Significantly Elevated FMR1 mRNA and Mosaicism for Methylated Premutation and Full Mutation Alleles in Two Brothers with Autism Features Referred for Fragile X Testing

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Medical History
2.2. Extended FMR1 CGG Testing
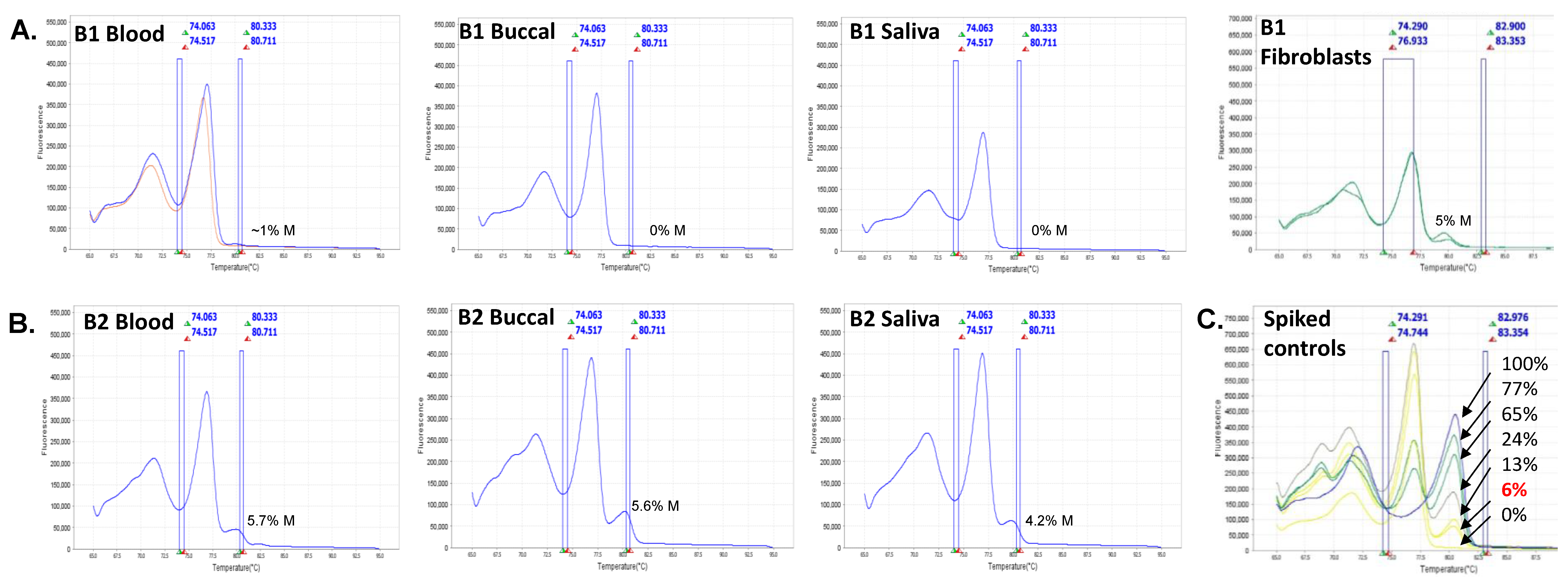
2.3. FMR1 Methylation and Gene Expression Analyses
2.4. Neurodevelopmental Outcomes at the Time of Recruitment
3. Discussion
4. Materials and Methods
4.1. Ethics Approval and Consent
4.2. Recruitment and Assessments
4.3. Sample Processing
4.4. Methylation Specific-Quantitative Melt Analysis (MS-QMA)
4.5. Methylation Analysis and CGG Sizing Using Southern Blot and AmplideX PCR
4.6. Melting Curve Analysis (MCA) to Determine the Presence of Expanded FMR1 Alleles
4.7. Sanger Sequencing
4.8. FMR1 mRNA Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007, 37, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.S.; Lightbody, A.A.; Reiss, A.L. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am. J. Ment. Retard. 2008, 113, 44–53. [Google Scholar] [CrossRef]
- Mazzocco, M.M.; Kates, W.R.; Baumgardner, T.L.; Freund, L.S.; Reiss, A.L. Autistic behaviors among girls with fragile X syndrome. J. Autism Dev. Disord. 1997, 27, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Merenstein, S.A.; Sobesky, W.E.; Taylor, A.K.; Riddle, J.E.; Tran, H.X.; Hagerman, R.J. Molecular-clinical correlations in males with an expanded FMR1 mutation. Am. J. Med. Genet. 1996, 64, 388–394. [Google Scholar] [CrossRef]
- Baker, E.K.; Arpone, M.; Aliaga, S.M.; Bretherton, L.; Kraan, C.M.; Bui, M.; Slater, H.R.; Ling, L.; Francis, D.; Hunter, M.F.; et al. Incomplete silencing of full mutation alleles in males with fragile X syndrome is associated with autistic features. Mol. Autism. 2019, 10, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraan, C.M.; Godler, D.E.; Amor, D.J. Epigenetics of fragile X syndrome and fragile X-related disorders. Dev. Med. Child Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Heitz, D.; Rousseau, F.; Devys, D.; Saccone, S.; Abderrahim, H.; Le Paslier, D.; Cohen, D.; Vincent, A.; Toniolo, D.; Della Valle, G.; et al. Isolation of sequences that span the fragile X and identification of a fragile X-related CpG island. Science 1991, 251, 1236–1239. [Google Scholar] [CrossRef] [PubMed]
- Oberle, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boue, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Rousseau, F.; Heitz, D.; Biancalana, V.; Blumenfeld, S.; Kretz, C.; Boué, J.; Tommerup, N.; Hagen, C.V.D.; DeLozier-Blanchet, C.; Croquette, M.-F.; et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 1991, 325, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.-P.; Mandel, J.-L. The FMR–1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, F.; Lokey, L.; Chastain, J.; Lakkis, L.; Eberhart, D.; Warren, S. Translational suppression by trinucleotide repeat expansion at FMR1. Science 1995, 268, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.; Yrigollen, C.M.; Tang, H.T.; Williamson, J.; Espinal, G.; Iwahashi, C.K.; Durbin-Johnson, B.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 2014, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Smits, A.; Mohkamsing, S.; van Beerendonk, H.; de Haan, A.; de Vries, B.; van den Ouweland, A.; Sistermans, E.; Galjaard, H.; Oostra, B.A. Rapid antibody test for diagnosing fragile X syndrome: A validation of the technique. Hum. Genet. 1997, 99, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, D.; Furfaro Joyce, A.; Hoeft, F.; Eckert Mark, A.; Hall Scott, S.; O’Hara, R.; Erba Heather, W.; Ringel, J.; Hayashi Kiralee, M.; Patnaik, S.; et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP). Ann. Neurol. 2007, 63, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 2000, 10, 1038–1044. [Google Scholar] [CrossRef]
- Jeanne, W.I.; Greenough, W.T. Synaptic synthesis of the fragile X protein: Possible involvement in synapse maturation and elimination. Am. J. Med. Genet. 1999, 83, 248–252. [Google Scholar]
- Pfeiffer, B.E.; Huber, K.M. The state of synapses in fragile X syndrome. Neuroscientist 2009, 15, 549–567. [Google Scholar] [CrossRef]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Revenga, L.; Madrigal, I.; Badenas, C.; Xuncla, M.; Jimenez, L.; Mila, M. Premature ovarian failure and fragile X female premutation carriers: No evidence for a skewed X-chromosome inactivation pattern. Menopause 2009, 16, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.L. Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 2000, 97, 189–194. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Houck, G.E., Jr.; Brown, W.T.; Dobkin, C.S. Mosaicism in fragile X affected males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Heitz, D.; Tarleton, J.; MacPherson, J.; Malmgren, H.; Dahl, N.; Barnicoat, A.; Mathew, C.; Mornet, E.; Tejada, I.; et al. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: The first 2,253 cases. Am. J. Hum. Genet. 1994, 55, 225–237. [Google Scholar]
- Aliaga, S.M.; Slater, H.R.; Francis, D.; Du Sart, D.; Li, X.; Amor, D.J.; Alliende, A.M.; Santa Maria, L.; Faundes, V.; Morales, P.; et al. Identification of Males with Cryptic Fragile X Alleles by Methylation-Specific Quantitative Melt Analysis. Clin. Chem. 2016, 62, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonarrigo, F.A.; Russo, S.; Vizziello, P.; Menni, F.; Cogliati, F.; Giorgini, V.; Monti, F.; Milani, D. Think about it: FMR1 gene mosaicism. J. Child Neurol. 2014, 29, NP74–NP77. [Google Scholar] [CrossRef]
- Jiraanont, P.; Kumar, M.; Tang, H.-T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with fragile X syndrome. Expert Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef]
- Orrico, A.; Galli, L.; Dotti, M.T.; Plewnia, K.; Censini, S.; Federico, A. Mosaicism for full mutation and normal-sized allele of the FMR1 gene: A new case. Am. J. Med. Genet. 1998, 78, 341–344. [Google Scholar] [CrossRef]
- Hwang, T.Y.; Aliaga, S.; Arpone, M.V.; Francis, D.; Li, X.; Chong, B.; Slater, H.R.; Rogers, C.; Bretherton, L.; Hunter, M.; et al. Partially Methylated Alleles, Microdeletion and Tissue Mosaicism in a Fragile X Male with Tremor and Ataxia at 30 Years of Age: A Case Report. Am. J. Med. Genet. 2016, 170, 3327–3332. [Google Scholar] [CrossRef]
- Roid, G.H. Stanford-Binet Intelligence Scales-Fifth Edition (SB5) Examiner’s Manual; Riverside Publishing: Itasca, IL, USA, 2003. [Google Scholar]
- Hagerman, R.J.; Hull, C.E.; Safanda, J.F.; Carpenter, I.; Staley, L.W.; O’Connor, R.A.; Seydel, C.; Mazzocco, M.M.; Snow, K.; Thibodeau, S.N.; et al. High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 1994, 51, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Basuta, K.; Schneider, A.; Gane, L.; Polussa, J.; Woodruff, B.; Pretto, D.; Hagerman, R.; Tassone, F. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. A 2015, 167, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Teo, C.R.; Law, H.Y.; Lee, C.G.; Chong, S.S. Screening for CGG repeat expansion in the FMR1 gene by melting curve analysis of combined 5′ and 3′ direct triplet-primed PCRs. Clin. Chem. 2012, 58, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Bishop, S.L.; Guthrie, W.; Coffing, M.; Lord, C. Convergent validity of the Mullen Scales of Early Learning and the differential ability scales in children with autism spectrum disorders. Am. J. Intellect. Dev. Disabil. 2011, 116, 331–343. [Google Scholar] [CrossRef]
- Santa Maria, L.; Pugin, A.; Alliende, M.; Aliaga, S.; Curotto, B.; Aravena, T.; Tang, H.T.; Mendoza-Morales, G.; Hagerman, R.; Tassone, F. FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.T.; Dudding, T.; Aliaga, S.M.; Arpone, M.; Francis, D.; Li, X.; Slater, H.R.; Rogers, C.; Bretherton, L.; du Sart, D.; et al. Molecular Inconsistencies in a Fragile X Male with Early Onset Ataxia. Genes 2016, 7, 68. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; DiLavore, P.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule, Second Edition: ADOS-2; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Wechsler, D. Wechsler Preschool & Primary Scale of Intelligence–Third Edition Australian Standardised Edition (WPPSI-III Australian); Pearson Clinical and Talent Assessment Australia and New Zealand: Sydney, NSW, Australia, 2004. [Google Scholar]
- Mullen, E.M. Mullen Scales of Early Learning: AGS Edition (MSEL:AGS); American Guidance Services, Inc.: Circle Pines, MN, USA, 1995. [Google Scholar]
- Achenbach, T.M.; Rescorla, L.A. Manual for the ASEBA Preschool Forms & Profiles; University of Vermont, Research Center for Children, Youth, & Families: Burlington, VT, USA, 2000. [Google Scholar]
- Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early Detection of Fragile X Syndrome: Applications of a Novel Approach for Improved Quantitative Methylation Analysis in Venous Blood and Newborn Blood Spots. Clin. Chem. 2014. [Google Scholar] [CrossRef]
- Godler, D.E.; Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Shi, E.Z.; Li, X.; Herlihy, A.S.; Skinner, C.; Hagerman, R.J.; Francis, D.; et al. Detection of skewed X-chromosome inactivation in Fragile X syndrome and X chromosome aneuploidy using quantitative melt analysis. Expert Rev. Mol. Med. 2015, 17, e13. [Google Scholar] [CrossRef] [Green Version]
- Arpone, M.; Baker, E.K.; Bretherton, L.; Bui, M.; Li, X.; Whitaker, S.; Dissanayake, C.; Cohen, J.; Hickerton, C.; Rogers, C.; et al. Intragenic DNA methylation in buccal epithelial cells and intellectual functioning in a paediatric cohort of males with fragile X. Sci. Rep. 2018, 8, 3644. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Godler, D.E.; Evans, A.; Bui, Q.M.; Gehling, F.; Kotschet, K.E.; Trost, N.; Storey, E.; Stimpson, P.; Kinsella, G.; et al. Evidence for the toxicity of bidirectional transcripts and mitochondrial dysfunction in blood associated with small CGG expansions in the FMR1 gene in patients with parkinsonism. Genet. Med. 2011, 13, 392–399. [Google Scholar] [CrossRef]
- Godler, D.E.; Loesch, D.Z.; Huggins, R.; Gordon, L.; Slater, H.R.; Gehling, F.; Burgess, T.; Choo, K.H. Improved methodology for assessment of mRNA levels in blood of patients with FMR1 related disorders. BMC Clin. Pathol. 2009, 9, 5. [Google Scholar] [CrossRef]
- Francis, D.; Burgess, T.; Mitchell, J.; Slater, H. Identification of small FRAXA premutations. Mol. Diagn. 2000, 5, 221–225. [Google Scholar] [CrossRef]
- Khaniani, M.S.; Kalitsis, P.; Burgess, T.; Slater, H.R. An improved Diagnostic PCR Assay for identification of Cryptic Heterozygosity for CGG Triplet Repeat Alleles in the Fragile X Gene (FMR1). Mol. Cytogenet. 2008, 1, 5. [Google Scholar] [CrossRef]
- Chen, L.; Hadd, A.G.; Sah, S.; Houghton, J.F.; Filipovic-Sadic, S.; Zhang, W.; Hagerman, P.J.; Tassone, F.; Latham, G.J. High-resolution methylation polymerase chain reaction for fragile X analysis: Evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet. Med. 2011, 13, 528–538. [Google Scholar] [CrossRef]
- Weckx, S.; Del-Favero, J.; Rademakers, R.; Claes, L.; Cruts, M.; De Jonghe, P.; Van Broeckhoven, C.; De Rijk, P. novoSNP, a novel computational tool for sequence variation discovery. Genome Res. 2005, 15, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Kraan, C.M.; Cornish, K.M.; Bui, Q.M.; Li, X.; Slater, H.R.; Godler, D.E. β-glucuronidase mRNA levels are correlated with gait and working memory in premutation females: Understanding the role of FMR1 premutation alleles. Sci. Rep. 2016, 6, 29366. [Google Scholar] [CrossRef]
- Kraan, C.M.; Cornish, K.M.; Bui, Q.M.; Li, X.; Slater, H.R.; Godler, D.E. beta-glucuronidase use as a single internal control gene may confound analysis in FMR1 mRNA toxicity studies. PLoS ONE 2018, 13, e0192151. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Field, M.; Dudding-Byth, T.; Arpone, M.; Baker, E.K.; Aliaga, S.M.; Rogers, C.; Hickerton, C.; Francis, D.; Phelan, D.G.; Palmer, E.E.; et al. Significantly Elevated FMR1 mRNA and Mosaicism for Methylated Premutation and Full Mutation Alleles in Two Brothers with Autism Features Referred for Fragile X Testing. Int. J. Mol. Sci. 2019, 20, 3907. https://doi.org/10.3390/ijms20163907
Field M, Dudding-Byth T, Arpone M, Baker EK, Aliaga SM, Rogers C, Hickerton C, Francis D, Phelan DG, Palmer EE, et al. Significantly Elevated FMR1 mRNA and Mosaicism for Methylated Premutation and Full Mutation Alleles in Two Brothers with Autism Features Referred for Fragile X Testing. International Journal of Molecular Sciences. 2019; 20(16):3907. https://doi.org/10.3390/ijms20163907
Chicago/Turabian StyleField, Michael, Tracy Dudding-Byth, Marta Arpone, Emma K. Baker, Solange M. Aliaga, Carolyn Rogers, Chriselle Hickerton, David Francis, Dean G. Phelan, Elizabeth E. Palmer, and et al. 2019. "Significantly Elevated FMR1 mRNA and Mosaicism for Methylated Premutation and Full Mutation Alleles in Two Brothers with Autism Features Referred for Fragile X Testing" International Journal of Molecular Sciences 20, no. 16: 3907. https://doi.org/10.3390/ijms20163907