholar] [CrossRef]
- Schrezenmeier, E.V.; Barasch, J.; Budde, K.; Westhoff, T.; Schmidt-Ott, K.M. Biomarkers in acute kidney injury—Pathophysiological basis and clinical performance. Acta Physiol. (Oxf.) 2017, 219, 554–572. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis. 2009, 53, 961–973. [Google Scholar] [CrossRef]
- Li, S.; Krawczeski, C.D.; Zappitelli, M.; Devarajan, P.; Thiessen-Philbrook, H.; Coca, S.G.; Kim, R.W.; Parikh, C.R.; Consortium, T.-A. Incidence, risk factors, and outcomes of acute kidney injury after pediatric cardiac surgery: A prospective multicenter study. Crit. Care Med. 2011, 39, 1493–1499. [Google Scholar] [CrossRef]
- Zappitelli, M.; Greenberg, J.H.; Coca, S.G.; Krawczeski, C.D.; Li, S.; Thiessen-Philbrook, H.R.; Bennett, M.R.; Devarajan, P.; Parikh, C.R.; Translational Research Investigating Biomarker Endpoints in Acute Kidney Injury (TRIBE-AKI) Consortium. Association of definition of acute kidney injury by cystatin C rise with biomarkers and clinical outcomes in children undergoing cardiac surgery. JAMA Pediatr. 2015, 169, 583–591. [Google Scholar] [CrossRef]
- Coca, S.G.; Garg, A.X.; Thiessen-Philbrook, H.; Koyner, J.L.; Patel, U.D.; Krumholz, H.M.; Shlipak, M.G.; Parikh, C.R.; Consortium, T.-A. Urinary biomarkers of AKI and mortality 3 years after cardiac surgery. J. Am. Soc. Nephrol. 2014, 25, 1063–1071. [Google Scholar] [CrossRef]
- Feltes, C.M.; Van Eyk, J.; Rabb, H. Distant-organ changes after acute kidney injury. Nephron Physiol. 2008, 109, p80–84. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J. Distant effects of experimental renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2003, 14, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Puthumana, J.; Shlipak, M.G.; Koyner, J.L.; Thiessen-Philbrook, H.; McArthur, E.; Kerr, K.; Kavsak, P.; Whitlock, R.P.; Garg, A.X.; et al. Relationship of Kidney Injury Biomarkers with Long-Term Cardiovascular Outcomes after Cardiac Surgery. J. Am. Soc. Nephrol. 2017, 28, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Hulthe, J.; McPheat, W.; Samnegård, A.; Tornvall, P.; Hamsten, A.; Eriksson, P. Plasma interleukin (IL)-18 concentrations is elevated in patients with previous myocardial infarction and related to severity of coronary atherosclerosis independently of C-reactive protein and IL-6. Atherosclerosis 2006, 188, 4450–4454. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leséche, G.; Chvatchko, Y.; Tedgui, A. Expression of Interleukin-18 in human atherosclerotic plaque and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht, H.J.; AtheroGene, I. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Luc, G.; Ducimetiére, P.; Arveiler, D.; Ferriéres, J.; Amouyel, P.; Evans, A.; Cambien, F.; Tiret, L. Interleukin-18 and the risk of coronary heart disease in european men: The prospective epidemiological study of myocardial infarction (PRIME). Circulation 2003, 108, 2453–2459. [Google Scholar] [CrossRef]
- Jefferis, B.J.M.H.; Papacosta, O.; Owen, C.G.; Toya Wannamethee, S.; Humphries, S.E.; Woodward, M.; Lennon, L.T.; Thomson, A.; Welsh, P.; Rumley, A.; et al. Interleukin 18 and coronary heart disease: Prospective study and systematic review. Atherosclerosis 2011, 217, 227–233. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaray, S.; Zhao, L.; Hwang, D.H.; Jialal, I. High Glucose Induces Toll-Like Receptor Expression in Human Monocytes. Diabetes 2008, 57, 3090–3098. [Google Scholar] [CrossRef]
- Okamura, H.; Kashiwamura, S.; Tsutsui, H.; Yoshimoto, T.; Nakanishi, K. Regulation of interferon-g production by IL-12 and IL-18. Curr. Opin. Immunol. 1998, 10, 259–264. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Granzin, M.; Wagner, J.; Kohl, U.; Cerwenka, A.; Huppert, V.; Ullrich, E. Shaping of Natural Killer Cell Antitumor Activity by Ex Vivo Cultivation. Front. Immunol. 2017, 8, 458. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Cheon, S.; Cho, D. The Dual Effects of Interleukin-18 in Tumor Progression. Cell. Mol. Immunol 2007, 4, 329–335. [Google Scholar] [PubMed]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Anderson, J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1409. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Merli, P.; Rutella, S. At the Bedside: Innate immunity as an immunotherapy tool for hematological malignancies. J. Leukoc. Biol. 2013, 94, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kubo, S.; Okuda, A.; Yamamoto, H.; Ueda, H.; Tanaka, T.; Nakamura, H.; Yamanishi, H.; Terada, N.; Okamura, H. Effect of IL-18 on Expansion of gd T Cells Stimulated by Zoledronate and IL-2. J. Immunother. 2010, 33, 287–296. [Google Scholar] [CrossRef]
- Tsuda, J.; Li, W.; Yamanishi, H.; Yamamoto, H.; Okuda, A.; Kubo, S.; Ma, Z.; Terada, N.; Tanaka, Y.; Okamura, H. Involvement of CD56brightCD11c+ Cells in IL-18–Mediated Expansion of Human gd T Cells. J. Immunol. 2011, 186, 2003–2012. [Google Scholar] [CrossRef]
- Li, W.; Okuda, A.; Yamamoto, H.; Yamanishi, K.; Terada, N.; Yamanishi, H.; Tanaka, Y.; Okamura, H. Regulation of Development of CD56brightCD11c+ NK-like Cells with Helper Function by IL-18. PLoS ONE 2013, 8, e82586. [Google Scholar] [CrossRef]
- Sugie, T.; Murata-Hirai, K.; Iwasaki, M.; Morita, C.T.; Li, W.; Okamura, H.; Minato, N.; Toi, M.; Tanaka, Y. Zoledronic acid-induced expansion of gammadelta T cells from early-stage breast cancer patients: Effect of IL-18 on helper NK cells. Cancer Immunol. Immunother. 2013, 62, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T Cell Receptor gamma delta Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Jauhiainen, M.; Monkkonen, H.; Rogers, M.J.; Monkkonen, J.; Thompson, K. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 2009, 144, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Guimont-Desrochers, F.; Boucher, G.; Dong, Z.; Dupuis, M.; Veillette, A.; Lesage, S. Redefining interferon-producing killer dendritic cells as a novel intermediate in NK-cell differentiation. Blood 2012, 119, 4349–4357. [Google Scholar] [CrossRef] [PubMed]
- Guimont-Desrochers, F.; Lesage, S. Revisiting the prominent anti-tumoral potential of pre-mNK cells. Front. Immunol. 2013, 4, Article–446. [Google Scholar] [CrossRef]
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Malireddi, R.K.S.; Guy, C.S.; Chang, T.-C.; Olsen, S.R.; Neale, G.A.; Vogel, P.; Kanneganti, T.-D. SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity 2018, 49, 515–530. [Google Scholar] [CrossRef]
- Sartor, R.B.; Wu, G.D. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 2017, 152, 327–339. [Google Scholar] [CrossRef]
- Yamasaki, S.; Matsumoto, M.; Takeuchi, O.; Matsuzawa, T.; Ishikawa, E.; Sakuma, M.; Tateno, M.; Uno, J.; Hirabayashi, J.; Mikami, Y.; et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl. Acad. Sci. USA 2009, 106, 1897–1902. [Google Scholar] [CrossRef]
- Saijo, S.; Fujikado, N.; Furuta, T.; Chung, S.-h.; Kotaki, H.; seki, K.; Sudo, K.; Akira, S.; Adachi, Y.; Ohno, N.; et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2007, 8, 39–46. [Google Scholar] [CrossRef]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.-h.; et al. Dectin-2 Recognition of a-Mannans and Induction of Th17 Cell Differentiation Is Essential for Host Defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-L.; Zhao, X.-Q.; Jiang, C.; You, Y.; Chen, X.-P.; Jiang, Y.-Y.; Jia, X.-M.; Lin, X. C-Type Lectin Receptors Dectin-3 and Dectin-2 Form a Heterodimeric Pattern-Recognition Receptor for Host Defense against Fungal Infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Franco, L.M.; Lionakis, M.S. Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front. Immunol. 2018, 9, 1836. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, Z.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Takatori, H.; Makita, S.; Ito, T.; Matsuki, A.; Nakajima, H. Regulatory Mechanisms of IL-33-ST2-Mediated Allergic Inflammation. Front. Immunol. 2018, 9, 2004. [Google Scholar] [CrossRef]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 2012, 287, 6941–6948. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, Z. The enigmatic processing and secretion of interleukin-33. Cell. Mol. Immunol. 2010, 7, 260–262. [Google Scholar] [CrossRef]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef]
- Lefrancais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Muto, T.; Kawagoe, T.; Matsumoto, M.; Sasaki, Y.; Matsushita, K.; Taki, Y.; Futatsugi-Yumikura, S.; Tsutsui, H.; Ishii, K.J.; et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3451–3456. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Adachi, T.; Koida, A.; Nakanishi, K. Nematode-Infected Mice Acquire Resistance to Subsequent Infection With Unrelated Nematode by Inducing Highly Responsive Group 2 Innate Lymphoid Cells in the Lung. Front. Immunol. 2018, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Fehlings, M.; Kloverpris, H.N.; McGovern, N.; Koo, S.L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.L.; et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 2018, 48, 1060. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Bacchi, M.; Bertolino, M. Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur. J. Drug Metab. Pharmacokinet. 2006, 31, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 4265–4273. [Google Scholar] [CrossRef]
- Robertson, M.J.; Kirkwood, J.M.; Logan, T.F.; Koch, K.M.; Kathman, S.; Kirby, L.C.; Bell, W.N.; Thurmond, L.M.; Weisenbach, J.; Dar, M.M. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008, 14, 3462–3469. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Kline, J.; Struemper, H.; Koch, K.M.; Bauman, J.W.; Gardner, O.S.; Murray, S.C.; Germaschewski, F.; Weisenbach, J.; Jonak, Z.; et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J. Immunother. 2013, 36, 331–341. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842. [Google Scholar] [CrossRef] [PubMed]
- Motavaf, M.; Safari, S.; Moayed Alavian, S. Interleukin-18 Gene Promoter Polymorphisms and Susceptibility to Chronic Hepatitis B Infection: A Review Study. Hepat. Mon. 2014, 14, e19879. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H. Association between interleukin-18 gene promoter (−607C/A and −137G/C) polymorphisms and chronic hepatitis C virus infections: A meta-analysis. Meta Gene 2015, 5, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; Li, J.-J.; Sun, C.-A.; Jin, Y.; Wu, W.-W. Interleukin-18 promoter polymorphisms and plasma levels are associated with increased risk of periodontitis: A meta-analysis. Inflamm. Res. 2014, 63, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.K.; de Carvalho, A.C.G.; Alves, E.H.P.; da Silva, F.R.P.; Pessoa, L.D.S.; Vasconcelos, D.F.P. Genetic Factors and the Risk of Periodontitis Development: Findings from a Systematic Review Composed of 13 Studies of Meta-Analysis with 71,531 Participants. Int. J. Dent. 2017, 2017, 1914073. [Google Scholar] [CrossRef]
- Pan, H.-F.; Leng, R.-X.; Ye, D.-Q. Lack of association of interleukin-18 gene promoter -607 A/C polymorphism with susceptibility to autoimmune diseases: A meta-analysis. Lupus 2011, 20, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Association between interleukin-18 polymorphisms and systemic lupus erythematosus: A meta-analysis. Mol. Biol. Rep. 2013, 40, 2581–2587. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, F.; Ren, J.; Liu, J.; Meng, W. Association of IL-18 polymorphisms with rheumatoid arthritis and systemic lupus erythematosus in Asian populations: A meta-analysis. BMC Med. Genet. 2012, 13, 107. [Google Scholar] [CrossRef]
- Guo, B.R.; Rong, J.J.; Wei, Y.H.; Zhong, W.Y.; Li, C.S.; Liu, M.; Li, W.; Wang, X.B.; Wang, L.; Zheng, H.F. Ethnicity-stratified analysis of the association between IL-18 polymorphisms and systemic lupus erythematosus in a European population: A meta-analysis. Arch. Dermatol. Res. 2015, 307, 747–755. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, K.; Yang, Z.; Li, F.; Wang, Z.; Xu, F.; He, C. Association of cytokine gene polymorphisms (IL6, IL12B, IL18) with Behcet’s disease: A meta-analysis. Z. Rheumatol. 2016, 75, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.-P.; Zhou, L.-J.; Lu, S.-Y.; Liang, Y.-E.; Chen, X.-Y.; Liu, L.; Lin, J. Association of IL-18 promoter gene polymorphisms with rheumatoid arthritis: A meta-analysis. Mol. Biol. Rep. 2014, 41, 8211–8217. [Google Scholar] [CrossRef] [PubMed]
- Khodaeian, M.; Enayati, S.; Tabatabaei-Malazy, O.; Amoli, M.M. Association between Genetic Variants and Diabetes Mellitus in Iranian Populations: A Systematic Review of Observational Studies. J. Daibetes Res. 2015, 2015, 585917. [Google Scholar] [CrossRef] [PubMed]
- Ho Lee, Y.; Kim, J.-H.; Gyu Song, G. Interleukin-18 promoter -607 C/A and -137 G/C polymorphisms and susceptibility to type 1 diabetes: A meta-analysis. Hum. Immunol. 2015, 76, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-J.; Zhang, L.; Lu, W.J.; Wang, L.; Chen, L.; Zhu, Z.; Zhu, H.-H. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to Crohn’s disease. World J. Gastroenterol. 2015, 21, 8711–8722. [Google Scholar] [CrossRef]
- Zhang, M.-J.; Zhou, Y.; Wang, X.; Chen, X.; Pi, Y.; Guo, L.; Gan, C.-Y.; Li, J.-C.; Zhang, L.-L. Interleukin-18 gene promoter 607A polymorphism, but not 137C polymorphism, is a protective factor for ischemic stroke in the Chinese population: A meta-analysis. Meta Gene 2016, 9, 165–172. [Google Scholar] [CrossRef]
- Mi, Y.-Y.; Yu, Q.-Q.; Yu, M.-L.; Xu, B.; Zhang, L.-F.; Cheng, W.; Zhang, W.; Hua, L.-X.; Feng, N.-H. Review and pooled analysis of studies on -607(C/A) and -137(G/C) polymorphisms in IL-18 and cancer risk. Med. Oncol. 2011, 28, 1107–1115. [Google Scholar] [CrossRef]
- Yang, X.; Qiu, M.-T.; Hu, J.-W.; Jiang, F.; Li, M.; Wang, J.; Zhang, Q.; Yin, R.; Xu, L. Association of Interleukin-18 Gene Promoter 2607 C.A and 2137G.C Polymorphisms with Cancer Risk: A Meta-Analysis of 26 Studies. PLoS ONE 2013, 8, e72671. [Google Scholar]
- Wang, M.; Zhu, X.-Y.; Wang, L.; Lin, Y. The-607C/A Polymorphisms in Interleukin-18 Gene Promoter Contributes to Cancer Risk: Evidence from A Meta-Analysis of 22 Case-Control Studies. PLos ONE 2013, 8, e76915. [Google Scholar] [CrossRef]
- Liang, T.J.; Ma, H.; Wang, C.X.; Liu, Y.R.; Wang, X.G. The-137G>C polymorphism in interleukin-18 promoter region and cancer risk: Evidence from a meta-analysis of 21 studies. Tumour Biol. 2013, 34, 3483–3490. [Google Scholar] [CrossRef]
- Guo, X.-Y.; Xia, Y. The Interleukin-18 Promoter-607C>A Polymorphism Contributes to Nasopharyngeal Carcinoma Risk: Evidence from a Meta-analysis Including 1,886 Subjects. Asian Pac. J. Cancer Prev. 2013, 14, 7577–7581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, Z.M.; Huang, H.B.; Sun, L.S.; Sun, A.Q.; Li, K. Association of IL-8 gene promoter -251 A/T and IL-18 gene promoter -137 G/C polymorphisms with head and neck cancer risk: A comprehensive meta-analysis. Cancer Manag. Res. 2018, 10, 2589–2604. [Google Scholar] [CrossRef]
- Zhu, S.-L.; Zhao, Y.; Hu, X.-Y.; Luo, T.; Chen, Z.-S.; Zhang, Y.; Yang, S.-H.; Zhou, L.; Li, L.-Q. Genetic polymorphisms-137 (rs187238) and -607 (rs1946518) in the interleukin-18 promoter may not be associated with development of hepatocellular carcinoma. Sci. Rep. 2016, 6, 39404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, J.; Bao, X.; Niu, W.; Wang, L.; Du, L.; Zhang, N.; Sun, Y. Association between Genetic Polymorphisms in Interleukin Genes and Recurrent Pregnancy Loss ± A Systematic Review and Meta-Analysis. PLos ONE 2017, 12, e0169891. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, X.; Du, J.; Lu, M. Interleukin-18 gene polymorphisms and risk of recurrent pregnancy loss: A systematic review and meta-analysis. J. Obstet. Gynecol. Res. 2015, 41, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, K.; Yang, Z. Associations between TNF-alpha and interleukin gene polymorphisms with polycystic ovary syndrome risk: A systematic review and meta-analysis. J. Assist. Reprod. Genet. 2015, 32, 625–634. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pleiotropic action of IL-18 depends on its cytokine milieu.

Figure 2.
Representative inflammasomes. Caspase-1, an IL-1β/IL-18-converting enzyme, is produced as an enzymatically inactive precursor (pro). Upon the appropriate stimulation, many cell types including macrophages and epithelial cells, form cytoplasmic machinery termed the inflammasome for the activation of pro-caspase-1. Several cytoplasmic pattern-recognition receptors including NLR and AIM2-like receptors, serve as a scaffold molecule for individual inflammasome activation. (A) The NLRP3 inflammasome. After recognition of oxidized mitochondrial (ox-mit) DNA, NLRP3, a member of the NLR, assembles a caspase activating adaptor, ASC, and pro-caspase-1, which eventually leads to active caspase-1. (B) The NLRC4 inflammasome. After recognition of corresponding bacterial secretion system III, components from human NAIP or murine NAIP family members associate with NLRC4, which results in formation of the NLRC4 inflammasome. (C) The AIM2 inflammasome. After the recognition of double-stranded (ds) DNA, AIM2 similarly generates the AIM2 inflammasome. Active caspase-1 then cleaves pro-IL-18 and pro-IL-1β into IL-18 and IL-1β, respectively. Caspase-1 also cleaves gasdermin D into a pore-forming N-terminal protein. IL-18 and IL-1β as well as other cytoplasmic proteins including HMGB1, are extracellularly released through membrane pores generated by the N-terminal protein. Blue line indicated the cell membrane.
Figure 2.
Representative inflammasomes. Caspase-1, an IL-1β/IL-18-converting enzyme, is produced as an enzymatically inactive precursor (pro). Upon the appropriate stimulation, many cell types including macrophages and epithelial cells, form cytoplasmic machinery termed the inflammasome for the activation of pro-caspase-1. Several cytoplasmic pattern-recognition receptors including NLR and AIM2-like receptors, serve as a scaffold molecule for individual inflammasome activation. (A) The NLRP3 inflammasome. After recognition of oxidized mitochondrial (ox-mit) DNA, NLRP3, a member of the NLR, assembles a caspase activating adaptor, ASC, and pro-caspase-1, which eventually leads to active caspase-1. (B) The NLRC4 inflammasome. After recognition of corresponding bacterial secretion system III, components from human NAIP or murine NAIP family members associate with NLRC4, which results in formation of the NLRC4 inflammasome. (C) The AIM2 inflammasome. After the recognition of double-stranded (ds) DNA, AIM2 similarly generates the AIM2 inflammasome. Active caspase-1 then cleaves pro-IL-18 and pro-IL-1β into IL-18 and IL-1β, respectively. Caspase-1 also cleaves gasdermin D into a pore-forming N-terminal protein. IL-18 and IL-1β as well as other cytoplasmic proteins including HMGB1, are extracellularly released through membrane pores generated by the N-terminal protein. Blue line indicated the cell membrane.

Figure 3.
IL-18 signal transduction. When IL-18 binds to IL-18Rα (black arrow), IL-18Rβ binds to the complex. IL-18 signaling activates the transcription factors NF-κB and AP-1 via signal transduction molecules including MyD88, IRAKs, and TRAF6 (white arrows). IL-18BP competes IL-18 binding to IL-18Rα. When IL-18Rα binds to IL-37 it prevents binding to IL-18Rβ, which binds to IL-1R8 to send an inhibitory signal via STAT3.
Figure 3.
IL-18 signal transduction. When IL-18 binds to IL-18Rα (black arrow), IL-18Rβ binds to the complex. IL-18 signaling activates the transcription factors NF-κB and AP-1 via signal transduction molecules including MyD88, IRAKs, and TRAF6 (white arrows). IL-18BP competes IL-18 binding to IL-18Rα. When IL-18Rα binds to IL-37 it prevents binding to IL-18Rβ, which binds to IL-1R8 to send an inhibitory signal via STAT3.

Figure 4.
Involvement of the NLRP1 inflammasome in non-hematopoietic cells on metabolic homeostasis. (A) Requirement of NLRP1 for metabolic homeostasis: Nlrp1−/− mice spontaneously develop obesity and related metabolic syndromes when fed a normal diet (ND) that does not induce obesity in wild type (WT) mice. Nlrp1−/− mice fed a high-fat diet (HFD) had a more severe illness compared with WT mice. (B) IL-18 is released after activation of NLRP1 in non-hematopoietic cells. Nlrp1−/− mice reconstituted with WT bone marrow cells (Nlrp1−/− > WT) showed a phenotype similar to Nlrp1−/− mice. In contrast, WT mice reconstituted with Nlrp1−/− bone marrow cells (WT > Nlrp1−/−) exhibited a phenotype comparable with WT mice. (C) The protective role of IL-18 against cachexia. Il1r−/−Nlrp1MUT mice harboring a mutant gene from human patients with familial Mediterranean fever but lacking the IL-1R gene have an almost intact phenotype when fed a ND. However, when fed an HFD, Il1r−/−Nlrp1MUT mice suffered from fatal illness with a marked increase in circulating IL-18 levels. By contrast, Il18−/−Il1r−/−Nlrp1MUT mice were resistant to HFD.
Figure 4.
Involvement of the NLRP1 inflammasome in non-hematopoietic cells on metabolic homeostasis. (A) Requirement of NLRP1 for metabolic homeostasis: Nlrp1−/− mice spontaneously develop obesity and related metabolic syndromes when fed a normal diet (ND) that does not induce obesity in wild type (WT) mice. Nlrp1−/− mice fed a high-fat diet (HFD) had a more severe illness compared with WT mice. (B) IL-18 is released after activation of NLRP1 in non-hematopoietic cells. Nlrp1−/− mice reconstituted with WT bone marrow cells (Nlrp1−/− > WT) showed a phenotype similar to Nlrp1−/− mice. In contrast, WT mice reconstituted with Nlrp1−/− bone marrow cells (WT > Nlrp1−/−) exhibited a phenotype comparable with WT mice. (C) The protective role of IL-18 against cachexia. Il1r−/−Nlrp1MUT mice harboring a mutant gene from human patients with familial Mediterranean fever but lacking the IL-1R gene have an almost intact phenotype when fed a ND. However, when fed an HFD, Il1r−/−Nlrp1MUT mice suffered from fatal illness with a marked increase in circulating IL-18 levels. By contrast, Il18−/−Il1r−/−Nlrp1MUT mice were resistant to HFD.

Figure 5.
Importance of IL-18 for gut homeostasis. Intestinal epithelial cells utilize various inflammasomes for the constitutive production of IL-18. Under SPF conditions, colonic epithelial cells release IL-18 dependent on the NLRP6 inflammasome, which in turn activates epithelial cells to produce antimicrobial peptides (AMPs) to maintain microbiome homeostasis. Nlrp6−/− mice spontaneously develop dysbiosis resulting in colitis. The NLRP6 inflammasome is involved in the generation of the homeostatic mucin layer by intestinal epithelial cells. IL-18 derived from pyrin inflammasome activation is involved in gut homeostasis through the generation of intact tight junction (Tj) formation. Furthermore, pyrin inflammation-mediated IL-18 protects against colitis-associated colorectal cancer by reducing the stemness of colon epithelial cells.
Figure 5.
Importance of IL-18 for gut homeostasis. Intestinal epithelial cells utilize various inflammasomes for the constitutive production of IL-18. Under SPF conditions, colonic epithelial cells release IL-18 dependent on the NLRP6 inflammasome, which in turn activates epithelial cells to produce antimicrobial peptides (AMPs) to maintain microbiome homeostasis. Nlrp6−/− mice spontaneously develop dysbiosis resulting in colitis. The NLRP6 inflammasome is involved in the generation of the homeostatic mucin layer by intestinal epithelial cells. IL-18 derived from pyrin inflammasome activation is involved in gut homeostasis through the generation of intact tight junction (Tj) formation. Furthermore, pyrin inflammation-mediated IL-18 protects against colitis-associated colorectal cancer by reducing the stemness of colon epithelial cells.

Figure 6.
IL-18 protection against cancer. (A) Robust proliferation of tumoricidal human γδT cells. IL-18 in combination with IL-2 activates and induces the proliferation of human CD56+CD11c+ precursor NK cells, which in turn robustly proliferate and activate γδT cells stimulated with γδT cell antigen produced by zoledronate (Zol)-treated monocytes (Mo). (B) Involvement of CLRs-mediated CARD9 inflammasome activation in the induction of colon cancer. Fungi in the intestinal microbial flora activate macrophages through C-type lectin receptors (CLRs), which promote Syk to activate caspase-1 via the CARD9 inflammasome. The resultant IL-18 is required for protection against colitis and colitis-associated cancer.
Figure 6.
IL-18 protection against cancer. (A) Robust proliferation of tumoricidal human γδT cells. IL-18 in combination with IL-2 activates and induces the proliferation of human CD56+CD11c+ precursor NK cells, which in turn robustly proliferate and activate γδT cells stimulated with γδT cell antigen produced by zoledronate (Zol)-treated monocytes (Mo). (B) Involvement of CLRs-mediated CARD9 inflammasome activation in the induction of colon cancer. Fungi in the intestinal microbial flora activate macrophages through C-type lectin receptors (CLRs), which promote Syk to activate caspase-1 via the CARD9 inflammasome. The resultant IL-18 is required for protection against colitis and colitis-associated cancer.


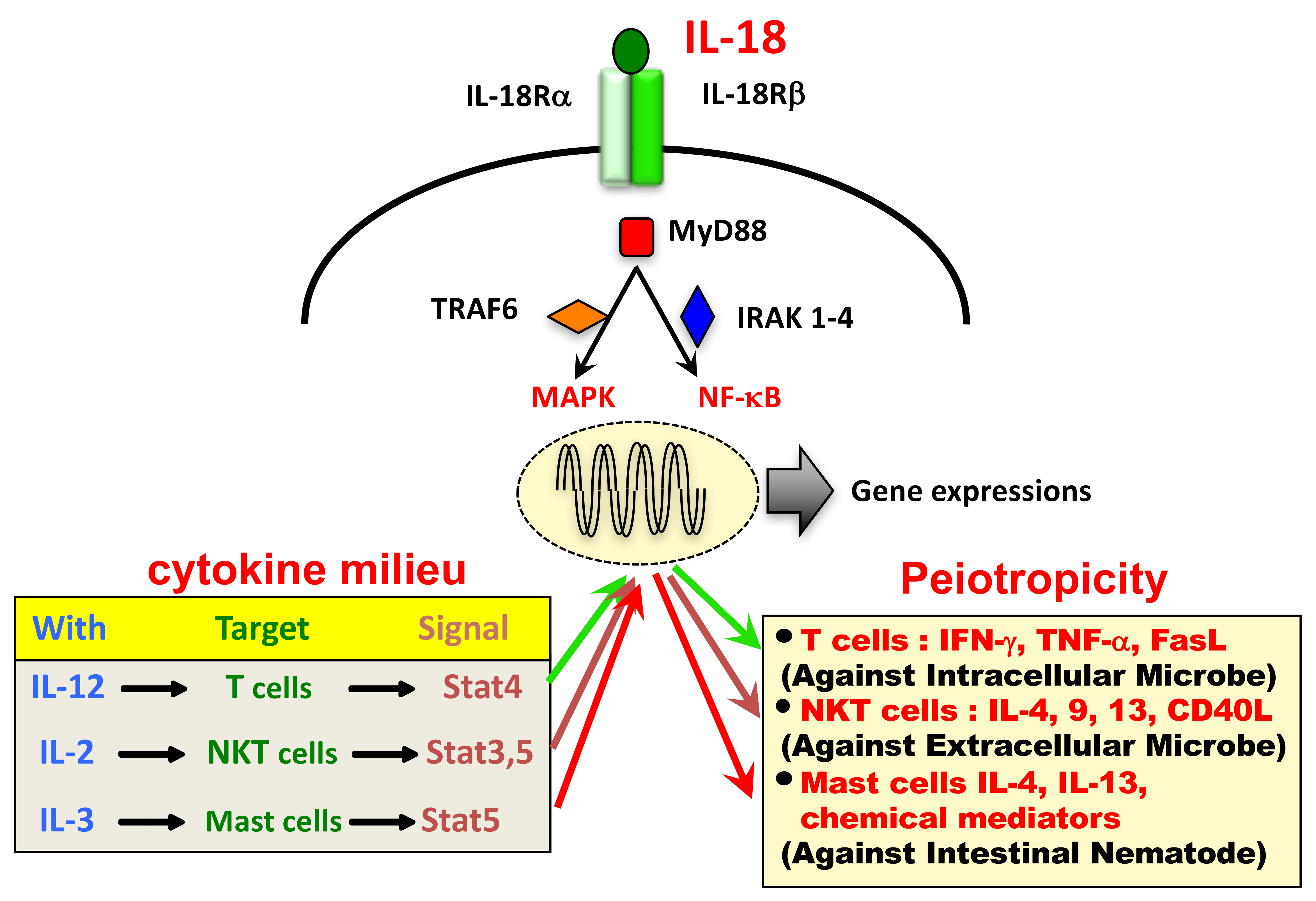{kind=link}
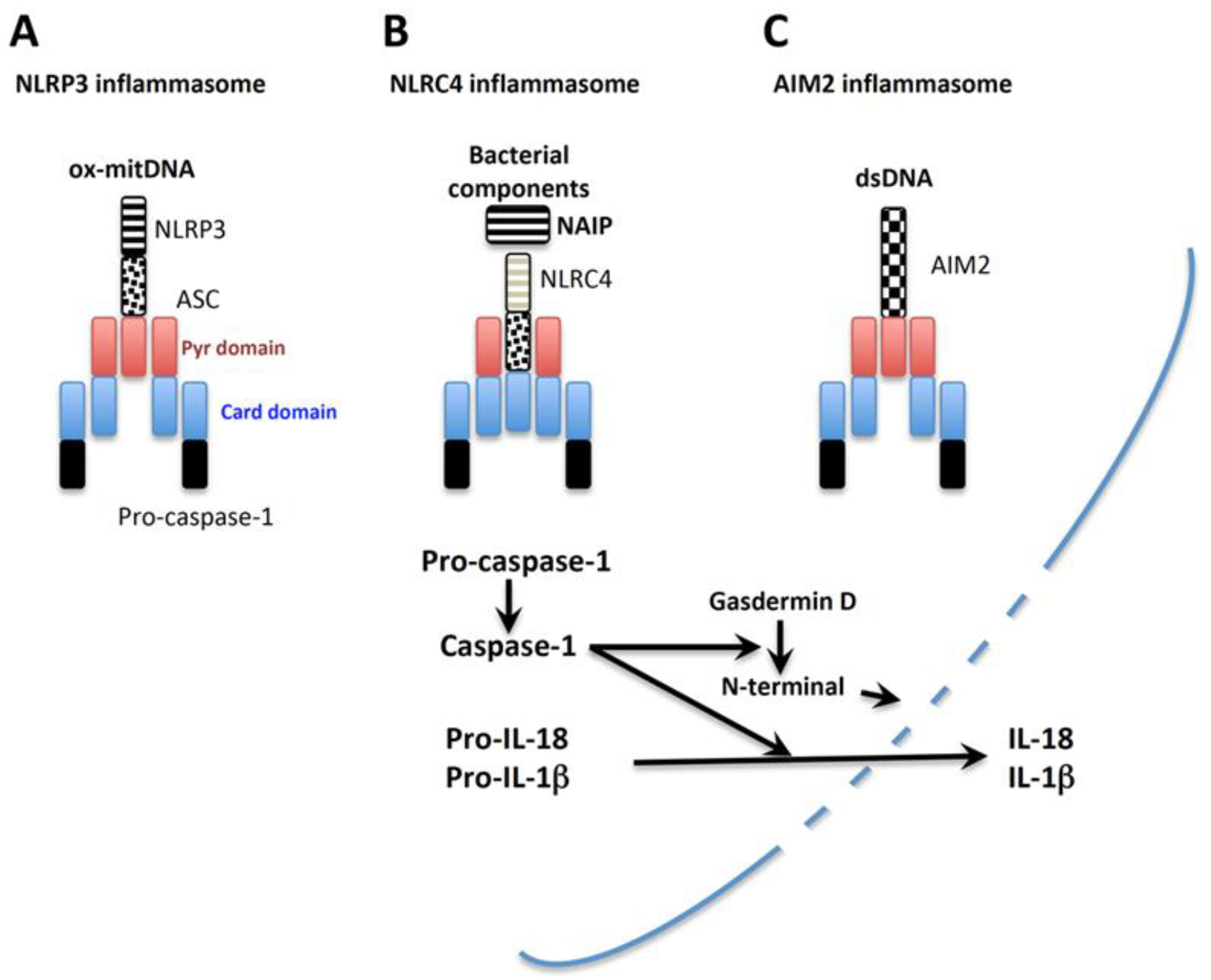{kind=link}
{kind=link}
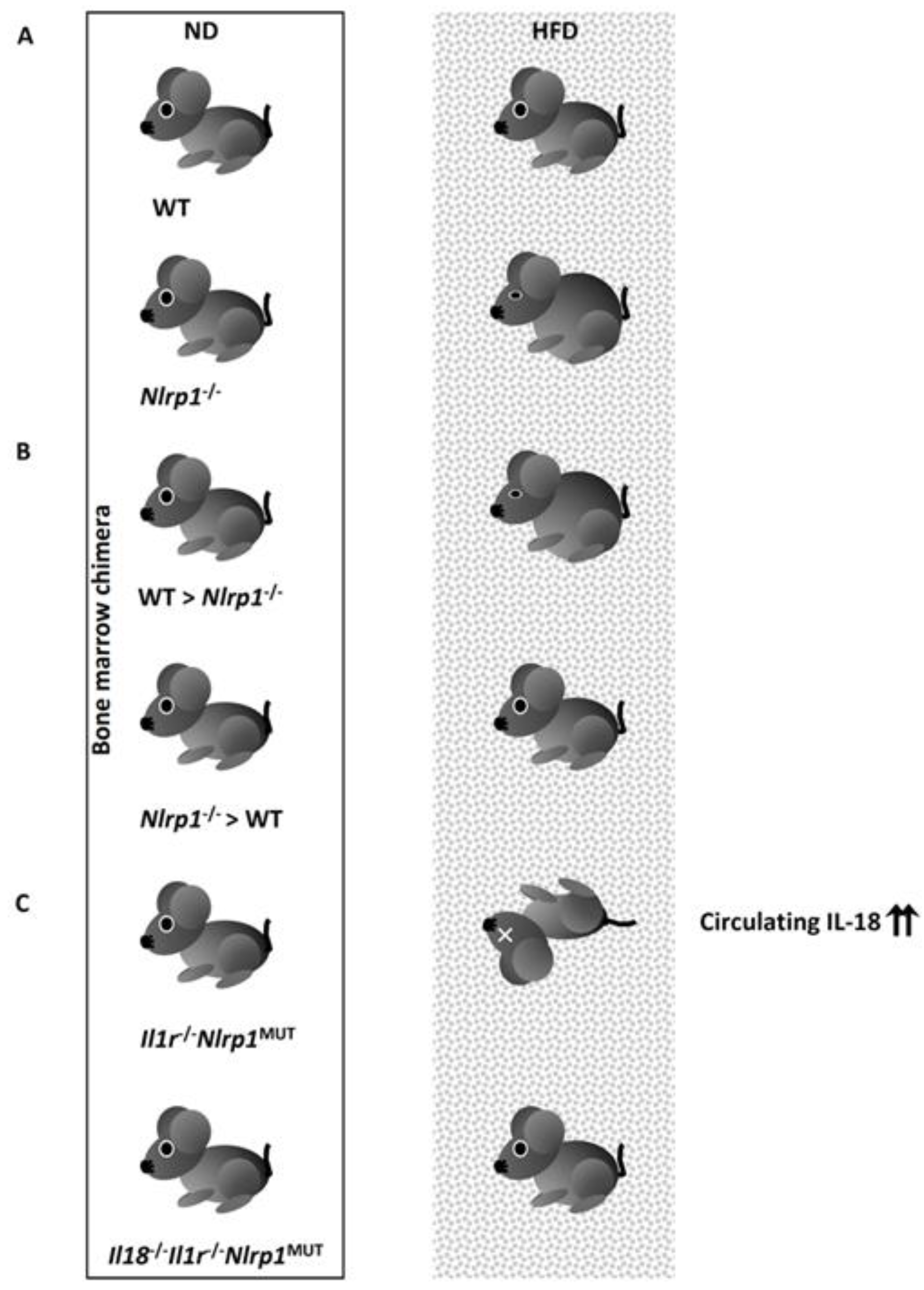{kind=link}
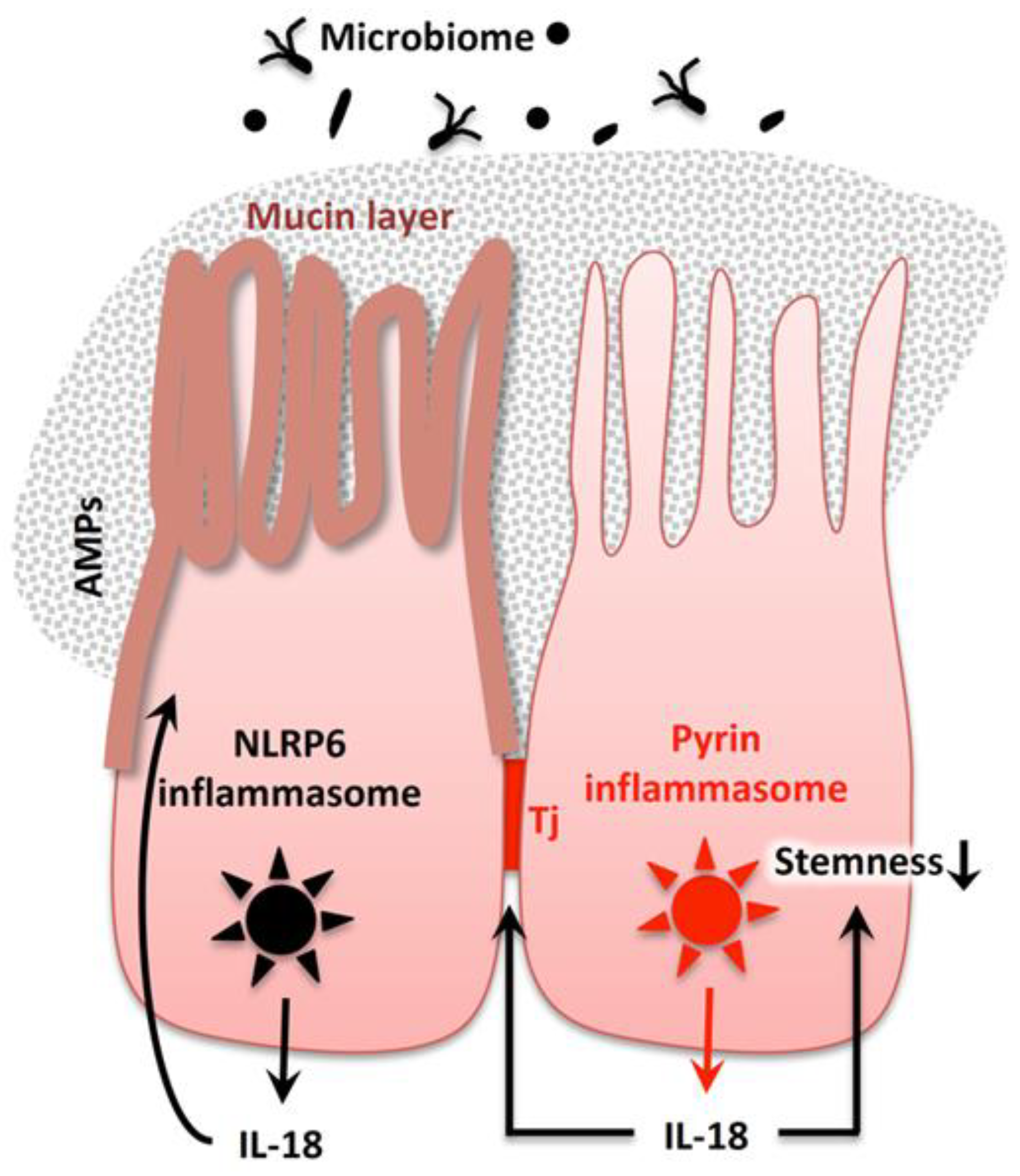{kind=link}
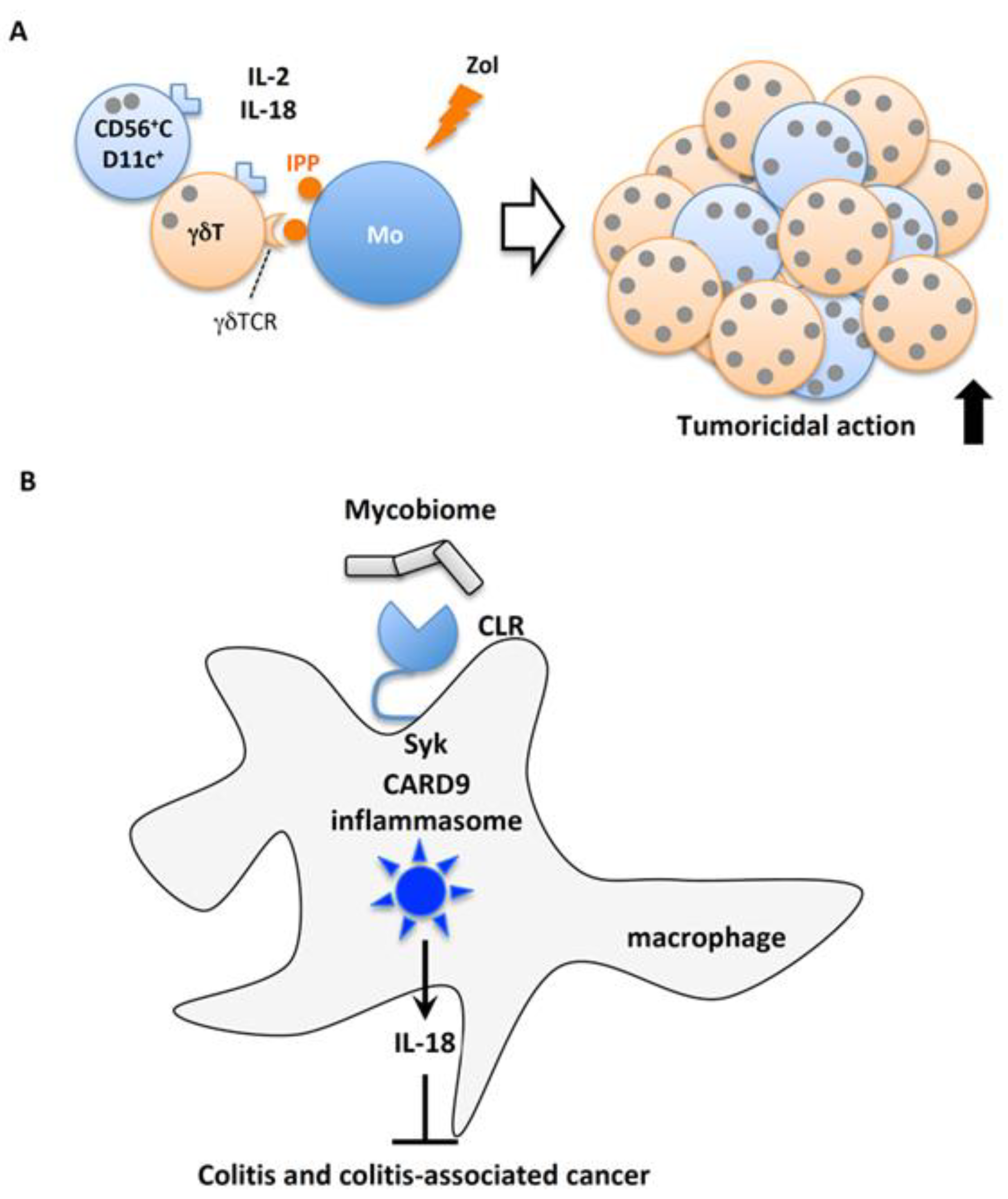{kind=link}
Table 1.
IL-18 gene promoter polymorphisms (meta-analysis and/or systematic review).
| Disease Association | SNP Polymorphism | Ref | |||||
|---|---|---|---|---|---|---|---|
| Promoters | 5’-UTR | ||||||
| −1297 C/T | −656 G/T | −607 C/A | −137 G/C | +113 T/T | |||
| rs360719 | rs1946519 | rs1946518 | rs187238 | rs360718 | |||
| Chronic viral infection | HBV | ND | ND | + | + | ND | [373] |
| HCV | ND | ND | − | + | ND | [374] | |
| Periodontitis | ND | ND | + | + | ND | [375] | |
| ND | ND | − | − | ND | [376] | ||
| Autoimmune diseases | SLE | ND | ND | − | ND | ND | [377] |
| +a | ND | +a | +b | ND | [378] | ||
| ND | ND | +c | − | ND | [379] | ||
| + | ND | +a | − | ND | [380] | ||
| Behcet’s disease | ND | ND | + | ND | ND | [381] | |
| RA | ND | ND | − | ND | ND | [377] | |
| ND | ND | +c | − | ND | [379] | ||
| ND | ND | +b | − | ND | [382] | ||
| T1D | ND | ND | − | ND | ND | [377] | |
| ND | ND | + | − | ND | [383] | ||
| ND | ND | +b | − | ND | [384] | ||
| CD | ND | − | + | + | + | [385] | |
| ND | ND | − | ND | ND | [377] | ||
| UC | ND | ND | − | ND | ND | [377] | |
| Ischemic stroke | ND | ND | + | − | ND | [386] | |
| Cancer | Total cancer | ND | ND | + | − | ND | [387] |
| ND | ND | − | +b | ND | [388] | ||
| ND | ND | +b | ND | ND | [389] | ||
| ND | ND | ND | +b | ND | [390] | ||
| Nasopharyngeal cancer | ND | ND | + | + | ND | [388] | |
| ND | ND | ND | + | ND | [387] | ||
| ND | ND | ND | + | ND | [390] | ||
| ND | ND | + | ND | ND | [389] | ||
| ND | ND | + | ND | ND | [391] | ||
| Esophageal cancer | ND | ND | + | ND | ND | [389] | |
| Gastric cancer | ND | ND | + | ND | ND | [388] | |
| Head and neck cancer | ND | ND | ND | + | ND | [392] | |
| Hepatocellular carcinoma | ND | ND | − | − | ND | [393] | |
| Recurrent pregnancy loss | ND | ND | + | + | ND | [394] | |
| ND | ND | − | + | ND | [395] | ||
| Polycystic ovary syndrome | ND | ND | − | − | ND | [396] | |
+; significant association between the variant and disease. −; no significant association between the variant and disease. +a; association in European/Caucasian populations, but not in Asian populations. +b; association in Asian populations, but not in European/Caucasian populations. +c; association in Chinese populations, but not in Asian populations. ND; not determined. Abbreviations: HBV, hepatitis B virus; HCV, hepatitis C virus; SLE, systemic lupus erythematosus; RA, rheumatoid arthritis; T1D, type 1 diabetes mellitus; CD, Crohn’s disease; UC, ulcerative colitis.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. https://doi.org/10.3390/ijms20030649
AMA Style
Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in Health and Disease. International Journal of Molecular Sciences. 2019; 20(3):649. https://doi.org/10.3390/ijms20030649
Chicago/Turabian StyleYasuda, Koubun, Kenji Nakanishi, and Hiroko Tsutsui. 2019. "Interleukin-18 in Health and Disease" International Journal of Molecular Sciences 20, no. 3: 649. https://doi.org/10.3390/ijms20030649
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.