Kaempferia parviflora Extract Inhibits STAT3 Activation and Interleukin-6 Production in HeLa Cervical Cancer Cells

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
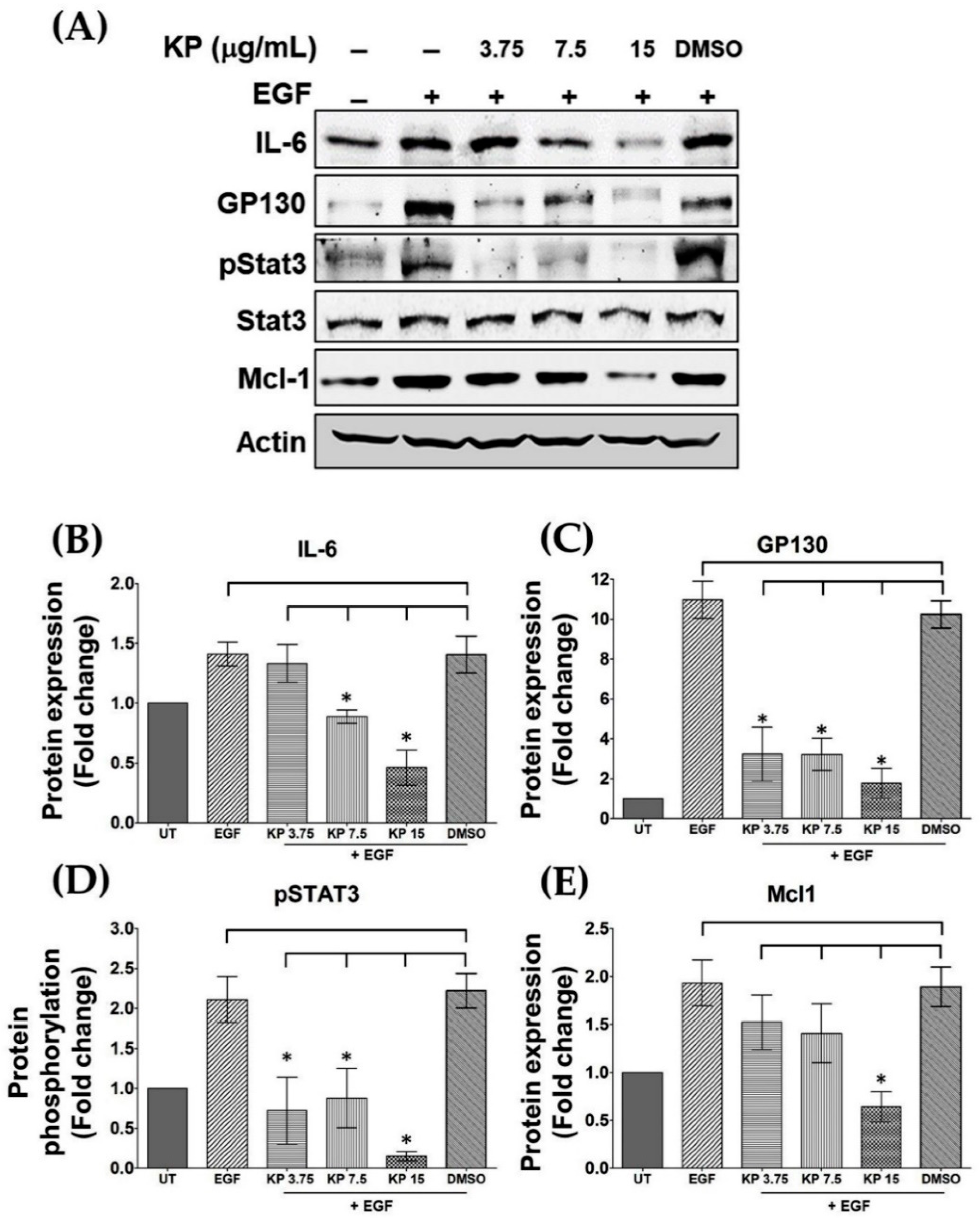
2.1. Chemical Profile of Methoxyflavones in KP Extract and Effects of KP on IL-6 Production
2.2. KP Inhibits EGF-Induced STAT3 Activation
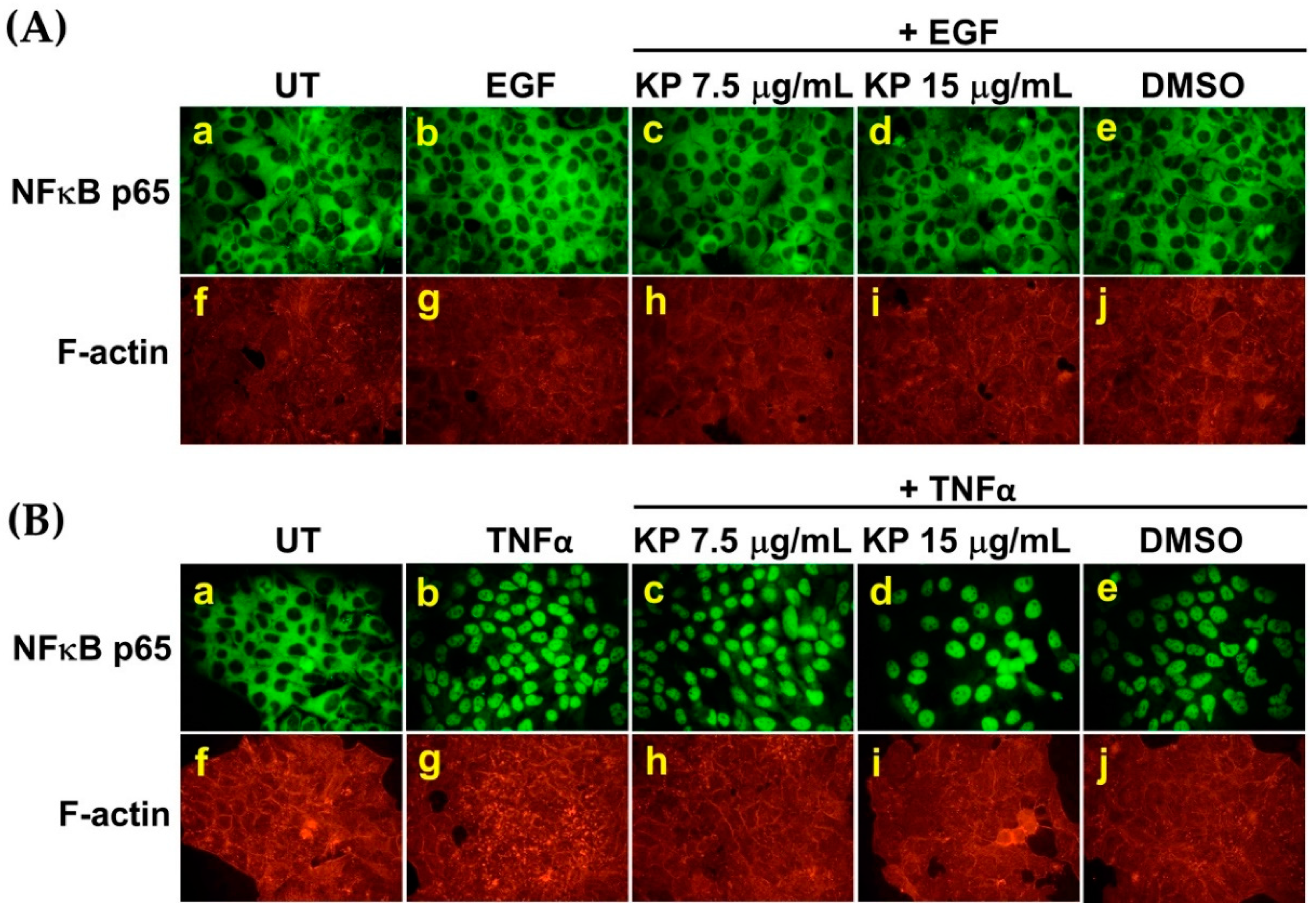
2.3. EGF has no Effects on Nuclear Factor Kappa B (NF-κB) Activation in HeLa Cells, and the Suppression of IL-6 Production by KP is not Regulated through NF-κB Inhibition
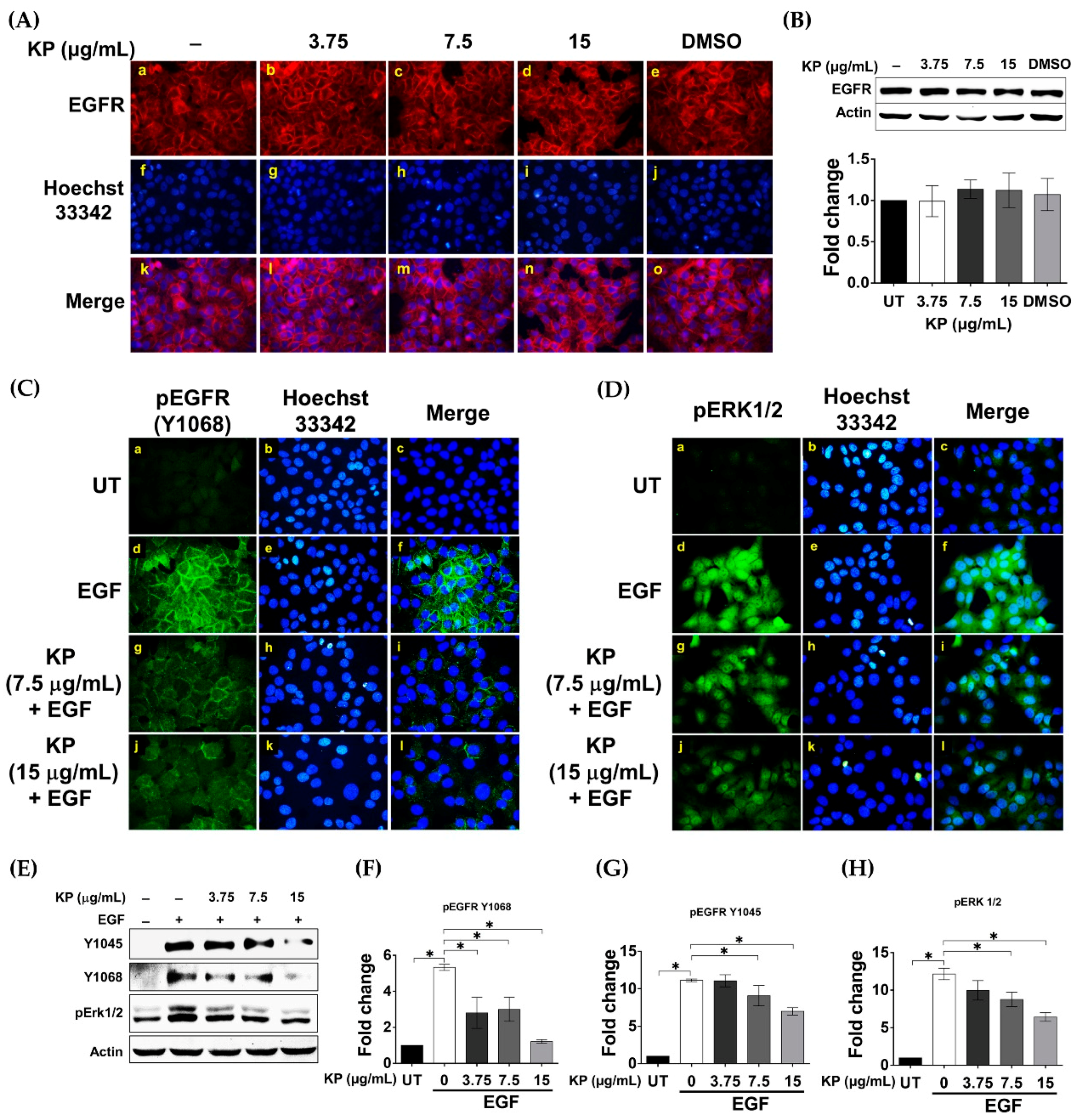
2.4. KP Inhibits EGF-Induced Activation of EGFR
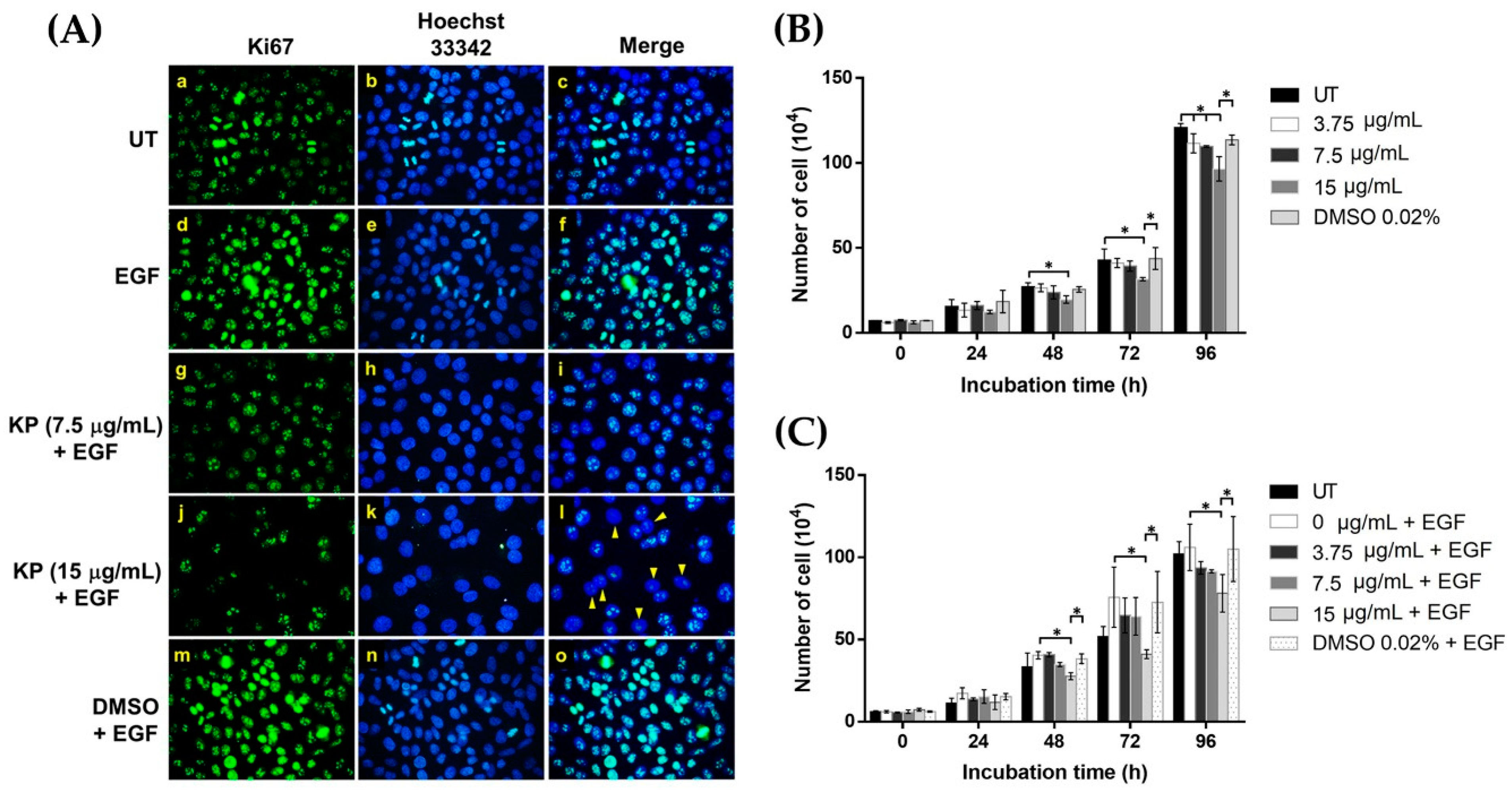
2.5. KP Exhibits Anti-Proliferative Effects over EGF Stimulation
2.6. KP Inhibits IL-6-Induced STAT3 Activation and Expression of the Anti-Apoptotic Protein Mcl-1
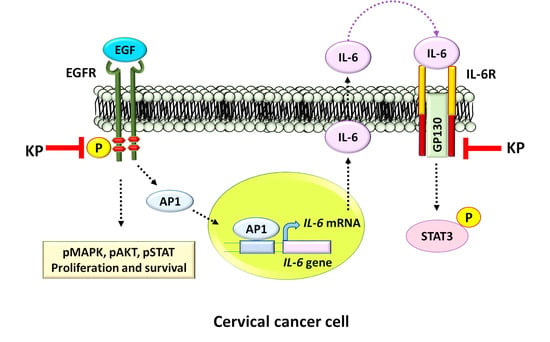
3. Discussion
4. Materials and Methods
4.1. Plant Material and Extraction of Kaempferia parviflora Rhizomes
4.2. High Performance Liquid Chromatograph Analysis
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Western Blot Analysis
4.7. Immunofluorescence Study
4.8. Cell-Counting Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMSO | Dimethyl sulfoxide |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| GP130 | Glycoprotein 130 |
| HPLC | High Performance Liquid Chromatograph |
| IL-6 | Interleukin 6 |
| KP | Kaempferia parviflora |
| STAT3 | Signal transducers and activators of transcription 3 |
| TNF-α | Tumor necrosis factor-alpha |
References
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, P.E.; Hillier, S.L.; Rabe, L.K.; Hildesheim, A.; Herrero, R.; Bratti, M.C.; Sherman, M.E.; Burk, R.D.; Rodriguez, A.C.; Alfaro, M.; et al. An association of cervical inflammation with high-grade cervical neoplasia in women infected with oncogenic human papillomavirus (HPV). Cancer Epidemiol. Biomark. Prev. 2001, 10, 1021–1027. [Google Scholar]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant expression and constitutive activation of STAT3 in cervical carcinogenesis: Implications in high-risk human papillomavirus infection. Mol. Cancer 2010, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1-NFkappaB-IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Hibi, M.; Nakagawa, N.; Nakagawa, T.; Yasukawa, K.; Yamanishi, K.; Taga, T.; Kishimoto, T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 1993, 260, 1808–1810. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Nakajima, K.; Hibi, M. Signaling mechanisms through gp130: A model of the cytokine system. Cytokine Growth Factor Rev. 1997, 8, 241–252. [Google Scholar] [CrossRef]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindler, C. STATs Dimerize in the Absence of Phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-L.; Hsieh, F.-C.; Lieblein, J.C.; Brown, J.; Chan, C.; A Wallace, J.; Cheng, G.; Hall, B.M.; Lin, J. Stat3 activation in human endometrial and cervical cancers. Br. J. Cancer 2007, 96, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.-W.; Liu, L.-J.; Huang, J. Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int. J. Oncol. 2014, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-H.; Kuo, M.-L.; Chen, C.-A.; Chou, C.-H.; Cheng, W.-F.; Chang, M.-C.; Su, J.-L.; Hsieh, C.-Y. The anti-apoptotic role of interleukin-6 in human cervical cancer is mediated by up-regulation of Mcl-1 through a PI 3-K/Akt pathway. Oncogene 2001, 20, 5799–5809. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, C.; Darnell, J.E. Transcriptional Responses to Polypeptide Ligands: The JAK-STAT Pathway. Annu. Rev. Biochem. 1995, 64, 621–652. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chapman, W.B.; Lorincz, A.T.; Willett, G.D.; Wright, V.C.; Kurman, R.J. Epidermal Growth Factor Receptor Expression and the Presence of Human Papillomavirus in Cervical Squamous Intraepithelial Lesions. Int. J. Gynecol. Pathol. 1992, 11, 221–276. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.P.; Mathur, R.S.; Young, R.C.; Rust, P.F. Human Papilloma Virus (HPV)-E6/E7 and Epidermal Growth Factor Receptor (EGF-R) Protein Levels in Cervical Cancer and Cervical Intraepithelial Neoplasia (CIN). Am. J. Reprod. Immunol. 2001, 46, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl–EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [Green Version]
- Tsai, T.-C.; Chen, S.-L. The biochemical and biological functions of human papillomavirus type 16 E5 protein. Arch. Virol. 2003, 148, 1445–1453. [Google Scholar] [CrossRef]
- Sizemore, N.; Choo, C.; Eckert, R.L.; Rorke, E.A. Transcriptional Regulation of the EGF Receptor Promoter by HPV16 and Retinoic Acid in Human Ectocervical Epithelial Cells. Exp. Cell Res. 1998, 244, 349–356. [Google Scholar] [CrossRef]
- Dellas, A.; Schultheiss, E.; Almendral, A.C.; Torhorst, J.; Gudat, F. Assessment of EGFR and TGF-alpha expression in relationship to HPV status and KI-67 distribution in cervical intraepithelial neoplasms. Int. J. Cancer 1996, 69, 165–169. [Google Scholar] [CrossRef]
- Wang, Y.; Van Boxel-Dezaire, A.H.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonomi, P.; A Blessing, J.; Stehman, F.B.; DiSaia, P.J.; Walton, L.; Major, F.J. Randomized trial of three cisplatin dose schedules in squamous-cell carcinoma of the cervix: A Gynecologic Oncology Group study. J. Clin. Oncol. 1985, 3, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.F.; Roman, L.D.; Garcia, A.A.; Muderspach, L.I.; Brader, K.R.; Morrow, C. A Phase II Study of Gemcitabine and Cisplatin in Patients with Advanced, Persistent, or Recurrent Squamous Cell Carcinoma of the Cervix. Gynecol. Oncol. 2000, 76, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Long, H.J.; Bundy, B.N.; Grendys, E.C.; Benda, J.A.; McMeekin, D.S.; Sorosky, J.; Miller, D.; Eaton, L.A.; Fiorica, J.V. Randomized Phase III Trial of Cisplatin with or Without Topotecan in Carcinoma of the Uterine Cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2005, 23, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Blessing, J.A.; Gershenson, D.M.; McGehee, R. Paclitaxel and Cisplatin as First-Line Therapy in Recurrent or Advanced Squamous Cell Carcinoma of the Cervix: A Gynecologic Oncology Group Study. J. Clin. Oncol. 1999, 17, 2676–2680. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar] [PubMed]
- Bellone, S.; Frera, G.; Landolfi, G.; Romani, C.; Bandiera, E.; Tognon, G.; Roman, J.J.; Burnett, A.F.; Pecorelli, S.; Santin, A.D. Overexpression of epidermal growth factor type-1 receptor (EGF-R1) in cervical cancer: Implications for Cetuximab-mediated therapy in recurrent/metastatic disease. Gynecol. Oncol. 2007, 106, 513–520. [Google Scholar] [CrossRef]
- Potikanond, S.; Sookkhee, S.; Na Takuathung, M.; Mungkornasawakul, P.; Wikan, N.; Smith, D.R.; Nimlamool, W. Kaempferia parviflora Extract Exhibits Anti-cancer Activity against HeLa Cervical Cancer Cells. Front. Pharmacol. 2017, 8, 630. [Google Scholar] [CrossRef]
- Tjiong, M.Y.; Van Der Vange, N.; Kate, F.J.T.; Tjong-A-Hung, S.P.; Ter Schegget, J.; Burger, M.P.; Out, T.A. Increased IL-6 and IL-8 Levels in Cervicovaginal Secretions of Patients with Cervical Cancer. Gynecol. Oncol. 1999, 73, 285–291. [Google Scholar] [CrossRef]
- Malejczyk, J.; Malejczyk, M.; Urbanski, A.; Köck, A.; Jablonska, S.; Orth, G.; Luger, T.A. Constitutive release of IL6 by human papillomavirus type 16 (HPV16)-harboring keratinocytes: A mechanism augmenting the NK-cell-mediated lysis of HPV-bearing neoplastic cells. Cell. Immunol. 1991, 136, 155–164. [Google Scholar] [CrossRef]
- Hausen, H.Z. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.H.; Brinton, L.A. The epidemiology of cervical carcinogenesis. Cancer 1995, 76, 1888–1901. [Google Scholar] [CrossRef]
- Kastritis, E.; Charidimou, A.; Varkaris, A.; Dimopoulos, M.A. Targeted therapies in multiple myeloma. Target. Oncol. 2009, 4, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Yoshio-Hoshino, N.; Nishimoto, N. The blockade of IL-6 signaling in rational drug design. Curr. Pharm. Des. 2008, 14, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-H.; Kuo, M.-L.; Chen, C.-A.; Cheng, W.-F.; Cheng, S.-P.; Hsieh, F.-J.; Hsieh, C.-Y. Interleukin-6 in Cervical Cancer: The Relationship with Vascular Endothelial Growth Factor. Gynecol. Oncol. 2001, 82, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.-H.; Kuo, M.-L.; Chen, C.-A.; Chou, C.-H.; Lai, K.-B.; Lee, C.-N.; Hsieh, C.-Y. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene 2003, 22, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer 2013, 49, 3889–3899. [Google Scholar] [CrossRef]
- Lee, H.; Shiau, M.-Y.; Wu, T.-C.; Huang, T.-T.; Chang, Y.-H.; Cheng, Y.-W. Human Papillomavirus Type 16/18 Up-Regulates the Expression of Interleukin-6 and Antiapoptotic Mcl-1 in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 4705–4712. [Google Scholar]
- Alberti, C.; Pinciroli, P.; Valeri, B.; Ferri, R.; Ditto, A.; Umezawa, K.; Sensi, M.; Canevari, S.; Tomassetti, A. Ligand-dependent EGFR activation induces the co-expression of IL-6 and PAI-1 via the NFkB pathway in advanced-stage epithelial ovarian cancer. Oncogene 2012, 31, 4139–4149. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.T.; Cohen, P.; Rousseau, S. IL-1beta-stimulated activation of ERK1/2 and p38alpha MAPK mediates the transcriptional up-regulation of IL-6, IL-8 and GRO-alpha in HeLa cells. Cell Signal. 2008, 20, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Paramee, S.; Sookkhee, S.; Sakonwasun, C.; Na Takuathung, M.; Mungkornasawakul, P.; Nimlamool, W.; Potikanond, S. Anti-cancer effects of Kaempferia parviflora on ovarian cancer SKOV3 cells. BMC Complement. Altern. Med. 2018, 18, 178. [Google Scholar] [CrossRef] [PubMed]
- Taga, T. Gp130, a shared signal transducing receptor component for hematopoietic and neuropoietic cytokines. J. Neurochem. 1996, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Taga, T. The signal transducer gp130 is shared by interleukin-6 family of haematopoietic and neurotrophic cytokines. Ann. Med. 1997, 29, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Kishimoto, T. Gp 130 and the Interleukin-6 Family of Cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Lo, H.W. Landscape of EGFR signaling network in human cancers: Biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012, 318, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downward, J.; Parker, P.; Waterfield, M.D. Autophosphorylation sites on the epidermal growth factor receptor. Nature 1984, 311, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, M.; Veyrune, J.-L.; De Vos, J.; Redal, N.; Couderc, G.; Klein, B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene 2003, 22, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.Y.; Rios, A.C.; Pal, B.; Soetanto, R.; Lun, A.T.L.; Liu, K.; Beck, T.; Best, S.A.; Vaillant, F.; Bouillet, P.; et al. EGF-mediated induction of Mcl-1 at the switch to lactation is essential for alveolar cell survival. Nature 2015, 17, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Booy, E.P.; Henson, E.S.; Gibson, S.B. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene 2011, 30, 2367–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Cruz, A.P.; Gansberger, E.; Pardee, A.B. Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2000, 97, 8542–8547. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Rojas, M.; Lin, Y.-Z. Controlling Epidermal Growth Factor (EGF)-stimulated Ras Activation in Intact Cells by a Cell-permeable Peptide Mimicking Phosphorylated EGF Receptor. J. Biol. Chem. 1996, 271, 27456–27461. [Google Scholar] [Green Version]
- Gerdes, J.; Schwab, U.; Lemke, H.; Stein, H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int. J. Cancer 1983, 31, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Yerushalmi, R.; Woods, R.; Ravdin, P.M.; Hayes, M.M.; Gelmon, K.A. Ki67 in breast cancer: Prognostic and predictive potential. Lancet Oncol. 2010, 11, 174–183. [Google Scholar] [CrossRef]
- Sutthanut, K.; Sripanidkulchai, B.; Yenjai, C.; Jay, M. Simultaneous identification and quantitation of 11 flavonoid constituents in Kaempferia parviflora by gas chromatography. J. Chromatogr. A 2007, 1143, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Yenjai, C.; Prasanphen, K.; Daodee, S.; Wongpanich, V.; Kittakoop, P. Bioactive flavonoids from Kaempferia parviflora. Fitoterapia 2004, 75, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suradej, B.; Sookkhee, S.; Panyakaew, J.; Mungkornasawakul, P.; Wikan, N.; Smith, D.R.; Potikanond, S.; Nimlamool, W. Kaempferia parviflora Extract Inhibits STAT3 Activation and Interleukin-6 Production in HeLa Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 4226. https://doi.org/10.3390/ijms20174226
Suradej B, Sookkhee S, Panyakaew J, Mungkornasawakul P, Wikan N, Smith DR, Potikanond S, Nimlamool W. Kaempferia parviflora Extract Inhibits STAT3 Activation and Interleukin-6 Production in HeLa Cervical Cancer Cells. International Journal of Molecular Sciences. 2019; 20(17):4226. https://doi.org/10.3390/ijms20174226
Chicago/Turabian StyleSuradej, Benjamart, Siriwoot Sookkhee, Jukreera Panyakaew, Pitchaya Mungkornasawakul, Nitwara Wikan, Duncan R. Smith, Saranyapin Potikanond, and Wutigri Nimlamool. 2019. "Kaempferia parviflora Extract Inhibits STAT3 Activation and Interleukin-6 Production in HeLa Cervical Cancer Cells" International Journal of Molecular Sciences 20, no. 17: 4226. https://doi.org/10.3390/ijms20174226