Ceftriaxone Treatment Affects EAAT2 Expression and Glutamatergic Neurotransmission and Exerts a Weak Anticonvulsant Effect in Young Rats

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
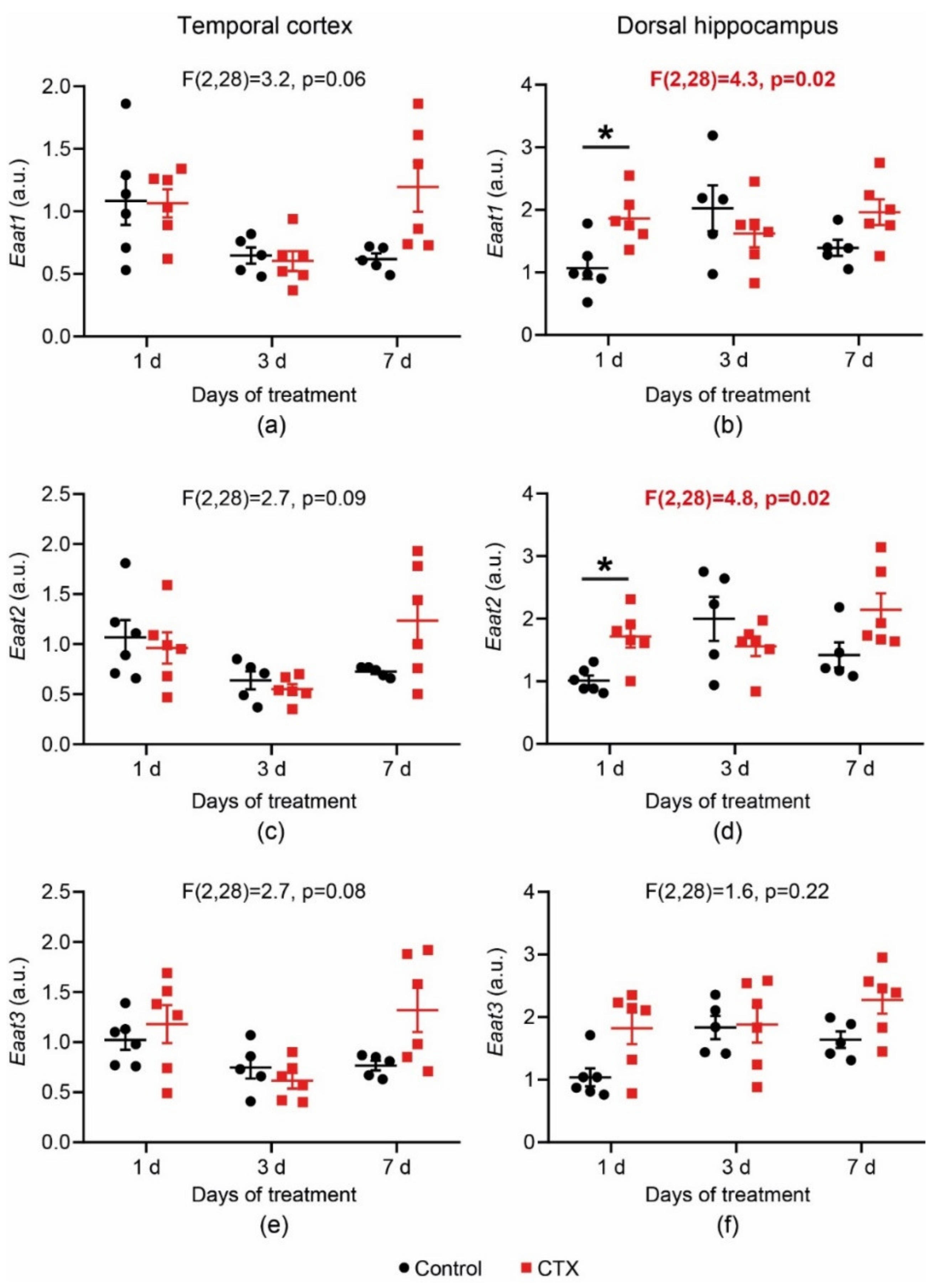
2.1. CTX-Treatment-Induced Changes in Eaats’ mRNA Production
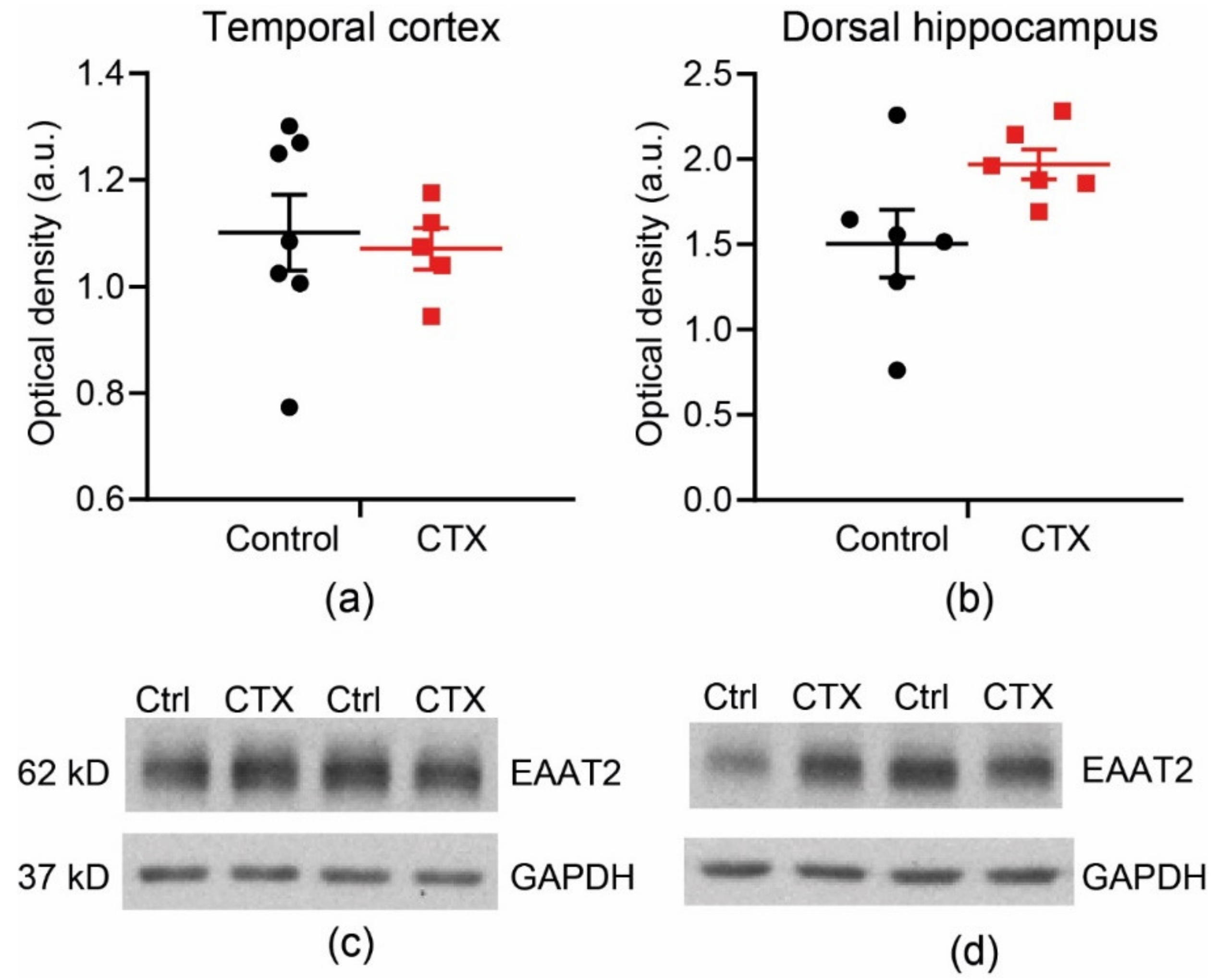
2.2. CTX Treatment Did Not Significantly Change the Protein Expression of EAAT2 in the Temporal Cortex and Dorsal Hippocampus
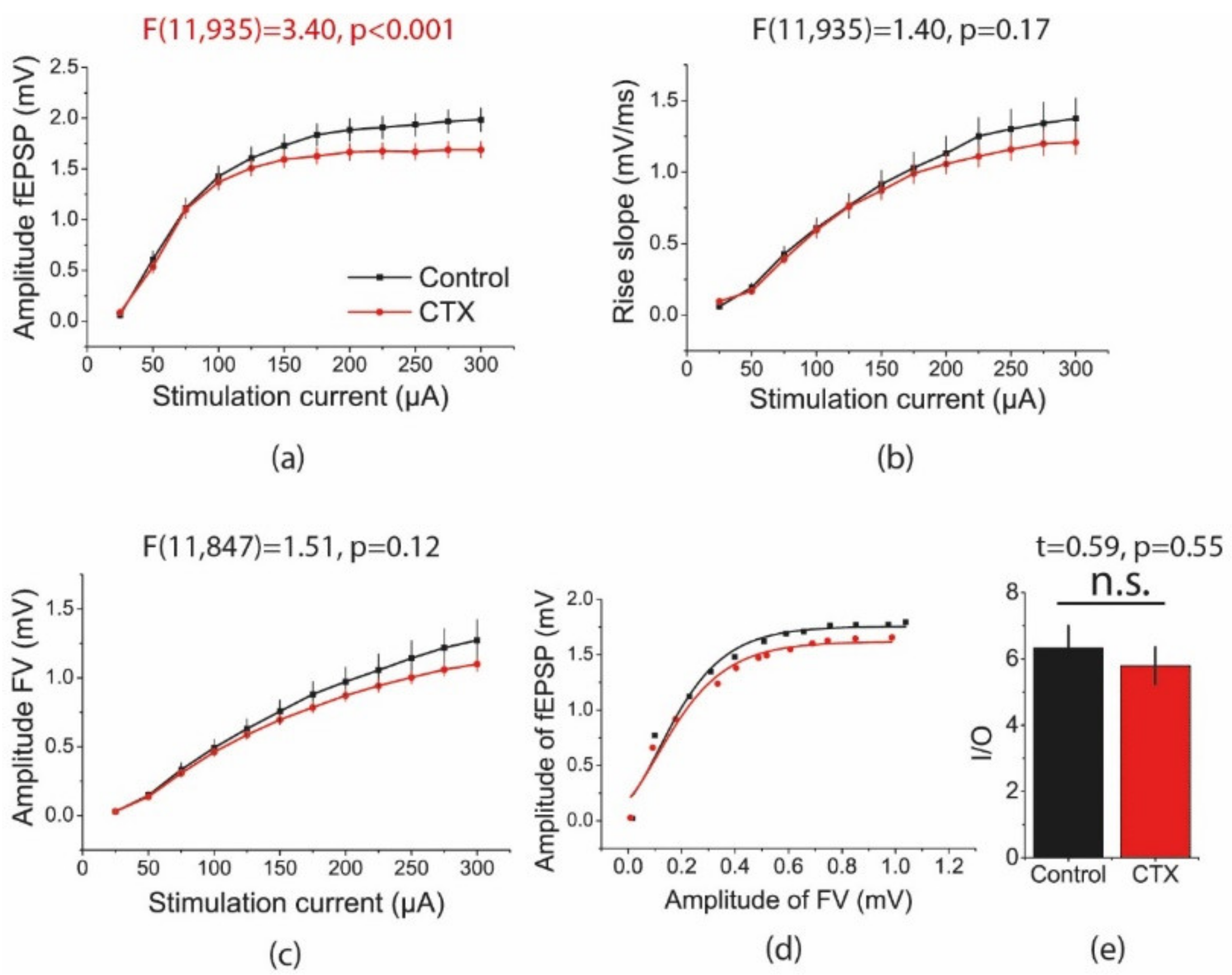
2.3. CTX Treatment Decreased the Amplitude of Field Excitatory Postsynaptic Potentials (fEPSPs) in the Hippocampus Evoked by Intense Electrical Stimulation
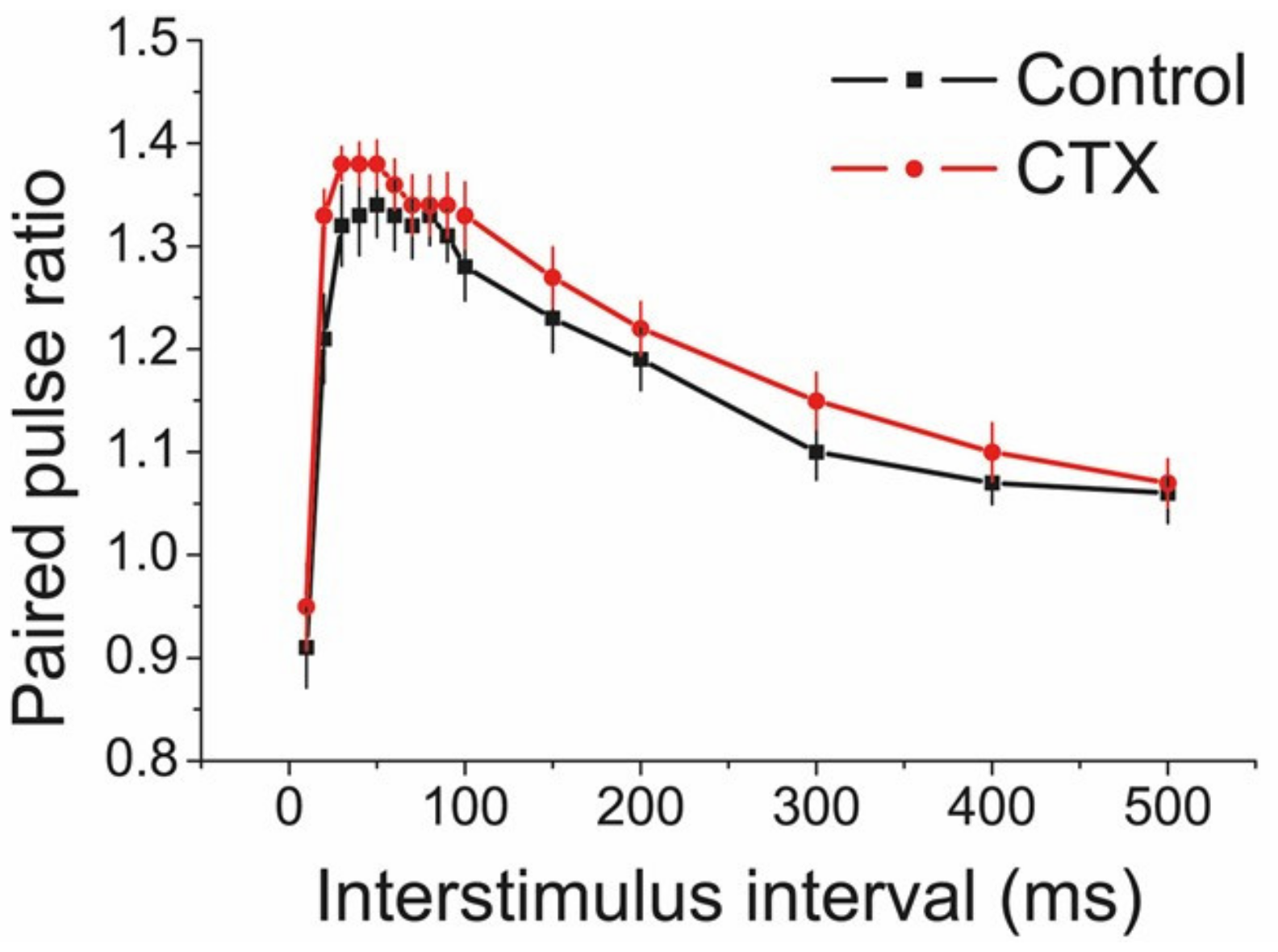
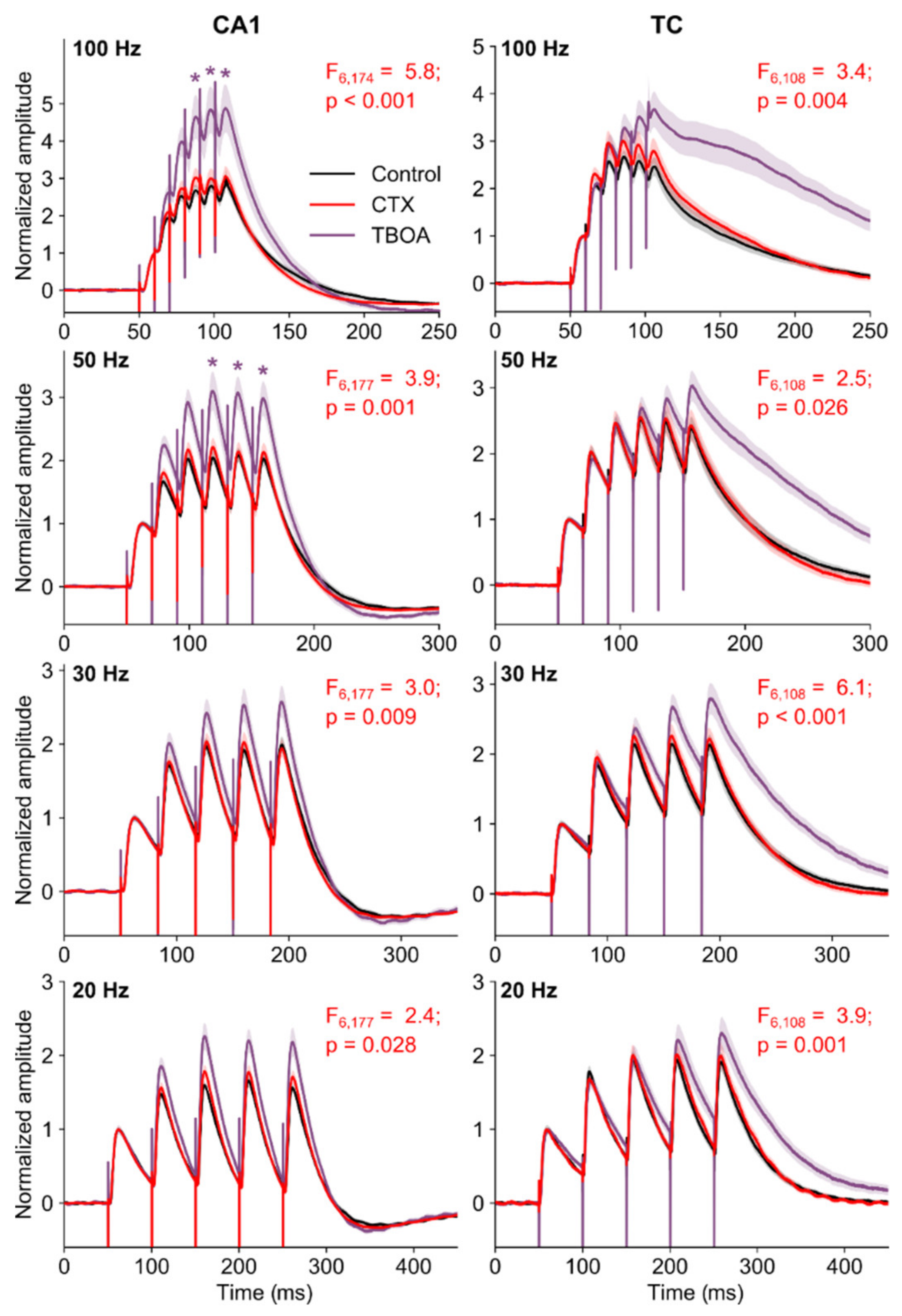
2.4. CTX Treatment Did Not Affect Short-Term Facilitation in the CA1 Area of the Hippocampus
2.5. The Summation of EPSPs in Pyramidal Neurons of the CA1 Hippocampus and Temporal Cortex Did Not Change after CTX Treatment
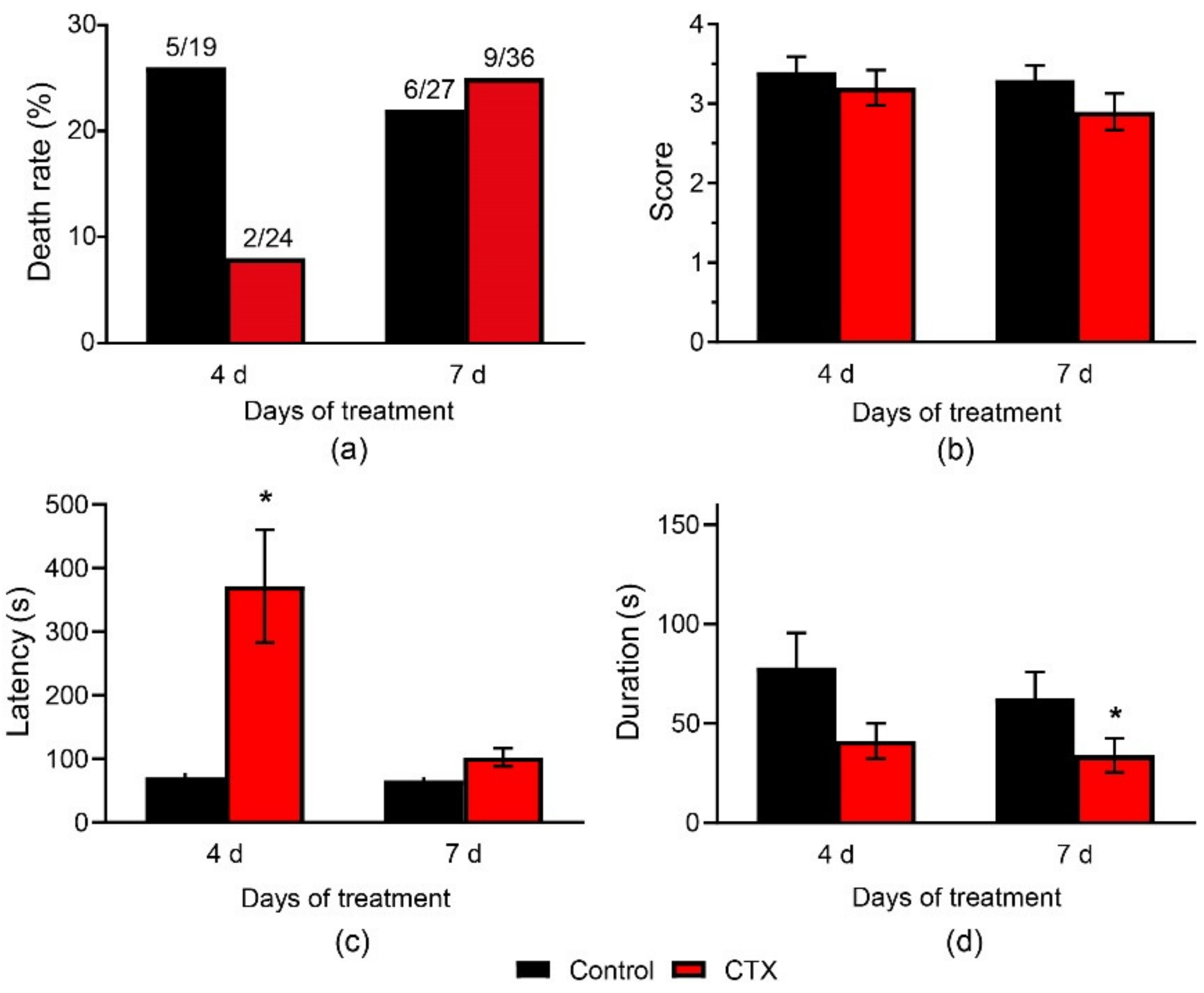
2.6. Effects of CTX Administration on Convulsions in a PTZ and MEST Tests
2.6.1. CTX Treatment Had a Weak Anticonvulsant Effect in the PTZ Test
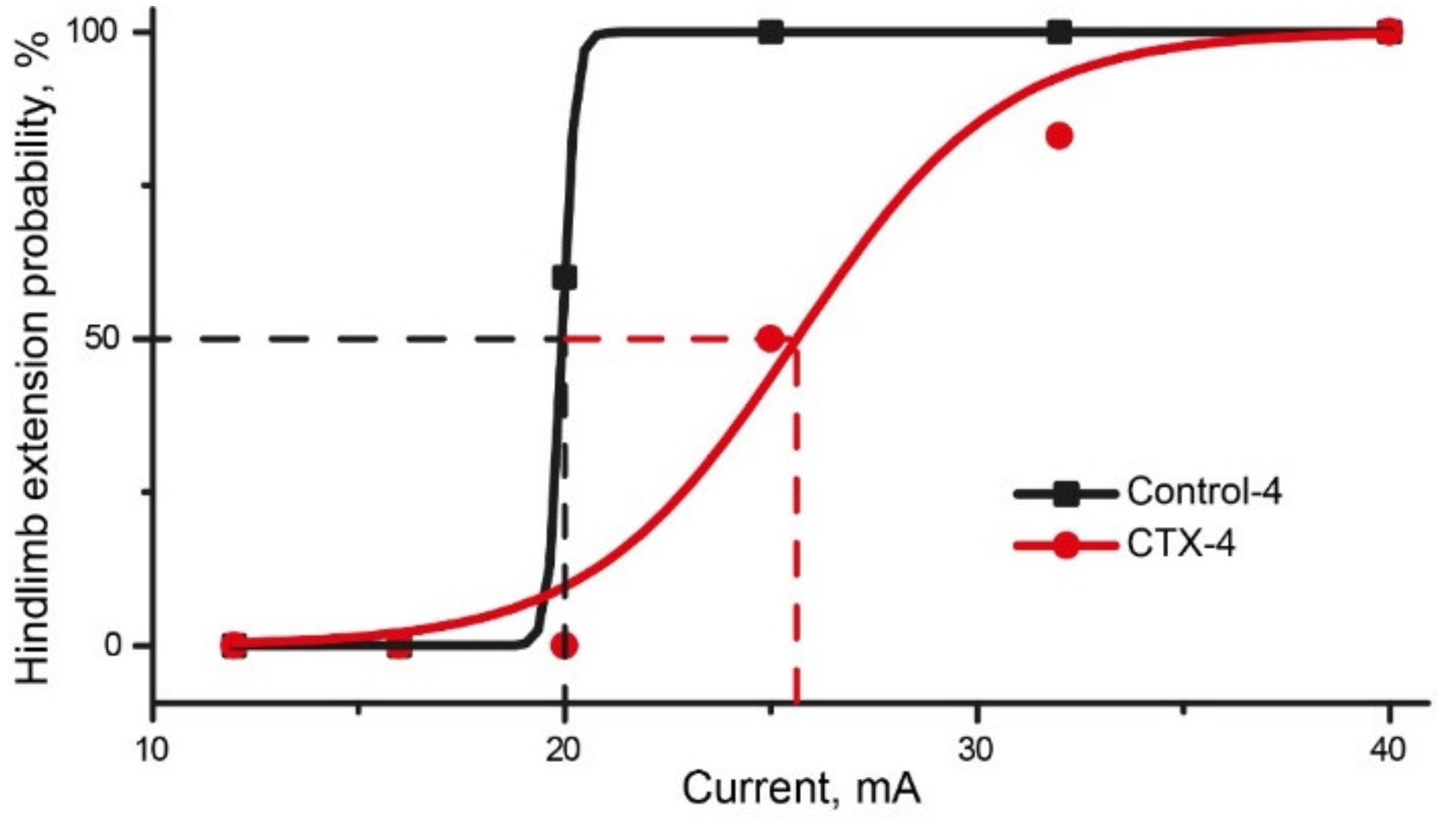
2.6.2. CTX Treatment Had Significant Effect in MEST Test
3. Discussion
4. Materials and Methods
4.1. Animals and CTX Administration
4.2. RNA Extraction and Real-Time Quantitative PCR Assay
4.3. Western Blot
4.4. Slice Preparation
4.5. Field Potential Recordings
4.6. Whole-Cell Recordings in Brain Slices
4.7. PTZ Test
4.8. MEST Test
4.9. Data Analysis and Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MEST | Maximal Electroshock Threshold |
| EPSP | Excitatory Postsynaptic Potential |
| EAAT | Excitatory Amino Acid Transporter |
| FV | Fiber Volley |
| CTX | Ceftriaxone |
| PTZ | Pentylenetetrazol |
| PPR | Paired-Pulse Ratio |
| PCR | Polymerase Chain Reaction |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Current, mA | Rats | Probability | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| Control group | |||||||||||||
| 12 | - | - | - | - | 0% | ||||||||
| 16 | 0 | - | - | 0 | 0% | ||||||||
| 20 | x | 0 | 0 | x | x | 60% | |||||||
| 25 | x | * | x | x | * | * | 100% | ||||||
| 32 | x | * | * | * | * | * | * | 100% | |||||
| 40 | x | * | * | * | * | * | * | * | 100% | ||||
| CTX-treated group | |||||||||||||
| 12 | - | - | - | - | - | - | 0% | ||||||
| 16 | - | - | - | - | - | - | 0% | ||||||
| 20 | 0 | - | - | 0 | 0 | - | 0% | ||||||
| 25 | x | 0 | - | x | x | 0 | 50% | ||||||
| 32 | * | 0 | x | * | * | x | 83% | ||||||
| 40 | * | x | * | * | * | * | 100% | ||||||
| Properties | Effect | Method |
|---|---|---|
| Eaats’ mRNA production | Increase in Eaat1 and Eaat2 mRNA level in the dorsal hippocampus but not in temporal cortex No significant changes in Eaat3 expression | Real-time PCR |
| Protein expression of EAAT1 and EAAT2 | No significant changes in Eaat1-2 expression in the hippocampus and temporal cortex | Western blot |
| The amplitude of field excitatory postsynaptic potentials (fEPSPs) in the hippocampus | Decreased at intense electrical stimulation | Local field potential recording |
| Short-term facilitation in the CA1 area of the hippocampus | Unaffected | Local field potential recording |
| The summation of EPSPs in pyramidal neurons of the CA1 hippocampus and temporal cortex | Unaffected | Whole-cell patch-clamp |
| Convulsions and mortality in a PTZ test | Mortality rate and the mean maximal score of convulsions are unaffected Seizure duration decreased after 7-day treatment with CTX, seizure latency increased after 4-day treatment with CTX | PTZ test |
| Maximal electroshock threshold (MEST) | Increased | MEST test |
References
- Meyer, A.C.; Dua, T.; Ma, J.; Saxena, S.; Birbeck, G. Global disparities in the epilepsy treatment gap: A systematic review. Bull. World Health Organ. 2010, 88, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Moshe, S.L.; Perucca, E.; Ryvlin, P.; Tomson, T. Epilepsy: New advances. Lancet 2015, 385, 884–898. [Google Scholar] [CrossRef]
- Loscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Rosenow, F.; van Alphen, N.; Becker, A.; Chiocchetti, A.; Deichmann, R.; Deller, T.; Freiman, T.; Freitag, C.M.; Gehrig, J.; Hermsen, A.M.; et al. Personalized translational epilepsy research—Novel approaches and future perspectives: Part I: Clinical and network analysis approaches. Epilepsy Behav. 2017, 76, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.; Ronne-Engstrom, E.; Ungerstedt, U.; Hillered, L. Seizure related elevations of extracellular amino acids in human focal epilepsy. Neurosci. Lett. 1992, 140, 30–32. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Petroff, O.A.; Rothman, D.L.; Behar, K.L.; Hyder, F.; Mattson, R.H. Effects of valproate and other antiepileptic drugs on brain glutamate, glutamine, and gaba in patients with refractory complex partial seizures. Seizure 1999, 8, 120–127. [Google Scholar] [CrossRef]
- Wilson, C.L.; Maidment, N.T.; Shomer, M.H.; Behnke, E.J.; Ackerson, L.; Fried, I.; Engel, J., Jr. Comparison of seizure related amino acid release in human epileptic hippocampus versus a chronic, kainate rat model of hippocampal epilepsy. Epilepsy Res. 1996, 26, 245–254. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters glt-1 and glast in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 107559. [Google Scholar] [CrossRef]
- Tanaka, K. Cloning and expression of a glutamate transporter from mouse brain. Neurosci. Lett. 1993, 159, 183–186. [Google Scholar] [CrossRef]
- Pines, G.; Danbolt, N.C.; Bjørås, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain l-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Lehre, K.P.; Levy, L.M.; Ottersen, O.P.; Storm-Mathisen, J.; Danbolt, N.C. Differential expression of two glial glutamate transporters in the rat brain: Quantitative and immunocytochemical observations. J. Neurosci. 1995, 15, 1835–1853. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Lee, E.; Aschner, M. Manganese neurotoxicity: A focus on glutamate transporters. Ann. Occup. Environ. Med. 2013, 25, 4. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, eaat1 and eaat2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51–63. [Google Scholar] [CrossRef]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef]
- Fairman, W.; Vandenberg, R.; Arriza, J.; Kavanaught, M.; Amara, S. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Bjorn-Yoshimoto, W.E.; Underhill, S.M. The importance of the excitatory amino acid transporter 3 (eaat3). Neurochem. Int. 2016, 98, 4–18. [Google Scholar] [CrossRef]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef]
- Otis, T.S.; Kavanaugh, M.P. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J. Neurosci. 2000, 20, 2749–2757. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.G.; Kegelman, T.P.; Su, Z.Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of excitatory amino acid transporter-2 (eaat2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell. Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, P.; Green, P.S.; Watson, G.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G. Glt-1 loss accelerates cognitive deficit onset in an alzheimer′s disease animal model. J. Alzheimer’s Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Mathern, G.W.; Mendoza, D.; Lozada, A.; Pretorius, J.K.; Dehnes, Y.; Danbolt, N.C.; Nelson, N.; Leite, J.P.; Chimelli, L.; Born, D.E.; et al. Hippocampal gaba and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 1999, 52, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.; Schrama, L.H.; van Veelen, C.W.; van Rijen, P.C.; van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 2002, 125, 32–43. [Google Scholar] [CrossRef]
- Lopes, M.W.; Soares, F.M.; de Mello, N.; Nunes, J.C.; Cajado, A.G.; de Brito, D.; de Cordova, F.M.; da Cunha, R.M.; Walz, R.; Leal, R.B. Time-dependent modulation of ampa receptor phosphorylation and mrna expression of nmda receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp. Brain Res. 2013, 226, 153–163. [Google Scholar] [CrossRef]
- Samuelsson, C.; Kumlien, E.; Flink, R.; Lindholm, D.; Ronne-Engstrom, E. Decreased cortical levels of astrocytic glutamate transport protein glt-1 in a rat model of posttraumatic epilepsy. Neurosci. Lett. 2000, 289, 185–188. [Google Scholar] [CrossRef]
- Ueda, Y.; Doi, T.; Tokumaru, J.; Yokoyama, H.; Nakajima, A.; Mitsuyama, Y.; Ohya-Nishiguchi, H.; Kamada, H.; Willmore, L.J. Collapse of extracellular glutamate regulation during epileptogenesis: Down-regulation and functional failure of glutamate transporter function in rats with chronic seizures induced by kainic acid. J. Neurochem. 2001, 76, 892–900. [Google Scholar] [CrossRef]
- Sarfi, M.; Elahdadi Salmani, M.; Goudarzi, I.; Lashkar Boluki, T.; Abrari, K. Evaluating the role of astrocytes on beta-estradiol effect on seizures of pilocarpine epileptic model. Eur. J. Pharmacol. 2017, 797, 32–38. [Google Scholar] [CrossRef]
- Crino, P.B.; Jin, H.; Shumate, M.D.; Robinson, M.B.; Coulter, D.A.; Brooks-Kayal, A.R. Increased expression of the neuronal glutamate transporter (eaat3/eaac1) in hippocampal and neocortical epilepsy. Epilepsia 2002, 43, 211–218. [Google Scholar] [CrossRef]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter glt-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Takahashi, K.; Schulte, D.; Stouffer, N.; Lin, Y.; Lin, C.L. Increased glial glutamate transporter eaat2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2012, 47, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Dykes Hoberg, M.; Vidensky, S.; Chung, D.S.; et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Y.; Xu, J.; Zhang, M.; Wang, D.; Li, L.; Li, W.B. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J. Neurochem. 2015, 132, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Soni, N.; Koushal, P.; Reddy, B.V.; Deshmukh, R.; Kumar, P. Effect of glt-1 modulator and p2x7 antagonists alone and in combination in the kindling model of epilepsy in rats. Epilepsy Behav. 2015, 48, 4–14. [Google Scholar] [CrossRef]
- Jelenkovic, A.V.; Jovanovic, M.D.; Stanimirovic, D.D.; Bokonjic, D.D.; Ocic, G.G.; Boskovic, B.S. Beneficial effects of ceftriaxone against pentylenetetrazole-evoked convulsions. Exp. Biol. Med. (Maywood) 2008, 233, 1389–1394. [Google Scholar] [CrossRef]
- Uyanikgil, Y.; Ozkeskek, K.; Cavusoglu, T.; Solmaz, V.; Tumer, M.K.; Erbas, O. Positive effects of ceftriaxone on pentylenetetrazol-induced convulsion model in rats. Int. J. Neurosci. 2016, 126, 70–75. [Google Scholar] [CrossRef]
- Loewen, J.L.; Albertini, G.; Dahle, E.J.; Sato, H.; Smolders, I.J.; Massie, A.; Wilcox, K.S. Genetic and pharmacological manipulation of glial glutamate transporters does not alter infection-induced seizure activity. Exp. Neurol. 2019, 318, 50–60. [Google Scholar] [CrossRef]
- Thone-Reineke, C.; Neumann, C.; Namsolleck, P.; Schmerbach, K.; Krikov, M.; Schefe, J.H.; Lucht, K.; Hortnagl, H.; Godes, M.; Muller, S.; et al. The beta-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J. Hypertens. 2008, 26, 2426–2435. [Google Scholar] [CrossRef]
- Hussein, A.M.; Ghalwash, M.; Magdy, K.; Abulseoud, O.A. Beta lactams antibiotic ceftriaxone modulates seizures, oxidative stress and connexin 43 expression in hippocampus of pentylenetetrazole kindled rats. J. Epilepsy Res. 2016, 6, 8–15. [Google Scholar] [CrossRef]
- Goodrich, G.S.; Kabakov, A.Y.; Hameed, M.Q.; Dhamne, S.C.; Rosenberg, P.A.; Rotenberg, A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, glt-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J. Neurotrauma 2013, 30, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Asztely, F.; Erdemli, G.; Kullmann, D.M. Extrasynaptic glutamate spillover in the hippocampus: Dependence on temperature and the role of active glutamate uptake. Neuron 1997, 18, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Smolensky, I.V.; Zaitsev, A.V. Changes in functional properties of rat hippocampal neurons following pentylenetetrazole-induced status epilepticus. Neuroscience 2019, 399, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Ann. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- Dobrunz, L.E.; Stevens, C.F. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 1997, 18, 995–1008. [Google Scholar] [CrossRef] [Green Version]
- Zaitsev, A.V.; Anwyl, R. Inhibition of the slow afterhyperpolarization restores the classical spike timing-dependent plasticity rule obeyed in layer 2/3 pyramidal cells of the prefrontal cortex. J. Neurophysiol. 2012, 107, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.S.; Jahr, C.E. Transporters buffer synaptically released glutamate on a submillisecond time scale. J. Neurosci. 1997, 17, 4672–4687. [Google Scholar] [CrossRef]
- Tzingounis, A.V.; Wadiche, J.I. Glutamate transporters: Confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci. 2007, 8, 935–947. [Google Scholar] [CrossRef]
- Shimamoto, K.; Sakai, R.; Takaoka, K.; Yumoto, N.; Nakajima, T.; Amara, S.G.; Shigeri, Y. Characterization of novel l-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol. Pharmacol. 2004, 65, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Doi, T.; Nagatomo, K.; Willmore, L.J.; Nakajima, A. Functional role for redox in the epileptogenesis: Molecular regulation of glutamate in the hippocampus of fecl3-induced limbic epilepsy model. Exp. Brain Res. 2007, 181, 571–577. [Google Scholar] [CrossRef]
- Lee, E.; Sidoryk-Wegrzynowicz, M.; Yin, Z.; Webb, A.; Son, D.S.; Aschner, M. Transforming growth factor-alpha mediates estrogen-induced upregulation of glutamate transporter glt-1 in rat primary astrocytes. Glia 2012, 60, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.G.; Su, Z.Z.; Emdad, L.; Gupta, P.; Sarkar, D.; Borjabad, A.; Volsky, D.J.; Fisher, P.B. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem. 2008, 283, 13116–13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzyzanowska, W.; Pomierny, B.; Budziszewska, B.; Filip, M.; Pera, J. N-acetylcysteine and ceftriaxone as preconditioning strategies in focal brain ischemia: Influence on glutamate transporters expression. Neurotox. Res. 2016, 29, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, P.C.; Huang, Y.T.; Wu, C.C.; Lai, C.J.; Wang, P.J.; Chiu, T.H. Ceftriaxone attenuates hypoxic-ischemic brain injury in neonatal rats. J. Biomed Sci. 2011, 18, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilev, D.S.; Tumanova, N.L.; Kim, K.K.; Lavrentyeva, V.V.; Lukomskaya, N.Y.; Zhuravin, I.A.; Magazanik, L.G.; Zaitsev, A.V. Transient morphological alterations in the hippocampus after pentylenetetrazole-induced seizures in rats. Neurochem. Res. 2018, 43, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Zubareva, O.E.; Kovalenko, A.A.; Kalemenev, S.V.; Schwarz, A.P.; Karyakin, V.B.; Zaitsev, A.V. Alterations in mrna expression of glutamate receptor subunits and excitatory amino acid transporters following pilocarpine-induced seizures in rats. Neurosci. Lett. 2018, 686, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Szu, J.I.; Yonan, J.M.; Binder, D.K. Regulation of astrocyte glutamate transporter-1 (glt1) and aquaporin-4 (aqp4) expression in a model of epilepsy. Exp. Neurol. 2016, 283, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timofeev, I.; Steriade, M. Neocortical seizures: Initiation, development and cessation. Neuroscience 2004, 123, 299–336. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.R.; Park, Y.D.; Lee, M.R.; Sharma, S.; Harris, K.M.; Kirov, S.A. Three-dimensional relationships between perisynaptic astroglia and human hippocampal synapses. Glia 2010, 58, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, R.; Harris, K.M. Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Witcher, M.R.; Kirov, S.A.; Harris, K.M. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 2007, 55, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, Y.; Muller, D.; Nikonenko, I. Astrocyte-synapse structural plasticity. Neural Plast. 2014, 2014, 232105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haber, M.; Zhou, L.; Murai, K.K. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 2006, 26, 8881–8891. [Google Scholar] [CrossRef] [PubMed]
- Plata, A.; Lebedeva, A.; Denisov, P.; Nosova, O.; Postnikova, T.Y.; Pimashkin, A.; Brazhe, A.; Zaitsev, A.V.; Rusakov, D.A.; Semyanov, A. Astrocytic atrophy following status epilepticus parallels reduced ca(2+) activity and impaired synaptic plasticity in the rat hippocampus. Front. Mol. Neurosci. 2018, 11, 215. [Google Scholar] [CrossRef]
- Barbour, B.; Keller, B.U.; Llano, I.; Marty, A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar purkinje cells. Neuron 1994, 12, 1331–1343. [Google Scholar] [CrossRef]
- Tsukada, S.; Iino, M.; Takayasu, Y.; Shimamoto, K.; Ozawa, S. Effects of a novel glutamate transporter blocker, (2s, 3s)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate (tfb-tboa), on activities of hippocampal neurons. Neuropharmacology 2005, 48, 479–491. [Google Scholar] [CrossRef]
- Campbell, S.L.; Hablitz, J.J. Decreased glutamate transport enhances excitability in a rat model of cortical dysplasia. Neurobiol. Dis. 2008, 32, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.L.; Hablitz, J.J.; Olsen, M.L. Functional changes in glutamate transporters and astrocyte biophysical properties in a rodent model of focal cortical dysplasia. Front. Cell. Neurosci. 2014, 8, 425. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Lai, L.; Guo, H.; Lin, Y.; Butchbach, M.E.; Chang, Y.; Lin, C.L. Translational control of glial glutamate transporter eaat2 expression. J. Biol. Chem. 2007, 282, 1727–1737. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Chang, L.C.; Takahashi, K.; Liu, Q.; Schulte, D.A.; Lai, L.; Ibabao, B.; Lin, Y.; Stouffer, N.; Das Mukhopadhyay, C.; et al. Small-molecule activator of glutamate transporter eaat2 translation provides neuroprotection. J. Clin. Investig. 2014, 124, 1255–1267. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of rna, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–534, 536–537. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Swijsen, A.; Nelissen, K.; Janssen, D.; Rigo, J.M.; Hoogland, G. Validation of reference genes for quantitative real-time pcr studies in the dentate gyrus after experimental febrile seizures. BMC Res. Notes 2012, 5, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malkin, S.L.; Amakhin, D.V.; Veniaminova, E.A.; Kim, K.; Zubareva, O.E.; Magazanik, L.G.; Zaitsev, A.V. Changes of ampa receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016, 327, 146–155. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Hasselfeld, K.; Bauer, D.; Simmons, M.; Roussos, P.; Haroutunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Glutamate transporter splice variant expression in an enriched pyramidal cell population in schizophrenia. Transl. Psychiatry 2015, 5, e579. [Google Scholar] [CrossRef] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Postnikova, T.Y.; Zubareva, O.E.; Kovalenko, A.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status epilepticus impairs synaptic plasticity in rat hippocampus and is followed by changes in expression of nmda receptors. Biochemistry (Moscow) 2017, 82, 282–290. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation. Ii. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Zaitsev, A.V.; Kim, K.K.; Vasilev, D.S.; Lukomskaya, N.Y.; Lavrentyeva, V.V.; Tumanova, N.L.; Zhuravin, I.A.; Magazanik, L.G. N-methyl-d-aspartate receptor channel blockers prevent pentylenetetrazole-induced convulsions and morphological changes in rat brain neurons. J. Neurosci. Res. 2015, 93, 454–465. [Google Scholar] [CrossRef]
- Racusen, L.C.; McCrindle, B.W.; Christenson, M.; Fivush, B.; Fisher, R.S. Cyclosporine lowers seizure threshold in an experimental model of electroshock-induced seizures in munich-wistar rats. Life Sci. 1990, 46, 1021–1026. [Google Scholar] [CrossRef]







| Target Name and GeneBank Accession Number | Nucleotide Sequences (Forward, Reverse, TaqMan-Probe) | Reference |
|---|---|---|
| Ppia NM_017101 | Forward AGGATTCATGTGCCAGGGTG Reverse CTCAGTCTTGGCAGTGCAGA Probe CACGCCATAATGGCACTGGTGGCA | [75] |
| Eaat1 NM_019225.2 | Forward ACAAAAAGCAACGGAGAAGAGCC Reverse TACGGTCGGAGGGCAAATCC | [76] |
| Eaat2 NM_001302089.1 NM_017215.2 NM_001035233.1 | Forward CCAGTGCTGGAACTTTGCCT Reverse TAAAGGGCTGTACCATCCAT Probe AGCGTGTGACCAGATTCGTCCTCCCA | [76] |
| Eaat3 NM_013032.3 | Forward TTCTCCACCACCGTCATTGCT Reverse GCAGGCTTCACTTCTTCACGC | [76] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaitsev, A.V.; Malkin, S.L.; Postnikova, T.Y.; Smolensky, I.V.; Zubareva, O.E.; Romanova, I.V.; Zakharova, M.V.; Karyakin, V.B.; Zavyalov, V. Ceftriaxone Treatment Affects EAAT2 Expression and Glutamatergic Neurotransmission and Exerts a Weak Anticonvulsant Effect in Young Rats. Int. J. Mol. Sci. 2019, 20, 5852. https://doi.org/10.3390/ijms20235852
Zaitsev AV, Malkin SL, Postnikova TY, Smolensky IV, Zubareva OE, Romanova IV, Zakharova MV, Karyakin VB, Zavyalov V. Ceftriaxone Treatment Affects EAAT2 Expression and Glutamatergic Neurotransmission and Exerts a Weak Anticonvulsant Effect in Young Rats. International Journal of Molecular Sciences. 2019; 20(23):5852. https://doi.org/10.3390/ijms20235852
Chicago/Turabian StyleZaitsev, Aleksey V., Sergey L. Malkin, Tatyana Y. Postnikova, Ilya V. Smolensky, Olga E. Zubareva, Irina V. Romanova, Maria V. Zakharova, Vladimir B. Karyakin, and Vladimir Zavyalov. 2019. "Ceftriaxone Treatment Affects EAAT2 Expression and Glutamatergic Neurotransmission and Exerts a Weak Anticonvulsant Effect in Young Rats" International Journal of Molecular Sciences 20, no. 23: 5852. https://doi.org/10.3390/ijms20235852