Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases

 , and
, and 
Abstract
:1. Introduction
2. Structure of Extracellular Matrix in the Adipose Tissue and Obesity
2.1. Integrins and Other Receptors
2.2. Collagens
2.3. Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs)
2.4. Other Components: Osteopontin, Hyaluronan, and Thrombospondin
3. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Insulin Resistance
3.1. Angiogenesis
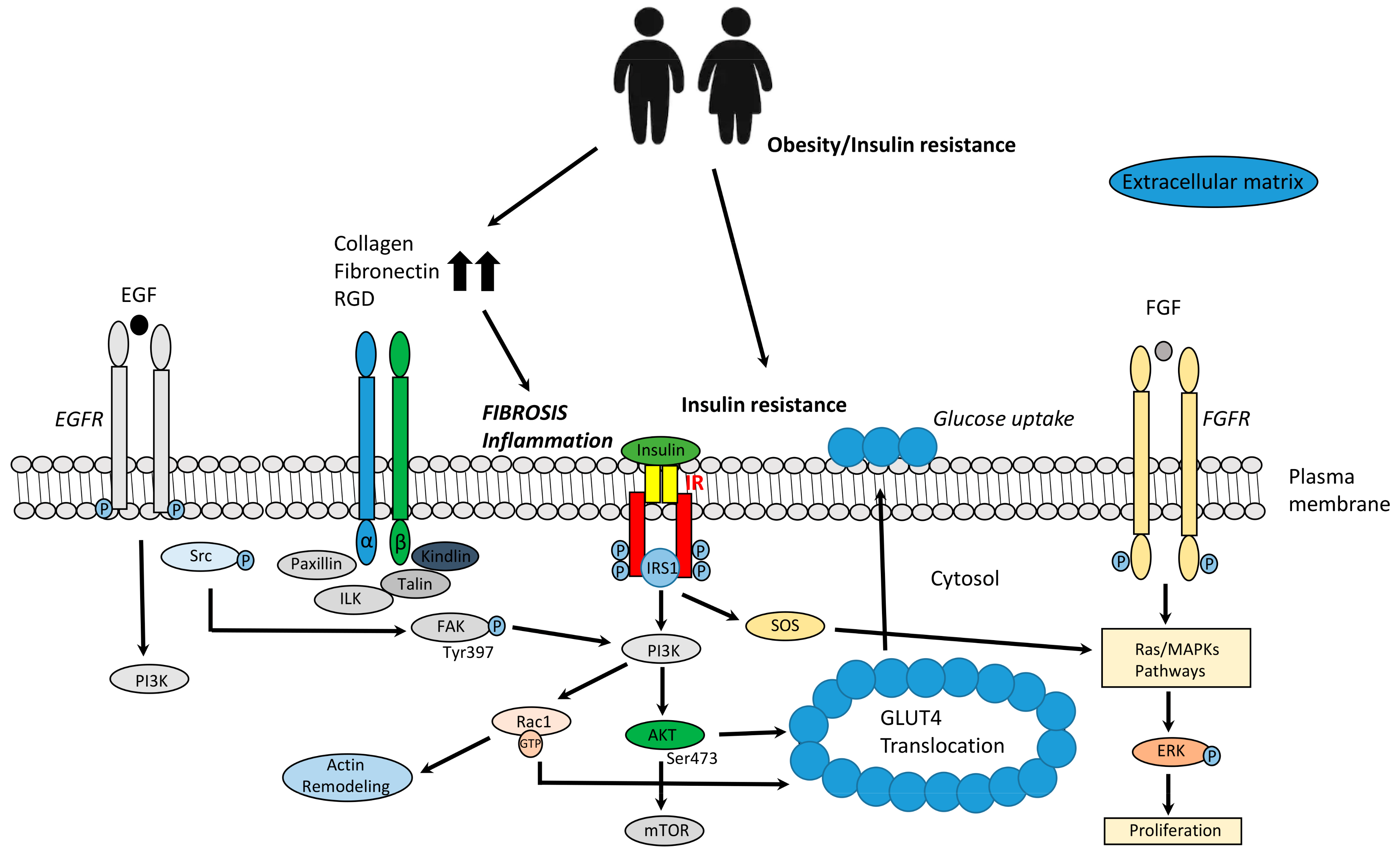
3.2. ECM Remodeling, Insulin Signaling, and Glucose Homeostasis
3.3. Potential Targets to Improve Adipose Fibrosis and Dysfunction in Obesity
4. Epigenetic
5. Clinical Studies
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- GBD 2015 Eastern Mediterranean Region Obesity Collaborators. Burden of obesity in the Eastern Mediterranean Region: Findings from the Global Burden of Disease 2015 study. Int. J. Public Health 2018, 63, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Forouzanfar, M.H.; Daoud, F.; El Bcheraoui, C.; Moradi-Lakeh, M.; Khalil, I.; Afshin, A.; Tuffaha, M.; Charara, R.; Barber, R.M.; et al. Health in times of uncertainty in the eastern Mediterranean region, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Glob. Health 2016, 4, e704–e713. [Google Scholar] [CrossRef]
- Mathew, H.; Farr, O.M.; Mantzoros, C.S. Metabolic health and weight: Understanding metabolically unhealthy normal weight or metabolically healthy obese patients. Metabolism 2016, 65, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.; Van Hulst, A.; Von Oettingen, J.E.; Benedetti, A.; Paradis, G.; Von Oettingenn, J.E. Normal weight metabolically unhealthy phenotype in youth: Do definitions matter? Pediatr. Diabetes 2019, 20, 143–151. [Google Scholar] [CrossRef]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Schoettl, T.; Fischer, I.P.; Ussar, S. Heterogeneity of adipose tissue in development and metabolic function. J. Exp. Biol. 2018, 221, jeb162958. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, I.; Aso, H.; Yamaguchi, T.; Ozutsumi, K. Adipose tissue extracellular matrix: Newly organized by adipocytes during differentiation. Differentiation 1998, 63, 193–200. [Google Scholar] [CrossRef]
- Mori, S.; Kiuchi, S.; Ouchi, A.; Hase, T.; Murase, T. Characteristic Expression of Extracellular Matrix in Subcutaneous Adipose Tissue Development and Adipogenesis; Comparison with Visceral Adipose Tissue. Int. J. Biol. Sci. 2014, 10, 825–833. [Google Scholar] [CrossRef] [Green Version]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Chun, T.H.; Kang, L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem. Pharmacol. 2016, 119, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Tordjman, J.; Clément, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R. Integrins: A family of cell surface receptors. Cell 1987, 48, 549–554. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and kindlins; partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef]
- Sun, Z.; Costell, M.; Fässler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef]
- Luk, C.T.; Shi, S.Y.; Cai, E.P.; Sivasubramaniyam, T.; Krishnamurthy, M.; Brunt, J.J.; Schroer, S.A.; Winer, D.A.; Woo, M. FAK signalling controls insulin sensitivity through regulation of adipocyte survival. Nat. Commun. 2017, 8, 14360. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Bastie, C.C.; Xu, J.; Fassler, R.; Campbell, K.P.; Kurland, I.J.; Pessin, J.E. Insulin resistance in striated muscle-specific integrin receptor beta1-deficient mice. J. Biol. Chem. 2009, 284, 4679–4688. [Google Scholar] [CrossRef]
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-Induced Muscle Insulin Resistance Is Associated with Extracellular Matrix Remodeling and Interaction with Integrin α2β1 in Mice. Diabetes 2011, 60, 416–426. [Google Scholar] [CrossRef]
- Gao, H.; Guo, Y.; Yan, Q.; Yang, W.; Li, R.; Lin, S.; Bai, X.; Liu, C.; Chen, D.; Cao, H.; et al. Lipoatrophy and metabolic disturbance in mice with adipose-specific deletion of kindlin-2. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Kang, L.; Mokshagundam, S.; Reuter, B.; Lark, D.S.; Sneddon, C.C.; Hennayake, C.; Williams, A.S.; Bracy, D.P.; James, F.D.; Pozzi, A.; et al. Integrin-Linked Kinase in Muscle Is Necessary for the Development of Insulin Resistance in Diet-Induced Obese Mice. Diabetes 2016, 65, 1590–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatem-Vaquero, M.; Griera, M.; García-Jerez, A.; Luengo, A.; Álvarez, J.; Rubio, J.A.; Calleros, L.; Rodríguez-Puyol, D.; Rodríguez-Puyol, M.; De Frutos, S. Peripheral insulin resistance in ILK-depleted mice by reduction of GLUT4 expression. J. Endocrinol. 2017, 234, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bilder, D.; Neufeld, T.P. Mechanical stress regulates insulin sensitivity through integrin-dependent control of insulin receptor localization. Genes Dev. 2018, 32, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Morrison, V.L.; Sneddon, C.C.; Savinko, T.; Uotila, L.; Jalicy, S.M.; Gabriel, J.L.; Kang, L.; Ashford, M.L.; Fagerholm, S.C. Mice Lacking beta2-Integrin Function Remain Glucose Tolerant in Spite of Insulin Resistance, Neutrophil Infiltration and Inflammation. PLoS ONE 2015, 10, e0138872. [Google Scholar] [CrossRef] [PubMed]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, Function and Association with the Malignant Process. Mol. Cell. Basis Metastasis 1997, 71, 241–319. [Google Scholar]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Kodama, K.; Horikoshi, M.; Toda, K.; Yamada, S.; Hara, K.; Irie, J.; Sirota, M.; Morgan, A.A.; Chen, R.; Ohtsu, H.; et al. Expression-based genome-wide association study links the receptor CD44 in adipose tissue with type 2 diabetes. Proc. Natl. Acad. Sci. USA 2012, 109, 7049–7054. [Google Scholar] [CrossRef]
- Rho, J.G.; Han, H.S.; Han, J.H.; Lee, H.; Nguyen, V.Q.; Lee, W.H.; Kwon, S.; Heo, S.; Yoon, J.; Shin, H.H.; et al. Self-assembled hyaluronic acid nanoparticles: Implications as a nanomedicine for treatment of type 2 diabetes. J. Control Release 2018, 279, 89–98. [Google Scholar] [CrossRef]
- Liu, L.F.; Kodama, K.; Wei, K.; Tolentino, L.L.; Choi, O.; Engleman, E.G.; Butte, A.J.; McLaughlin, T. The receptor CD44 is associated with systemic insulin resistance and proinflammatory macrophages in human adipose tissue. Diabetologia 2015, 58, 1579–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crujeiras, A.; Diaz-Lagares, A.; Moreno-Navarrete, J.; Sandoval, J.; Hervas, D.; Gomez, A.; Ricart, W.; Casanueva, F.; Esteller, M.; Fernández-Real, J.M. Genome-wide DNA methylation pattern in visceral adipose tissue differentiates insulin-resistant from insulin-sensitive obese subjects. Transl. Res. 2016, 178, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Mariman, E.C.M.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell. Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, Q.; Liu, W.; Yao, G.; Zhao, Y.; Xu, F.; Hayashi, T.; Fujisaki, H.; Hattori, S.; Tashiro, S.I.; et al. Enhanced migration of murine fibroblast-like 3T3-L1 preadipocytes on type I collagen-coated dish is reversed by silibinin treatment. Mol. Cell. Biochem. 2018, 441, 35–62. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Krautbauer, S.; Eisinger, K. Adipose tissue fibrosis. World J. Diabetes 2015, 6, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Gowronska-Kozak, B.; Burk, D.; Remedios, I.; Hymel, D.; Gimble, J.; Ravussin, E.; Bray, G.A.; Smith, S.R. Adipose tissue collagen VI in obesity. J. Clin. Endocrinol. Metab. 2009, 94, 5155–5162. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.; Loffler, M.; Bilban, M.; Reimers, M.; Kadl, A.; Todoric, J.; Zeyda, M.; Geyeregger, R.; Schreiner, M.; Weichhart, T.; et al. Prevention of high-fat diet-induced adipose tissue remodeling in obese diabetic mice by n-3 polyunsaturated fatty acids. Int. J. Obes. 2007, 31, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, K.; Franck, N.; Nitter, S.E.; Knight, B.; Mellgren, G.; Nyström, F.; Kos, K.; McCulloch, L.J.; Rawling, T.J.; Dankel, S.N.; et al. COL6A3 Is Regulated by Leptin in Human Adipose Tissue and Reduced in Obesity. Endocrinology 2015, 156, 134–146. [Google Scholar] [Green Version]
- Sun, K.; Park, J.; Gupta, O.T.; Holland, W.L.; Auerbach, P.; Zhang, N.; Marangoni, R.G.; Nicoloro, S.M.; Czech, M.P.; Varga, J.; et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat. Commun. 2014, 5, 3485. [Google Scholar] [CrossRef] [PubMed]
- Aikio, M.; Elamaa, H.; Vicente, D.; Izzi, V.; Kaur, I.; Seppinen, L.; Speedy, H.E.; Kaminska, D.; Kuusisto, S.; Sormunen, R.; et al. Specific collagen XVIII isoforms promote adipose tissue accrual via mechanisms determining adipocyte number and affect fat deposition. Proc. Natl. Acad. Sci. USA 2014, 111, E3043–E3052. [Google Scholar] [CrossRef] [Green Version]
- Bauters, D.; Cobbaut, M.; Geys, L.; Van Lint, J.; Hemmeryckx, B.; Lijnen, H.R. Loss of ADAMTS5 enhances brown adipose tissue mass and promotes browning of white adipose tissue via CREB signaling. Mol. Metab. 2017, 6, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Thrailkill, K.M.; Clay Bunn, R.; Fowlkes, J.L. Matrix metalloproteinases: Their potential role in the pathogenesis of diabetic nephropathy. Endocrine 2009, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Miksztowicz, V.; Schreier, L. Metalloproteinases in metabolic syndrome. Clin. Chim. Acta 2011, 412, 1731–1739. [Google Scholar] [CrossRef]
- Aldonyte, R.; Brantly, M.; Block, E.; Patel, J.; Zhang, J. Nuclear localization of active matrix metalloproteinase-2 in cigarette smoke-exposed apoptotic endothelial cells. Exp. Lung Res. 2009, 35, 59–75. [Google Scholar] [CrossRef]
- Tsai, J.-P.; Liou, J.-H.; Kao, W.-T.; Wang, S.-C.; Lian, J.-D.; Chang, H.-R. Increased Expression of Intranuclear Matrix Metalloproteinase 9 in Atrophic Renal Tubules Is Associated with Renal Fibrosis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Xie, Y.; Mustafa, A.; Yerzhan, A.; Merzhakupova, D.; Yerlan, P.; Orakov, A.N.; Wang, X.; Huang, Y.; Miao, L. Nuclear matrix metalloproteinases: Functions resemble the evolution from the intracellular to the extracellular compartment. Cell Death Discov. 2017, 3, 17036. [Google Scholar] [CrossRef]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, A.; Chhabra, A.; Malhotra, U.; Kohli, S.; Rani, V.R.V. Comparative analysis of human matrix metalloproteinases: Emerging therapeutic targets in diseases. Bioinformation 2011, 6, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Hopps, E.; Caimi, G. Matrix metalloproteases as a pharmacological target in cardiovascular diseases. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2583–2589. [Google Scholar] [PubMed]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Hopps, E.; Presti, R.L.; Montana, M.; Noto, D.; Averna, M.R.; Caimi, G. Gelatinases and Their Tissue Inhibitors in a Group of Subjects with Metabolic Syndrome. J. Investig. Med. 2013, 61, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Traurig, M.T.; Permana, P.A.; Nair, S.; Kobes, S.; Bogardus, C.; Baier, L.J. Differential Expression of Matrix Metalloproteinase 3 (MMP3) in Preadipocytes/Stromal Vascular Cells from Nonobese Nondiabetic Versus Obese Nondiabetic Pima Indians. Diabetes 2006, 55, 3160–3165. [Google Scholar] [CrossRef]
- Chun, T.-H.; Inoue, M.; Morisaki, H.; Yamanaka, I.; Miyamoto, Y.; Okamura, T.; Sato-Kusubata, K.; Weiss, S.J. Genetic Link Between Obesity and MMP14-Dependent Adipogenic Collagen Turnover. Diabetes 2010, 59, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Tinahones, F.J.; Coin-Araguez, L.; Mayas, M.D.; Garcia-Fuentes, E.; Hurtado-Del-Pozo, C.; Vendrell, J.; Cardona, F.; Calvo, R.-M.; Obregon, M.-J.; El Bekay, R. Obesity-associated insulin resistance is correlated to adipose tissue vascular endothelial growth factors and metalloproteinase levels. BMC Physiol. 2012, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.S.; Malaponte, G.; Libra, M.; Di Pino, L.; Celotta, G.; Bevelacqua, V.; Petrina, M.; Nicotra, G.S.; Indelicato, M.; Navolanic, P.M.; et al. Plasma levels and zymographic activities of matrix metalloproteinases 2 and 9 in type II diabetics with peripheral arterial disease. Vasc. Med. 2005, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-J.; Wu, Y.; Fried, S.K. Adipose tissue remodeling in pathophysiology of obesity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Maquoi, E.; Munaut, C.; Colige, A.; Collen, D.; Lijnen, H.R. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes 2002, 51, 1093–1101. [Google Scholar] [CrossRef]
- Miksztowicz, V.; Morales, C.; Zago, V.; Friedman, S.; Schreier, L.; Berg, G. Effect of insulin-resistance on circulating and adipose tissue MMP-2 and MMP-9 activity in rats fed a sucrose-rich diet. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 294–300. [Google Scholar] [CrossRef]
- Berg, G.; Barchuk, M.; Miksztowicz, V. Behavior of Metalloproteinases in Adipose Tissue, Liver and Arterial Wall: An Update of Extracellular Matrix Remodeling. Cells 2019, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, G.; Ferrari, I.; D’Angelo, A.; Tinelli, C.; Salvadeo, S.A.T.; Ciccarelli, L.; Piccinni, M.N.; Gravina, A.; Ramondetti, F.; Maffioli, P.; et al. Matrix Metalloproteinase-2 and -9 Levels in Obese Patients. Endothelium 2008, 15, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.M.; de Faria, A.P.; Barbaro, N.; Sabbatini, A.R.; Correa, N.B.; Brunelli, V.; Amorim, R.; Modolo, R.; Moreno, H. Crosstalk between obesity and MMP-9 in cardiac remodelling -a cross-sectional study in apparent treatment-resistant hypertension. Blood Press. 2017, 26, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, H.; Van Hoef, B.; Rodríguez, J.; Paramo, J.A. Stromelysin-2 (MMP-10) deficiency does not affect adipose tissue formation in a mouse model of nutritionally induced obesity. Biochem. Biophys. Res. Commun. 2009, 389, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Bourlier, V.; Zakaroff-Girard, A.; Miranville, A.; De Barros, S.; Maumus, M.; Sengenes, C.; Galitzky, J.; Lafontan, M.; Karpe, F.; Frayn, K.; et al. Remodeling Phenotype of Human Subcutaneous Adipose Tissue Macrophages. Circulation 2008, 117, 806–815. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Chiefari, E.; Laria, A.E.; Messineo, S.; Bilotta, F.L.; Britti, D.; Foti, D.P.; Foryst-Ludwig, A.; Kintscher, U.; Brunetti, A. Expression of matrix metalloproteinase-11 is increased under conditions of insulin resistance. World J. Diabetes 2017, 8, 422–428. [Google Scholar] [CrossRef]
- Amor, M.; Moreno-Viedma, V.; Sarabi, A.; Grün, N.G.; Itariu, B.; Leitner, L.; Steiner, I.; Bilban, M.; Kodama, K.; Butte, A.J.; et al. Identification of Matrix Metalloproteinase-12 as a Candidate Molecule for Prevention and Treatment of Cardiometabolic Disease. Mol. Med. 2016, 22, 487–496. [Google Scholar] [CrossRef]
- Lee, J.-T.; Pamir, N.; Liu, N.-C.; Kirk, E.A.; Averill, M.M.; Becker, L.; Larson, I.; Hagman, D.K.; Foster-Schubert, K.E.; Van Yserloo, B.; et al. Macrophage metalloelastase (MMP12) regulates adipose tissue expansion, insulin sensitivity, and expression of inducible nitric oxide synthase. Endocrinology 2014, 155, 3409–3420. [Google Scholar] [CrossRef]
- Abdelaziz, R.; Elbasel, M.; Esmat, S.; Essam, K.; Abdelaaty, S. Tissue Inhibitors of Metalloproteinase-1 and 2 and Obesity Related Non-Alcoholic Fatty Liver Disease: Is There a Relationship? Digestion 2015, 92, 130–137. [Google Scholar] [CrossRef]
- Yasmeen, S.; Khan, U.; Khan, G.M.; Fatima, S.S. Association of tissue inhibitor of metalloproteinase 2 with non-alcoholic fatty liver disease in metabolic syndrome. Arch. Physiol. Biochem. 2018. [Google Scholar] [CrossRef]
- Palladini, G.; Di Pasqua, L.G.; Berardo, C.; Siciliano, V.; Richelmi, P.; Perlini, S.; Ferrigno, A.; Vairetti, M. Animal Models of Steatosis (NAFLD) and Steatohepatitis (NASH) Exhibit Hepatic Lobe-Specific Gelatinases Activity and Oxidative Stress. Can. J. Gastroenterol. Hepatol. 2019, 2019, 5413461. [Google Scholar] [CrossRef] [PubMed]
- Vilmi-Kerälä, T.; Lauhio, A.; Tervahartiala, T.; Palomäki, O.; Uotila, J.; Sorsa, T.; Palomäki, A. Subclinical inflammation associated with prolonged TIMP-1 upregulation and arterial stiffness after gestational diabetes mellitus: A hospital-based cohort study. Cardiovasc. Diabetol. 2017, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhu, H.; Chen, X.; Peng, Y.; Wang, J.; Liu, F.; Shi, S.; Fu, B.; Lu, Y.; Hong, Q.; et al. TIMP-1 transgenic mice recover from diabetes induced by multiple low-dose streptozotocin. Diabetes 2007, 56, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Louis, G.W.; Zhang, X.; Prestwich, T.C.; Kumar, T.R.; Myers, M.G., Jr.; Macdougald, O.A.; Nothnick, W.B. Hyperphagia and obesity in female mice lacking tissue inhibitor of metalloproteinase-1. Endocrinology 2009, 150, 1697–1704. [Google Scholar] [CrossRef]
- Rebuffat, S.A.; Sidot, E.; Guzman, C.; Azay-Milhau, J.; Jover, B.; Lajoix, A.-D.; Peraldi-Roux, S. Adipose tissue derived-factors impaired pancreatic β-cell function in diabetes. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3378–3387. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, D.M.; Sideleva, O.; Stradecki, H.M.; Langlois, G.D.; Habibovic, A.; Satish, B.; Tharp, W.G.; Lausier, J.; LaRock, K.; Jetton, T.L.; et al. Sexually dimorphic diet-induced insulin resistance in obese tissue inhibitor of metalloproteinase-2 (TIMP-2)-deficient mice. Endocrinology 2011, 152, 1300–1313. [Google Scholar] [CrossRef]
- Duarte, F.O.; Gomes-Gatto, C.D.V.; Oishi, J.C.; Lino, A.D.D.S.; Stotzer, U.S.; Rodrigues, M.F.C.; Da Silva, G.H.G.; Selistre-De-Araújo, H.S. Physical training improves visceral adipose tissue health by remodelling extracellular matrix in rats with estrogen absence: A gene expression analysis. Int. J. Exp. Pathol. 2017, 98, 203–213. [Google Scholar] [CrossRef]
- Menghini, R.; Menini, S.; Amoruso, R.; Fiorentino, L.; Casagrande, V.; Marzano, V.; Tornei, F.; Bertucci, P.; Iacobini, C.; Serino, M.; et al. Tissue Inhibitor of Metalloproteinase 3 Deficiency Causes Hepatic Steatosis and Adipose Tissue Inflammation in Mice. Gastroenterology 2009, 136, 663–672. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Menini, S.; Marino, A.; Marzano, V.; Hribal, M.L.; Gentileschi, P.; Lauro, D.; Schillaci, O.; Pugliese, G.; et al. TIMP3 Overexpression in Macrophages Protects from Insulin Resistance, Adipose Inflammation, and Nonalcoholic Fatty Liver Disease in Mice. Diabetes 2012, 61, 454–462. [Google Scholar] [CrossRef]
- Wu, Y.; Lee, M.J.; Ido, Y.; Fried, S.K. High-fat diet-induced obesity regulates MMP3 to modulate depot- and sex-dependent adipose expansion in C57BL/6J mice. American journal of physiology. Endocrinol. Metab. 2017, 312, E58–E71. [Google Scholar] [CrossRef]
- Sakamuri, S.S.V.P.; Watts, R.; Takawale, A.; Wang, X.; Hernandez-Anzaldo, S.; Bahitham, W.; Fernandez-Patron, C.; Lehner, R.; Kassiri, Z. Absence of Tissue Inhibitor of Metalloproteinase-4 (TIMP4) ameliorates high fat diet-induced obesity in mice due to defective lipid absorption. Sci. Rep. 2017, 7, 6210. [Google Scholar] [CrossRef] [PubMed]
- Voros, G.; Maquoi, E.; Collen, D.; Lijnen, H. Differential expression of plasminogen activator inhibitor-1, tumor necrosis factor-α, TNF-α converting enzyme and ADAMTS family members in murine fat territories. Biochim. Biophys. Acta (BBA) Gene Struct. Expr. 2003, 1625, 36–42. [Google Scholar] [CrossRef]
- Voros, G.; Sandy, J.D.; Collen, D.; Lijnen, H.R. Expression of aggrecan(ases) during murine preadipocyte differentiation and adipose tissue development. Biochim. Biophys. Acta (BBA) Gen. Subj. 2006, 1760, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, X.; Pan, W.; Unger, R.H. Gene expression profile of rat adipose tissue at the onset of high-fat-diet obesity. Am. J. Physiol. Metab. 2002, 282, E1334–E1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koza, R.A.; Nikonova, L.; Hogan, J.; Rim, J.-S.; Mendoza, T.; Faulk, C.; Skaf, J.; Kozak, L.P. Changes in Gene Expression Foreshadow Diet-Induced Obesity in Genetically Identical Mice. PLoS Genet. 2006, 2, e81. [Google Scholar] [CrossRef] [PubMed]
- Bauters, D.; Scroyen, I.; Deprez-Poulain, R.; Lijnen, H.R. ADAMTS5 promotes murine adipogenesis and visceral adipose tissue expansion. Thromb. Haemost. 2016, 116, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.S.; Kim, B.S.; Kim, J.Y.; Kim, J.D.; Choi, Y.C.; Yang, H.-J.; Park, K.; Lee, H.Y.; Cho, Y.W. Decellularized extracellular matrix derived from human adipose tissue as a potential scaffold for allograft tissue engineering. J. Biomed. Mater. Res. Part A 2011, 97, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Filippov, S.; Caras, I.; Murray, R.; Matrisian, L.M.; Chapman, H.A.; Shapiro, S.; Weiss, S.J. Matrilysin-dependent Elastolysis by Human Macrophages. J. Exp. Med. 2003, 198, 925–935. [Google Scholar] [CrossRef]
- Martinez-Santibanez, G.; Singer, K.; Cho, K.W.; DelProposto, J.L.; Mergian, T.; Lumeng, C.N. Obesity-induced remodeling of the adipose tissue elastin network is independent of the metalloelastase MMP-12. Adipocyte 2015, 4, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Blaise, S.; Romier, B.; Kawecki, C.; Ghirardi, M.; Rabenoelina, F.; Baud, S.; Duca, L.; Maurice, P.; Heinz, A.; Schmelzer, C.E.; et al. Elastin-Derived Peptides Are New Regulators of Insulin Resistance Development in Mice. Diabetes 2013, 62, 3807–3816. [Google Scholar] [CrossRef]
- DeMarsilis, A.J.; Walji, T.A.; Maedeker, J.A.; Stoka, K.V.; Kozel, B.A.; Mecham, R.P.; Wagenseil, J.E.; Craft, C.S. Elastin Insufficiency Predisposes Mice to Impaired Glucose Metabolism. J. Mol. Genet. Med. 2014, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.W.; Zeyda, M.; Todoric, J.; Huber, J.; Geyeregger, R.; Weichhart, T.; Aszmann, O.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; et al. Osteopontin Expression in Human and Murine Obesity: Extensive Local Up-Regulation in Adipose Tissue but Minimal Systemic Alterations. Endocrinology 2008, 149, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschöp, M.H.; et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J. Clin. Investig. 2007, 117, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Miles, P.D.; Ofrecio, J.M.; Neels, J.G.; Yu, J.G.; Resnik, J.L.; Wilkes, J.; Talukdar, S.; Thapar, D.; Johnson, K.; et al. Osteopontin Is Required for the Early Onset of High Fat Diet-Induced Insulin Resistance in Mice. PLoS ONE 2010, 5, e13959. [Google Scholar] [CrossRef] [PubMed]
- Lancha, A.; Rodríguez, A.; Catalán, V.; Becerril, S.; Sáinz, N.; Ramírez, B.; Burrell, M.A.; Salvador, J.; Frühbeck, G.; Gómez-Ambrosi, J. Osteopontin Deletion Prevents the Development of Obesity and Hepatic Steatosis via Impaired Adipose Tissue Matrix Remodeling and Reduced Inflammation and Fibrosis in Adipose Tissue and Liver in Mice. PLoS ONE 2014, 9, e98398. [Google Scholar] [CrossRef]
- Kiefer, F.W.; Zeyda, M.; Gollinger, K.; Pfau, B.; Neuhofer, A.; Weichhart, T.; Säemann, M.D.; Geyeregger, R.; Schlederer, M.; Kenner, L.; et al. Neutralization of Osteopontin Inhibits Obesity-Induced Inflammation and Insulin Resistance. Diabetes 2010, 59, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, C.; Tang, Y.; Zhu, Q.; Li, Y.; Chen, H.; Bao, Y.; Xue, S.; Sun, L.; Tang, W.; et al. High glucose up-regulates osteopontin expression by FoxO1 activation in macrophage. J. Endocrinol. 2019, 242, 51–64. [Google Scholar] [CrossRef]
- Barchetta, I.; Ceccarelli, V.; Cimini, F.A.; Bertoccini, L.; Fraioli, A.; Alessandri, C.; Lenzi, A.; Baroni, M.G.; Cavallo, M.G. Impaired bone matrix glycoprotein pattern is associated with increased cardio-metabolic risk profile in patients with type 2 diabetes mellitus. J. Endocrinol. Investig. 2019, 42, 513–520. [Google Scholar] [CrossRef]
- Carbone, F.; Adami, G.; Liberale, L.; Bonaventura, A.; Bertolotto, M.; Andraghetti, G.; Scopinaro, N.; Camerini, G.B.; Papadia, F.S.; Cordera, R.; et al. Serum levels of osteopontin predict diabetes remission after bariatric surgery. Diabetes Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stern, R. Devising a pathway for hyaluronan catabolism: Are we there yet? Glycobiology 2003, 13, 105R–115R. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Subramanian, S.; Chan, C.K.; Omer, M.; Chiba, T.; Wight, T.N.; Chait, A. Adipocyte-Derived Serum Amyloid A3 and Hyaluronan Play a Role in Monocyte Recruitment and Adhesion. Diabetes 2007, 56, 2260–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, E.; Jung, M.Y.; Park, J.H.; Kim, S.; Seo, C.R.; Park, K.W.; Lee, E.K.; Yeom, C.H.; Lee, S. Inhibition of adipogenesis in 3T3-L1 cells and suppression of abdominal fat accumulation in high-fat diet-feeding C57BL/6J mice after downregulation of hyaluronic acid. Int. J. Obes. 2014, 38, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Lantier, L.; Kennedy, A.; Bonner, J.S.; Mayes, W.H.; Bracy, D.P.; Bookbinder, L.H.; Hasty, A.H.; Thompson, C.B.; Wasserman, D.H. Hyaluronan Accumulates with High-Fat Feeding and Contributes to Insulin Resistance. Diabetes 2013, 62, 1888–1896. [Google Scholar] [CrossRef]
- Prakash, J.; Gabdulina, G.; Trofimov, S.; Livshits, G. Quantitative genetics of circulating Hyaluronic Acid (HA) and its correlation with hand osteoarthritis and obesity-related phenotypes in a community-based sample. Ann. Hum. Biol. 2017, 44, 1–9. [Google Scholar] [CrossRef]
- Pothuraju, R.; Rachagani, S.; Junker, W.M.; Chaudhary, S.; Saraswathi, V.; Kaur, S.; Batra, S.K. Pancreatic cancer associated with obesity and diabetes: An alternative approach for its targeting. J. Exp. Clin. Cancer Res. 2018, 37, 319. [Google Scholar] [CrossRef]
- Wilson, N.; Steadman, R.; Muller, I.; Draman, M.; Rees, D.A.; Taylor, P.; Dayan, C.M.; Ludgate, M.; Zhang, L. Role of Hyaluronan in Human Adipogenesis: Evidence from in-Vitro and in-Vivo Studies. Int. J. Mol. Sci. 2019, 20, 2675. [Google Scholar] [CrossRef]
- Varma, V.; Yao-Borengasser, A.; Bodles, A.M.; Rasouli, N.; Phanavanh, B.; Nolen, G.T.; Kern, E.M.; Nagarajan, R.; Spencer, H.J., 3rd; Lee, M.J.; et al. Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes 2008, 57, 432–439. [Google Scholar] [CrossRef]
- Matsuo, Y.; Tanaka, M.; Yamakage, H.; Sasaki, Y.; Muranaka, K.; Hata, H.; Ikai, I.; Shimatsu, A.; Inoue, M.; Chun, T.-H.; et al. Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism 2015, 64, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Jiang, Y.; Barnes, R.H., 2nd; Tokunaga, M.; Martinez-Santibanez, G.; Geletka, L.; Lumeng, C.N.; Buchner, D.A.; Chun, T.H. Thrombospondin 1 mediates high-fat diet-induced muscle fibrosis and insulin resistance in male mice. Endocrinology 2013, 154, 4548–4559. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tong, X.; Rumala, C.; Clemons, K.; Wang, S. Thrombospondin1 Deficiency Reduces Obesity-Associated Inflammation and Improves Insulin Sensitivity in a Diet-Induced Obese Mouse Model. PLoS ONE 2011, 6, e26656. [Google Scholar] [CrossRef] [PubMed]
- Matsugi, K.; Hosooka, T.; Nomura, K.; Ogawa, W. Thrombospondin 1 Suppresses Insulin Signaling in C2C12 Myotubes. Kobe J. Med. Sci. 2016, 62, E13–E18. [Google Scholar] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; King, A.; Crombleholme, T.M.; Keswani, S.G. The Role of Endothelial Progenitor Cells in Postnatal Vasculogenesis: Implications for Therapeutic Neovascularization and Wound Healing. Adv. Wound Care 2013, 2, 283–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y. Angiogenesis and Vascular Functions in Modulation of Obesity, Adipose Metabolism, and Insulin Sensitivity. Cell Metab. 2013, 18, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiaens, V.; Lijnen, H. Angiogenesis and development of adipose tissue. Mol. Cell. Endocrinol. 2010, 318, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, A.Y.; LeDoux, S.; Larger, E. Adipose tissue angiogenesis in obesity. Thromb. Haemost. 2013, 110, 661–669. [Google Scholar] [CrossRef]
- Graupera, M.; Claret, M. Endothelial Cells: New Players in Obesity and Related Metabolic Disorders. Trends Endocrinol. Metab. 2018, 29, 781–794. [Google Scholar] [CrossRef]
- Cao, Y. Angiogenesis modulates adipogenesis and obesity. J. Clin. Investig. 2007, 117, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Brakenhielm, E.; Wahlestedt, C.; Thyberg, J.; Cao, Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc. Natl. Acad. Sci. USA 2001, 98, 6390–6395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Mepani, R.J.; Kleiner, S.; Lo, J.C.; Khandekar, M.J.; Cohen, P.; Frontini, A.; Bhowmick, D.C.; Ye, L.; Cinti, S.; et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012, 15, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Matulewicz, N.; Stefanowicz, M.; Nikołajuk, A.; Karczewska-Kupczewska, M. Markers of adipogenesis, but not inflammation in adipose tissue, are independently related to insulin sensitivity. J. Clin. Endocrinol. Metab. 2017, 102, 3040–3049. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Cao, Y. Angiogenesis as a therapeutic target for obesity and metabolic diseases. Chem. Immunol. Allergy 2014, 99, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Gealekman, O.; Burkart, A.; Chouinard, M.; Nicoloro, S.M.; Straubhaar, J.; Corvera, S. Enhanced angiogenesis in obesity and in response to PPARgamma activators through adipocyte VEGF and ANGPTL4 production. Am. J. Physiol. Metab. 2008, 295, E1056–E1064. [Google Scholar]
- Gealekman, O.; Gurav, K.; Chouinard, M.; Straubhaar, J.; Thompson, M.; Malkani, S.; Hartigan, C.; Corvera, S. Control of Adipose Tissue Expandability in Response to High Fat Diet by the Insulin-like Growth Factor-binding Protein-4. J. Biol. Chem. 2014, 289, 18327–18338. [Google Scholar] [CrossRef] [Green Version]
- Moens, S.; Goveia, J.; Stapor, P.C.; Cantelmo, A.R.; Carmeliet, P. The multifaceted activity of VEGF in angiogenesis – Implications for therapy responses. Cytokine Growth Factor Rev. 2014, 25, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, H.R. Angiogenesis and obesity. Cardiovasc. Res. 2008, 78, 286–293. [Google Scholar] [CrossRef]
- Miyazawa-Hoshimoto, S.; Takahashi, K.; Bujo, H.; Hashimoto, N.; Saito, Y. Elevated serum vascular endothelial growth factor is associated with visceral fat accumulation in human obese subjects. Diabetologia 2003, 46, 1483–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colina, I.; Silva, C.; Mugueta, M.D.C.; Cienfuegos, J.A.; Gómez-Ambrosi, J.; Catalán, V.; Ramírez, B.; Rodríguez, A.; Rotellar, F.; Gil, M.J.; et al. Plasma Osteopontin Levels and Expression in Adipose Tissue Are Increased in Obesity. J. Clin. Endocrinol. Metab. 2007, 92, 3719–3727. [Google Scholar] [Green Version]
- Loebig, M.; Klement, J.; Schmoller, A.; Betz, S.; Heuck, N.; Schweiger, U.; Peters, A.; Schultes, B.; Oltmanns, K.M. Evidence for a Relationship between VEGF and BMI Independent of Insulin Sensitivity by Glucose Clamp Procedure in a Homogenous Group Healthy Young Men. PLoS ONE 2010, 5, e12610. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Considine, R.V.; Bovenkerk, J.E.; Li, J.; Slavens, C.A.; Jones, R.M.; March, K.L. Obesity is associated with increased levels of circulating hepatocyte growth factor. J. Am. Coll. Cardiol. 2003, 41, 1408–1413. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.-K.; Doh, K.-O.; Son, J.E.; Park, J.G.; Bae, Y.; Choi, S.; Nelson, S.M.L.; Cowling, R.; Nagy, K.; Michael, I.P.; et al. Adipose Vascular Endothelial Growth Factor Regulates Metabolic Homeostasis through Angiogenesis. Cell Metab. 2013, 17, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-Inducible Factor 1α Induces Fibrosis and Insulin Resistance in White Adipose Tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef]
- Pasarica, M.; Sereda, O.R.; Redman, L.M.; Albarado, D.C.; Hymel, D.T.; Roan, L.E.; Rood, J.C.; Burk, D.H.; Smith, S.R. Reduced adipose tissue oxygenation in human obesity: Evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 2009, 58, 718–725. [Google Scholar] [CrossRef]
- Elias, I.; Franckhauser, S.; Ferre, T.; Vilà, L.; Tafuro, S.; Muñoz, S.; Roca, C.; Ramos, D.; Pujol, A.; Riu, E.; et al. Adipose Tissue Overexpression of Vascular Endothelial Growth Factor Protects Against Diet-Induced Obesity and Insulin Resistance. Diabetes 2012, 61, 1801–1813. [Google Scholar] [CrossRef] [Green Version]
- Romacho, T.; Elsen, M.; Röhrborn, D.; Eckel, J. Adipose tissue and its role in organ crosstalk. Acta Physiol. 2014, 210, 733–753. [Google Scholar] [CrossRef]
- Zafar, M.I.; Mills, K.; Ye, X.; Blakely, B.; Min, J.; Kong, W.; Zhang, N.; Gou, L.; Regmi, A.; Hu, S.Q.; et al. Association between the expression of vascular endothelial growth factors and metabolic syndrome or its components: A systematic review and meta-analysis. Diabetol. Metab. Syndr. 2018, 10, 62. [Google Scholar] [CrossRef]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose Tissue Hypoxia in Obesity and Its Impact on Adipocytokine Dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobellis, G. Epicardial adipose tissue in endocrine and metabolic diseases. Endocrine 2014, 46, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Hypoxia and Adipose Tissue Function and Dysfunction in Obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, J.M.; Davis, K.E.; Scherer, P.E. Mechanisms of obesity and related pathologies: The macro- and microcirculation of adipose tissue. FEBS J. 2009, 276, 5738–5746. [Google Scholar] [CrossRef] [Green Version]
- Regazzetti, C.; Peraldi, P.; Gremeaux, T.; Najem-Lendom, R.; Ben-Sahra, I.; Cormont, M.; Bost, F.; Le Marchand-Brustel, Y.; Tanti, J.F.; Giorgetti-Peraldi, S. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes 2009, 58, 95–103. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, Q. Therapeutic Targets of Multiple Angiogenic Factors for the Treatment of Cancer and Metastasis. Mol. Cell. Basis Metastasis 2007, 97, 203–224. [Google Scholar]
- Goossens, G.H.; Bizzarri, A.; Venteclef, N.; Essers, Y.; Cleutjens, J.P.; Konings, E.; Jocken, J.W.; Cajlakovič, M.; Ribitsch, V.; Clément, K.; et al. Increased Adipose Tissue Oxygen Tension in Obese Compared with Lean Men Is Accompanied by Insulin Resistance, Impaired Adipose Tissue Capillarization, and Inflammation. Circulation 2011, 124, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Pajvani, U.B.; Trujillo, M.E.; Combs, T.P.; Iyengar, P.; Jelicks, L.; Roth, K.A.; Kitsis, R.N.; Scherer, P.E. Fat apoptosis through targeted activation of caspase 8: A new mouse model of inducible and reversible lipoatrophy. Nat. Med. 2005, 11, 797–803. [Google Scholar] [CrossRef]
- Yin, J.; Gao, Z.; He, Q.; Zhou, D.; Guo, Z.; Ye, J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. American journal of physiology. Endocrinol. Metab. 2009, 296, E333–E342. [Google Scholar] [CrossRef]
- Kabon, B.; Nagele, A.; Reddy, D.; Eagon, C.; Fleshman, J.W.; Sessler, D.I.; Kurz, A. Obesity decreases perioperative tissue oxygenation. Anesthesiology 2004, 100, 274–280. [Google Scholar] [CrossRef]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, G.H.; Vogel, M.A.A.; Vink, R.G.; Mariman, E.C.; Van Baak, M.A.; Blaak, E.E. Adipose tissue oxygenation is associated with insulin sensitivity independently of adiposity in obese men and women. Diabetes Obes. Metab. 2018, 20, 2286–2290. [Google Scholar] [CrossRef] [PubMed]
- Hiltebrand, L.B.; Kaiser, H.A.; Niedhart, D.J.; Pestel, G.; Kurz, A. Subcutaneous oxygen pressure in spontaneously breathing lean and obese volunteers: A pilot study. Obes. Surg. 2008, 18, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Pasarica, M.; Xie, H.; Hymel, D.; Bray, G.; Greenway, F.; Ravussin, E.; Smith, S.R. Lower Total Adipocyte Number but No Evidence for Small Adipocyte Depletion in Patients with Type 2 Diabetes. Diabetes Care 2009, 32, 900–902. [Google Scholar] [CrossRef] [PubMed]
- Anthanont, P.; Ramos, P.; Jensen, M.D.; Hames, K.C. Family history of type 2 diabetes, abdominal adipocyte size and markers of the metabolic syndrome. Int. J. Obes. 2017, 41, 1621–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Arner, E.; Hammarstedt, A.; Smith, U. Genetic Predisposition for Type 2 Diabetes, but Not for Overweight/Obesity, Is Associated with a Restricted Adipogenesis. PLoS ONE 2011, 6, e18284. [Google Scholar] [CrossRef]
- Goossens, G.H.; Blaak, E.E. Adipose Tissue Dysfunction and Impaired Metabolic Health in Human Obesity: A Matter of Oxygen? Front. Endocrinol. 2015, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Petrus, P.; Fernandez, T.L.; Kwon, M.M.; Huang, J.L.; Lei, V.; Safikhan, N.S.; Karunakaran, S.; O’Shannessy, D.J.; Zheng, X.; Catrina, S.-B.; et al. Specific loss of adipocyte CD248 improves metabolic health via reduced white adipose tissue hypoxia, fibrosis and inflammation. EBioMedicine 2019, 44, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Kokai, L.E.; Schilling, B.K.; Chnari, E.; Huang, Y.-C.; Imming, E.A.; Karunamurthy, A.; Khouri, R.K.; D’Amico, R.A.; Coleman, S.R.; Marra, K.G.; et al. Injectable Allograft Adipose Matrix Supports Adipogenic Tissue Remodeling in the Nude Mouse and Human. Plast. Reconstr. Surg. 2019, 143, 299e–309e. [Google Scholar] [CrossRef]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen VI. Mol. Cell. Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Mayes, W.H.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Matrix metalloproteinase 9 opposes diet-induced muscle insulin resistance in mice. Diabetologia 2014, 57, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Frühbeck, G. Role of extracellular matrix remodelling in adipose tissue pathophysiology: Relevance in the development of obesity. Histol. Histopathol. 2012, 27, 1515–1528. [Google Scholar]
- Wang, B.; Wood, I.S.; Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflügers Arch. Eur. J. Physiol. 2007, 455, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keophiphath, M.; Achard, V.; Henegar, C.; Rouault, C.; Clément, K.; Lacasa, D. Macrophage-Secreted Factors Promote a Profibrotic Phenotype in Human Preadipocytes. Mol. Endocrinol. 2009, 23, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Consitt, L.A.; Bell, J.A.; Houmard, J.A. Intramuscular lipid metabolism, insulin action, and obesity. IUBMB Life 2009, 61, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A.; Lotan, Y.; Zhang, J.; Rosenthal, T.R.; Rogers, J.T.; Adams-Huet, B.; Sakhaee, K.; Moe, O.W. Triglycerides in the Human Kidney Cortex: Relationship with Body Size. PLoS ONE 2014, 9, e101285. [Google Scholar] [CrossRef]
- Catanzaro, R.; Cuffari, B.; Italia, A.; Marotta, F. Exploring the metabolic syndrome: Nonalcoholic fatty pancreas disease. World J. Gastroenterol. 2016, 22, 7660–7675. [Google Scholar] [CrossRef]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and nonalcoholic fatty liver disease: Current perspectives. Diabetes Metab. Syndr. Obesity Targets Ther. 2018, 11, 533–542. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nature reviews. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Milić, S.; Lulić, D.; Štimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J. Gastroenterol. 2014, 20, 9330–9337. [Google Scholar] [PubMed]
- Hassan, K.; Bhalla, V.; El Regal, M.E.; A-Kader, H.H. Nonalcoholic fatty liver disease: A comprehensive review of a growing epidemic. World J. Gastroenterol. 2014, 20, 12082–12101. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Barve, S.; Deaciuc, I. Good fat/bad fat. Hepatology 2007, 45, 1343–1346. [Google Scholar] [CrossRef]
- Lebensztejn, D.M.; Flisiak-Jackiewicz, M.; Białokoz-Kalinowska, I.; Bobrus-Chociej, A.; Kowalska, I. Hepatokines and non-alcoholic fatty liver disease. Acta Biochim. Pol. 2016, 63, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Liu, Z.-X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.-M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic Fatty Liver Disease: Cytokine-Adipokine Interplay and Regulation of Insulin Resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Brøns, C.; Grunnet, L.G. MECHANISMS IN ENDOCRINOLOGY: Skeletal muscle lipotoxicity in insulin resistance and type 2 diabetes: A causal mechanism or an innocent bystander? Eur. J. Endocrinol. 2017, 176, R67–R78. [Google Scholar] [CrossRef]
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Lim, S.; Oh, T.J.; Koh, K.K. Mechanistic link between nonalcoholic fatty liver disease and cardiometabolic disorders. Int. J. Cardiol. 2015, 201, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Vianna, C.R.; Fukuda, M.; Berglund, E.D.; Liu, C.; Tao, C.; Sun, K.; Liu, T.; Harper, M.J.; Lee, C.E.; et al. Hepatocyte Toll-like Receptor 4 Regulates Obesity-Induced Inflammation and Insulin Resistance. Nat. Commun. 2014, 5, 3878. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.A.; Lillioja, S.; Kriketos, A.D.; Milner, M.R.; Baur, L.A.; Bogardus, C.; Jenkins, A.B.; Storlien, L.H. Skeletal Muscle Triglyceride Levels Are Inversely Related to Insulin Action. Diabetes 1997, 46, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.S.; Sauers, S.E.; Goodpaster, B.H. Exercise-induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am. J. Physiol. Metab. 2008, 294, E882–E888. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Meigs, J.B. Links between ectopic fat and vascular disease in humans. Arter. Thromb. Vasc. Biol. 2014, 34, 1820–1826. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, A.; Hamilton, D.J.; Deng, T. Epicardial Fat in the Maintenance of Cardiovascular Health. Methodist DeBakey Cardiovasc. J. 2017, 13, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Guglielmi, V.; Sbraccia, P. Type 2 diabetes: Does pancreatic fat really matter? Diabetes 2018, 34. [Google Scholar] [CrossRef]
- Wang, C.; Ou, H.; Chen, M.; Chang, T.; Chang, C. Enigmatic Ectopic Fat: Prevalence of Nonalcoholic Fatty Pancreas Disease and Its Associated Factors in a Chinese Population. J. Am. Hear. Assoc. 2014, 3, e000297. [Google Scholar] [CrossRef] [Green Version]
- Adeva-Andany, M.M.; Castro-Quintela, E.; Fernández-Fernández, C.; Carneiro-Freire, N.; Vila-Altesor, M. The role of collagen homeostasis in the pathogenesis of vascular disease associated to insulin resistance. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.R.; Timms, P.M.; Chandran, S.; Gordon, D. Peripheral blood level alterations of TIMP-1, MMP-2 and MMP-9 in patients with Type 1 diabetes. Diabet. Med. 2001, 18, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, C.R.; Kohn, R.R.; Luschin, J.H. Apparent Accelerated Aging of Human Collagen in Diabetes Mellitus. Diabetes 1975, 24, 902–904. [Google Scholar] [CrossRef] [PubMed]
- James, V.J.; Delbridge, L.; McLennan, S.V.; Yue, D.K. Use of X-Ray Diffraction in Study of Human Diabetic and Aging Collagen. Diabetes 1991, 40, 391–394. [Google Scholar] [CrossRef]
- Siperstein, M.D. Capillary basement membranes and diabetic microangiopathy. Adv. Intern. Med. 1972, 18, 325–344. [Google Scholar] [PubMed]
- Williamson, J.R.; Kilo, C. Current Status of Capillary Basement-membrane Disease in Diabetes Mellitus. Diabetes 1977, 26, 65–75. [Google Scholar] [CrossRef]
- Ranger, T.A.; Wong, A.M.; Cook, J.L.; Gaida, J.E. Is there an association between tendinopathy and diabetes mellitus? A systematic review with meta-analysis. Br. J. Sports Med. 2016, 50, 982–989. [Google Scholar] [CrossRef]
- Alba, D.L.; Farooq, J.A.; Lin, M.Y.C.; Schafer, A.L.; Shepherd, J.; Koliwad, S.K. Subcutaneous Fat Fibrosis Links Obesity to Insulin Resistance in Chinese-Americans. J. Clin. Endocrinol. Metab. 2018, 103, 3194–3204. [Google Scholar] [CrossRef]
- Guglielmi, V.; Cardellini, M.; Cinti, F.; Corgosinho, F.; Cardolini, I.; D’Adamo, M.; Zingaretti, M.C.; Bellia, A.; Lauro, D.; Gentileschi, P.; et al. Omental adipose tissue fibrosis and insulin resistance in severe obesity. Nutr. Diabetes 2015, 5, e175. [Google Scholar] [CrossRef]
- Tam, C.S.; Covington, J.D.; Bajpeyi, S.; Tchoukalova, Y.; Burk, D.; Johannsen, D.L.; Zingaretti, C.M.; Cinti, S.; Ravussin, E. Weight gain reveals dramatic increases in skeletal muscle extracellular matrix remodeling. J. Clin. Endocrinol. Metab. 2014, 99, 1749–1757. [Google Scholar] [CrossRef]
- Spencer, M.; Unal, R.; Zhu, B.; Rasouli, N.; McGehee, R.E.; Peterson, C.A.; Kern, P.A. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2011, 96, E1990–E1998. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.; O’Reilly, P.; Antony, V.B.; Gaggar, A.; Thannickal, V.J. Matrix Remodeling in Pulmonary Fibrosis and Emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 54, 751–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W., 2nd; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef]
- Guzik, T.J.; Skiba, D.S.; Touyz, R.M.; Harrison, D.G. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res. 2017, 113, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Nocon, A.; Fry, J.; Sherban, A.; Rui, X.; Jiang, B.; Xu, X.J.; Han, J.; Yan, Y.; Yang, Q.; et al. AMPK Activation by Metformin Suppresses Abnormal Extracellular Matrix Remodeling in Adipose Tissue and Ameliorates Insulin Resistance in Obesity. Diabetes 2016, 65, 2295–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, J.; Wang, L.; Li, A.; Qiu, Z.; Qi, L.; Kou, J.; Liu, K.; Liu, B.; Huang, F. The role of metformin and resveratrol in the prevention of hypoxia-inducible factor 1α accumulation and fibrosis in hypoxic adipose tissue. Br. J. Pharmacol. 2016, 173, 2001–2015. [Google Scholar] [CrossRef]
- Zhong, J.; Rao, X.; Deiuliis, J.; Braunstein, Z.; Narula, V.; Hazey, J.; Mikami, D.; Needleman, B.; Satoskar, A.R.; Rajagopalan, S. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes 2013, 62, 149–157. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Tellam, R.L.; Morrison, J.L.; Muhlhausler, B.S.; Molloy, P.L. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin. Epigenetics 2015, 7, 66. [Google Scholar] [CrossRef]
- Davegårdh, C.; García-Calzón, S.; Bacos, K.; Ling, C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol. Metab. 2018, 14, 12–25. [Google Scholar] [CrossRef]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, A.; Sinha, I.; Dadvar, S.; Arner, P.; Dahlman, I. Epigenetic regulation of diabetogenic adipose morphology. Mol. Metab. 2019, 25, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Crujeiras, A.B.; Pissios, P.; Moreno-Navarrete, J.M.; Diaz-Lagares, A.; Sandoval, J.; Gomez, A.; Ricart, W.; Esteller, M.; Casanueva, F.F.; Fernandez-Real, J.M. An Epigenetic Signature in Adipose Tissue Is Linked to Nicotinamide N-Methyltransferase Gene Expression. Mol. Nutr. Food Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pietilainen, K.H.; Ismail, K.; Jarvinen, E.; Heinonen, S.; Tummers, M.; Bollepalli, S.; Lyle, R.; Muniandy, M.; Moilanen, E.; Hakkarainen, A.; et al. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int. J. Obes. 2016, 40, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Aslibekyan, S.; Demerath, E.W.; Mendelson, M.; Zhi, D.; Guan, W.; Liang, L.; Sha, J.; Pankow, J.S.; Liu, C.; Irvin, M.R.; et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity 2015, 23, 1493–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viguerie, N.; Poitou, C.; Cancello, R.; Stich, V.; Clement, K.; Langin, D. Transcriptomics applied to obesity and caloric restriction. Biochimie 2005, 87, 117–123. [Google Scholar] [CrossRef]
- Nho, Y.-K.; Ha, E.; Yu, K.-I.; Chung, J.-H.; Wook, N.-C.; Chung, I.-S.; Lee, M.-Y.; Shin, D.-H. Matrix metalloproteinase-1 promoter is associated with body mass index in Korean population with aged greater or equal to 50 years. Clin. Chim. Acta 2008, 396, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef]
- Tam, C.S.; Tordjman, J.; Divoux, A.; Baur, L.A.; Clément, K. Adipose Tissue Remodeling in Children: The Link between Collagen Deposition and Age-Related Adipocyte Growth. J. Clin. Endocrinol. Metab. 2012, 97, 1320–1327. [Google Scholar] [CrossRef] [Green Version]
- Kolehmainen, M.; Salopuro, T.; Schwab, U.S.; Kekalainen, J.; Kallio, P.; Laaksonen, D.E.; Pulkkinen, L.; Lindi, V.I.; Sivenius, K.; Mager, U.; et al. Weight reduction modulates expression of genes involved in extracellular matrix and cell death: The GENOBIN study. Int. J. Obes. 2008, 32, 292–303. [Google Scholar] [CrossRef]
- Roumans, N.J.; Vink, R.G.; Fazelzadeh, P.; Van Baak, M.A.; Mariman, E.C. A role for leukocyte integrins and extracellular matrix remodeling of adipose tissue in the risk of weight regain after weight loss. Am. J. Clin. Nutr. 2017, 105, 1054–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumans, N.J.T.N.J.; Wang, P.; Vink, R.G.R.G.; Van Baak, M.M.A.; Mariman, E.C.M.E.C. Combined Analysis of Stress- and ECM-Related Genes in Their Effect on Weight Regain. Obesity 2018, 26, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumans, N.J.T.; Vink, R.G.; Gielen, M.; Zeegers, M.P.; Holst, C.; Wang, P.; Astrup, A.; Saris, W.H.; Valsesia, A.; Hager, J.; et al. Variation in extracellular matrix genes is associated with weight regain after weight loss in a sex-specific manner. Genes Nutr. 2015, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Alligier, M.; Meugnier, E.; Debard, C.; Lambert-Porcheron, S.; Chanséaume, E.; Sothier, M.; Loizon, E.; Hssain, A.A.; Brožek, J.; Scoazec, J.-Y.; et al. Subcutaneous Adipose Tissue Remodeling during the Initial Phase of Weight Gain Induced by Overfeeding in Humans. J. Clin. Endocrinol. Metab. 2012, 97, E183–E192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, C.S.; Chaudhuri, R.; Hutchison, A.T.; Samocha-Bonet, D.; Heilbronn, L.K. Skeletal muscle extracellular matrix remodeling after short-term overfeeding in healthy humans. Metabolism 2017, 67, 26–30. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Measure/. Reference | Effect in Blood Levels/WAT | Fluid or Tissue | Sample | Other Effects |
|---|---|---|---|---|
| VEGF [131] | Elevated in patients with obesity | Serum | 10 men and 28 women, all of them with obesity | VEGF-A serum was reduced after weight reduction. VEGF-A was positively associated with visceral fat accumulation and BMI |
| VEGF [133] | Elevated in patients with obesity | Plasma | 15 obese and 15 normal-weight men | VEGF-A positively associated with BMI |
| VEGF [134] | No change | Serum | 21 (13 women/ 8 men) lean and 44 (32 women/ 12 men) obese | |
| VEGF-A [136] | Decreased in obesity | WAT | Obese mice | |
| VEGF [137] | Decreased in patients with obesity | WAT | 9 (5 men/4 women) lean and 12 (6/6) obese | VEGF-A expression negatively associated with capillary density |
| VEGF-A [138] | - | - | C57Bl6/SJL mice | VEGF-A overexpression protected mice from HFD inflammation and IR |
| VEGF-A [56] | Overexpression in patients with obesity | WAT | 26 obese and 17 normal-weight men | VEGF-A expression was higher in low IR obese than in high IR patients |
| PO2 [150] | Decreased in obesity | WAT | 23 obese and 21 lean men | |
| PO2 [151] | Decreased in obesity | WAT | 24 (20 women/4 men) obese and 10 lean (7 women/3 men) | |
| PO2 [153] | No differences | WAT | 7 lean (5 women/2 men), 7 obese women | |
| PO2 [137] | Decreased in obesity | WAT | 9 lean (4 women/5 men), 12 (6/6) overweight and obese | |
| PO2 [152] | No differences | WAT | 7 lean men, 28 (14 women/14 men) obese | Abdominal subcutaneous AT oxygenation is associated with insulin sensitivity |
| PO2 [147] | Elevated in obesity | WAT | 10 lean, 10 obese men |
| Reference | Population | Sample | Main Results |
|---|---|---|---|
| Nho et al. [217] | Population-based cohort study consisting of 530 subjects | One group with BMI <25.0 and the other BMI ≥25.0, and MMP-1 polymorphisms by pyrosequencing analysis were measured. | MMP-1 frequencies were significantly higher in subjects with BMI <25.0 |
| Henegar et al. [218] | Fifty five obese subjects and 15 lean controls were prospectively recruited | Transcriptomic signature of the subcutaneous WAT in obese human subjects was analyzed | Phenotypic alterations of human pre-adipocytes may lead to an excessive synthesis of ECM components |
| Tam et al. [219] | 65 otherwise healthy children having elective surgery were selected | Collagen (total and pericellular), and ECM gene expression markers were measured | Increased collagen in AT is associated with BMI z-score, suggesting dynamic interaction between ECM remodeling and immune cells even at an early age. |
| Kolehmainen et al. [220] | Forty-six subjects with metabolic syndrome were randomized either to a weight reduction (n=28) or a control (n=18) group lasting for 33 weeks. | Subcutaneous AT biopsies were performed using microarray technology | Genes regulating the ECM and cell death showed a strong downregulation after long-term weight reduction |
| Roumans et al. [221] | 61 healthy overweight or obese participants followed either a very-low-calorie diet or a low-calorie diet | Abdominal subcutaneous AT biopsy samples were collected for microarray analysis | ECM modification seems to be involved |
| Roumans et al. [222] | 31 participants with overweight or obesity followed a 5-week very-low-calorie diet with a subsequent 4-week weight-stable diet, and then an uncontrolled 9-month follow-up. | AT biopsies were collected for microarray analysis. | Interaction analysis between stress- and ECM-related genes revealed that several gene combinations were highly related to weight regain. |
| Roumans et al. [223] | 469 overweight and obese subjects were on an 8-week low-calorie diet with a 6-month follow-up. | AT biopsies were collected for microarray analysis. | Variants of ECM genes are associated with weight regain after weight loss in a sex-specific manner. |
| Alligier et al. [224] | Forty-four healthy men were involved in an overfeeding protocol with a lipid-enriched diet for 2 months. | Subcutaneous abdominal AT biopsies were taken | Reorganization of gene expression patterns occurred in AT with an upregulation of numerous genes involved in angiogenesis and ECM remodeling. |
| Tam et al. [225] | Forty healthy individuals were overfed by 1,250 kcal/day for 28days. | Skeletal muscle biopsies were taken | Skeletal muscle ECM remodeling occurs early in response to over-nutrition with as little as 3% body weight gain. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4888. https://doi.org/10.3390/ijms20194888
Ruiz-Ojeda FJ, Méndez-Gutiérrez A, Aguilera CM, Plaza-Díaz J. Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases. International Journal of Molecular Sciences. 2019; 20(19):4888. https://doi.org/10.3390/ijms20194888
Chicago/Turabian StyleRuiz-Ojeda, Francisco Javier, Andrea Méndez-Gutiérrez, Concepción María Aguilera, and Julio Plaza-Díaz. 2019. "Extracellular Matrix Remodeling of Adipose Tissue in Obesity and Metabolic Diseases" International Journal of Molecular Sciences 20, no. 19: 4888. https://doi.org/10.3390/ijms20194888