Disulfide-Linked Peptides for Blocking BTLA/HVEM Binding

 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Peptide Design
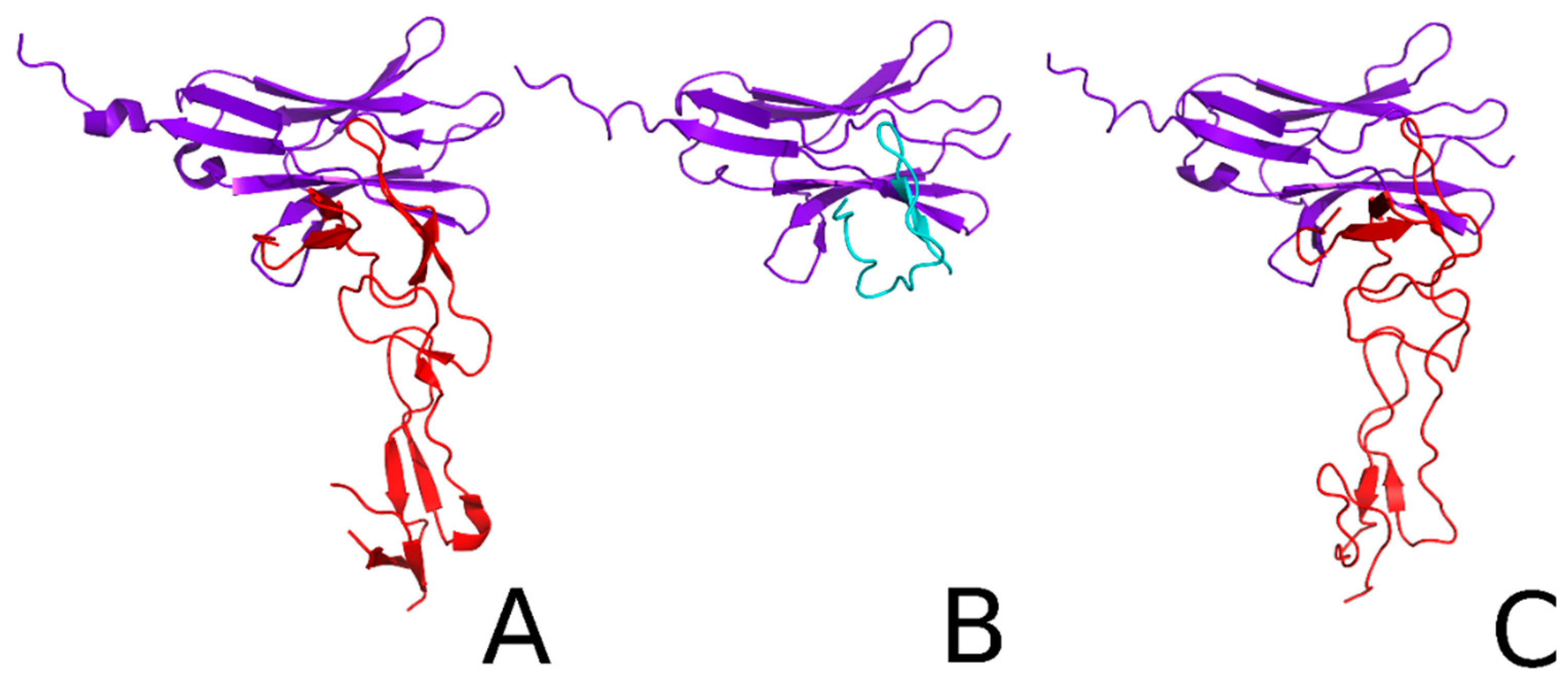
2.2. Docking and Kinetic Studies for the HVEM Protein and HVEM (14–39) Peptide Based on the Crystal Structure of BTLA/HVEM Complex
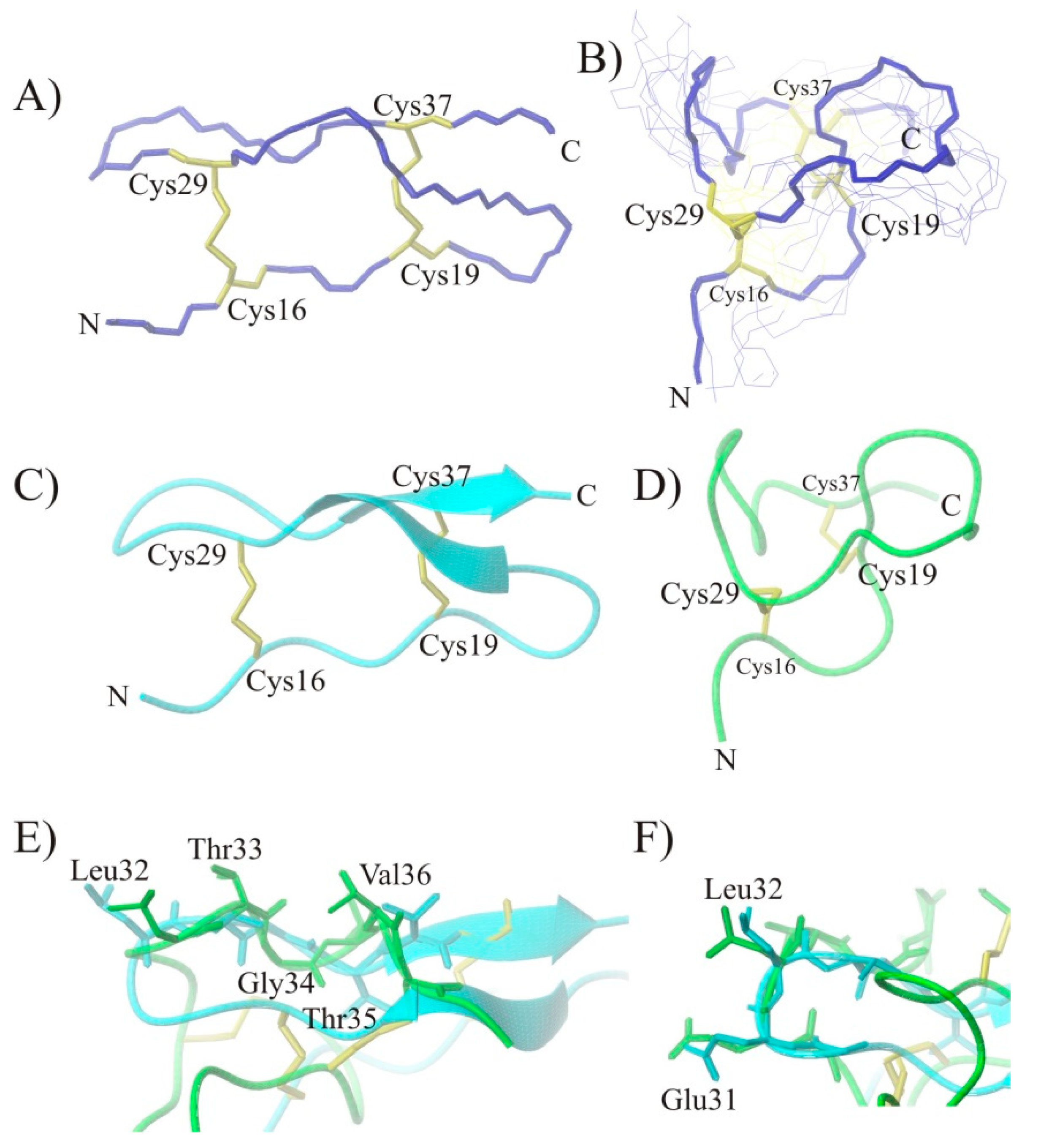
2.3. Conformational Studies of HVEM (14–39) Peptide Using NMR Techniques
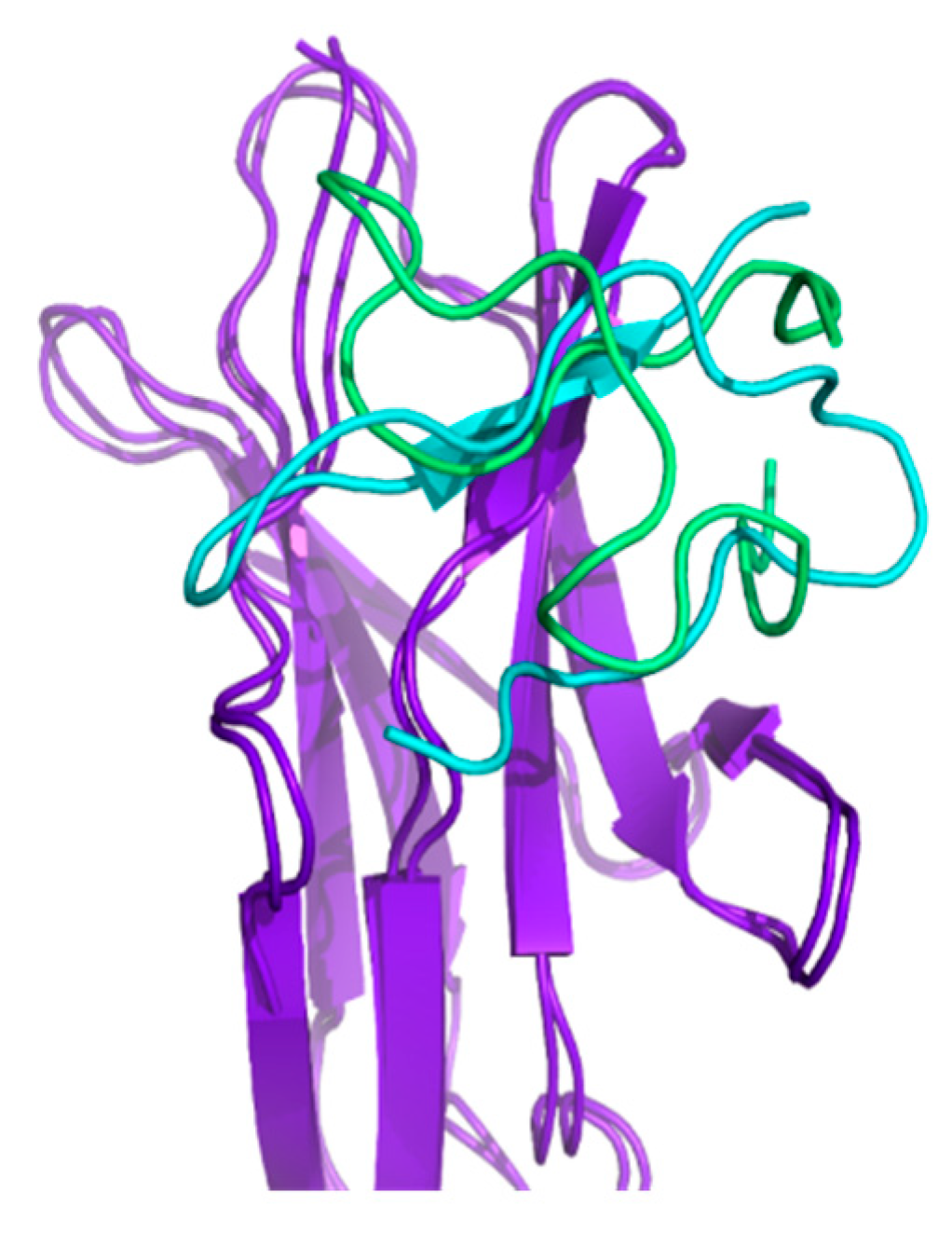
2.4. Docking and Determination of the Kinetic Constants of HVEM (14–39) Peptide Binding to BTLA Based on the NMR Structure of the Peptide
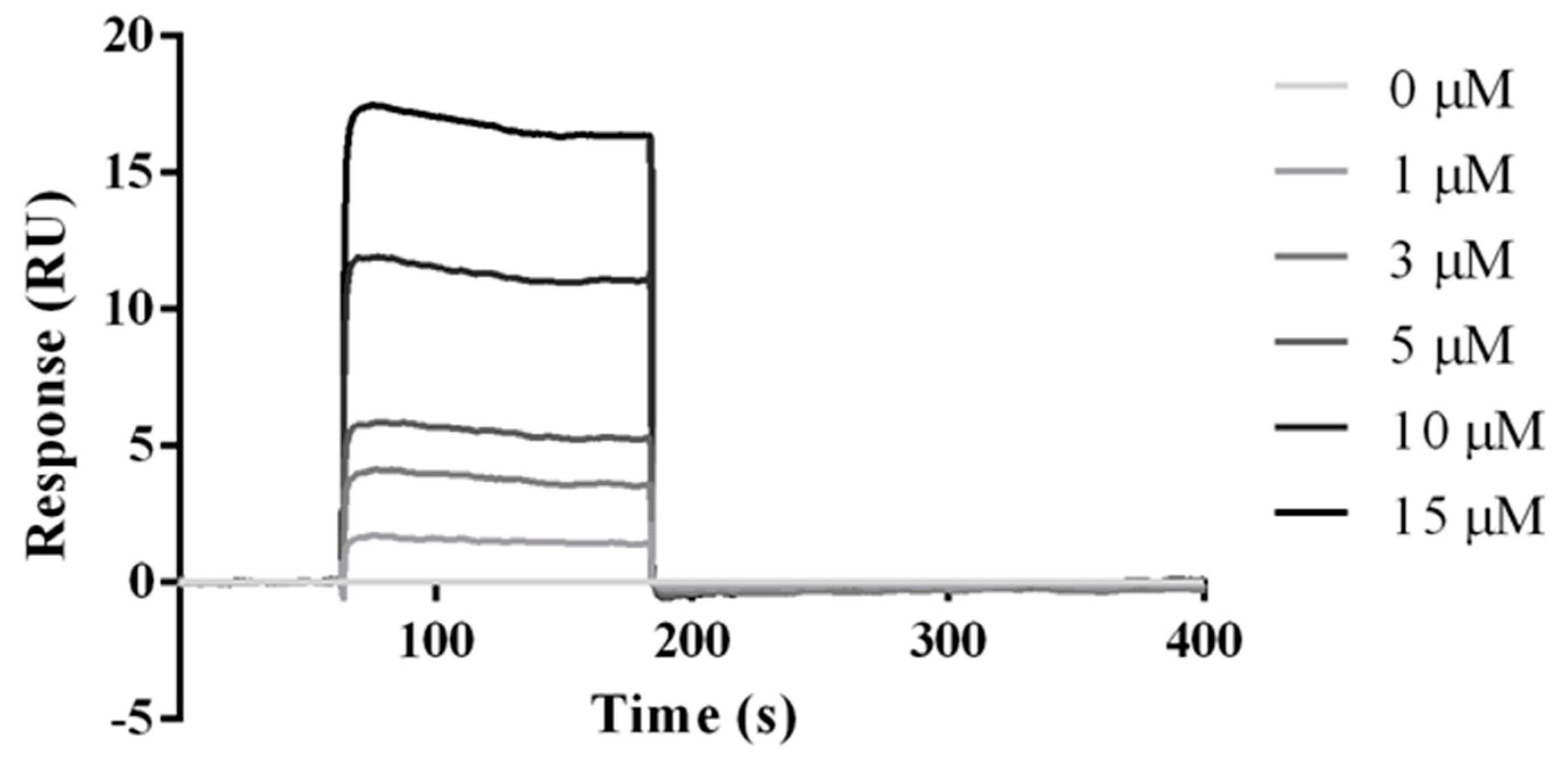
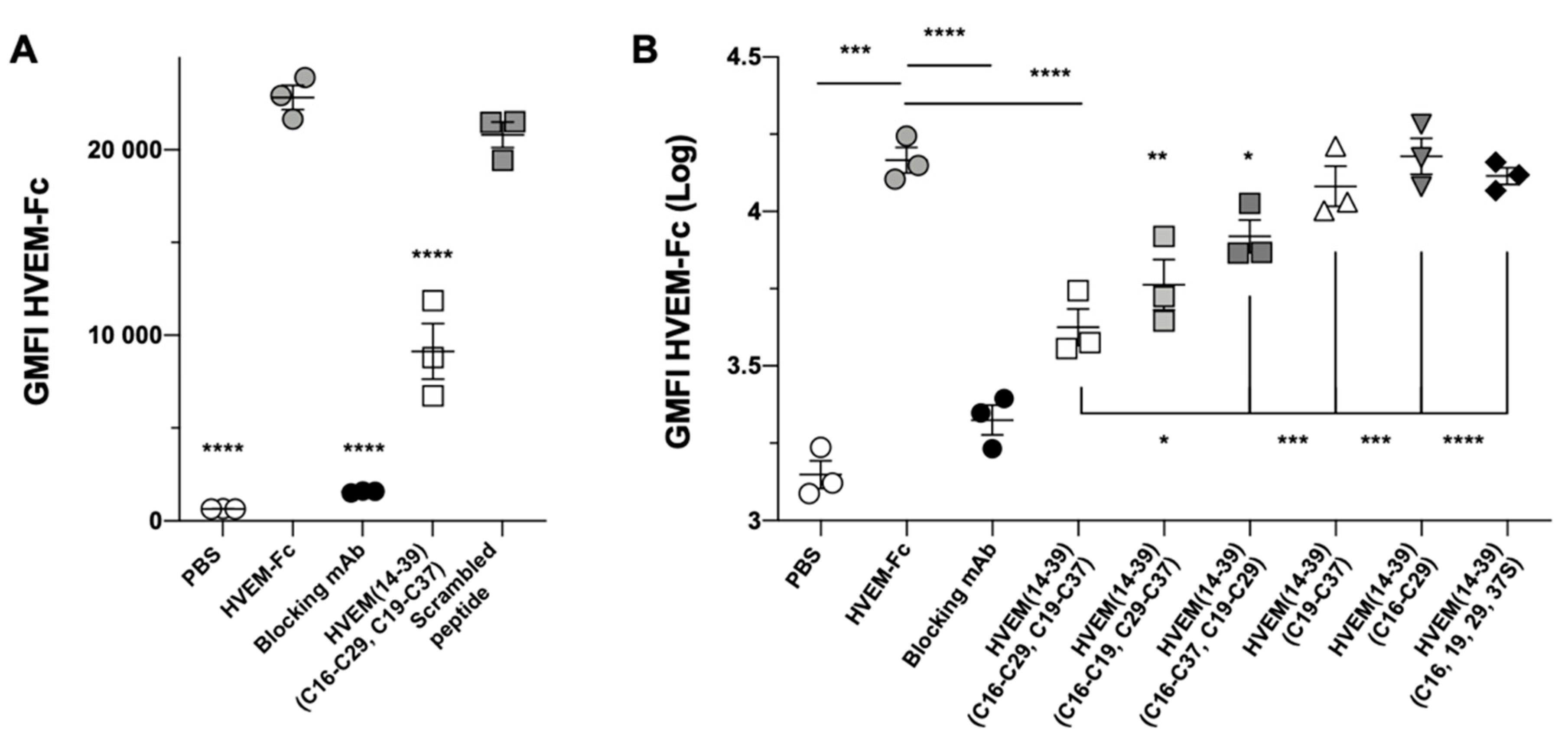
2.5. Experimental Studies of the Interactions Between the HVEM (14–39) Peptide and BTLA Protein
2.6. Studies of the Inhibitory Properties of the HVEM (14–39) Peptide in a Cell Line Assay
3. Discussion
4. Materials and Methods
4.1. Recombinant Molecules and Antibodies
4.2. Docking of the HVEM Protein or HVEM (14–39) Peptide to BTLA
4.3. Determination of the Kinetic Constants of HVEM or HVEM (14–39) Peptide Binding to BTLA
4.4. Peptide Synthesis
4.5. Peptide Purification
4.6. Formation of Disulfide Bonds
4.7. Nuclear Magnetic Resonance Spectroscopy and NMR Structure Calculation
4.8. Docking and Determination of the Kinetic Constants of the HVEM (14–39) Peptide to BTLA Based on the NMR Structure of the Peptide
4.9. Preparation of Microcolumn and Affinity Test
4.10. SPR Analysis
4.11. BTLA/HVEM Interaction—Cell Line Assay
4.12. Stability of the HVEM (14–39) Peptide
4.13. Cytotoxic Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BTLA | B-and T-lymphocyte attenuator |
| CHCA | α-cyano-4-hydroxycinnamic acid |
| CD160 | cluster of differentiation 160 |
| CRD | cysteine-rich domain |
| CSI | chemical shift index |
| CTLA-4 | cytotoxic T-lymphocyte antigen-4 |
| DAPI | 4′,6-diamidino-2-phenylindole dihydrochloride |
| DSS | sodium 2,2-dimethyl-2-silapentane-5-sulfonate |
| DTT | dithiothreitol |
| ELISA | enzyme-linked immunosorbent assay |
| Fmoc/tBu | 9-fluorenylmethoxycarbonyl/tert-butyl |
| HSQC | heteronuclear single quantum coherence |
| HVEM | herpes virus entry mediator |
| IgSF | immunoglobulin-like superfamily |
| ITIM | immunoreceptor tyrosine-based inhibitory motif |
| LC ESI–IT–TOF MS | liquid chromatography coupled with electrospray ionization, ion trap, and time-of-flight mass spectroscopy |
| LIGHT | homologous to lymphotoxins, exhibits inducible expression, and competes with HSV glycoprotein D for herpes virus entry mediator, a receptor expressed by T lymphocytes |
| LTα | lymphotoxin α |
| mAb | monoclonal antibody |
| MS | mass spectroscopy |
| MALDI-TOF | matrix-assisted laser desorption ionization, time-of-flight |
| MREMD | multiplexed-replica exchange molecular dynamics |
| MD | molecular dynamics |
| NOE | nuclear Overhauser effect |
| NOESY | nuclear Overhauser effect spectroscopy |
| NMR | nuclear magnetic resonance |
| PBS | phosphate-buffered saline |
| PD-1 | programmed cell death 1 |
| PDB | protein data bank |
| PD-L1 | programmed cell death-ligand 1 |
| PBMC | peripheral blood mononuclear cells |
| PPIs | protein–protein interactions |
| RP-HPLC | reverse-phase high-performance liquid chromatography |
| RMSD | root-mean-square deviation |
| SPPS | solid-phase peptide synthesis |
| SHP-1 and 2 | Src homology-2-containing protein 1 and 2 |
| SPR | surface plasmon resonance |
| TFA | trifluoroacetic acid |
| TNFR | tumor necrosis factor receptor |
| TOCSY | total correlation spectroscopy |
| UNRES | UNited RESidue |
| WHAM | weighted histogram analysis method |
References
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Dömling, A. Inhibitors of programmed cell death 1 (PD-1): a patent review (2010-2015). Expert Opin. Ther. Pat. 2016, 26, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Constantinidou, A.; Alifieris, C.; Trafalis, D.T. Targeting Programmed Cell Death -1 (PD-1) and Ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol. Ther. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2017, 32, 317–326. [Google Scholar]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef]
- Gonzalez, L.C.; Loyet, K.M.; Calemine-Fenaux, J.; Chauhan, V.; Wranik, B.; Ouyang, W.; Eaton, D.L. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. PNAs 2005, 102, 1116–1121. [Google Scholar] [CrossRef] [Green Version]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef]
- Pasero, C.; Olive, D. Interfering with coinhibitory molecules: BTLA/HVEM as new targets to enhance anti-tumor immunity. Immunol. Lett. 2013, 151, 71–75. [Google Scholar] [CrossRef]
- Lan, X.; Li, S.; Gao, H.; Nanding, A.; Quan, L.; Yang, C.; Ding, S.; Xue, Y. Increased BTLA and HVEM in gastric cancer are associated with progression and poor prognosis. Onco. Targets. Ther. 2017, 10, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, M.F.; Bohner, P.; Pieraerts, C.; Lhermitte, B.; Gourmaud, J.; Nobile, A.; Rotman, S.; Cesson, V.; Martin, V.; Legris, A.S.; et al. Immunoregulation of Dendritic Cell Subsets by Inhibitory Receptors in Urothelial Cancer. Eur. Urol. 2017, 71, 854–857. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, J.; He, M.; Zhang, G.L.; Zhao, Q. Distinct Changes of BTLA and HVEM Expressions in Circulating CD4+ and CD8+ T Cells in Hepatocellular Carcinoma Patients. J. Immunol. Res. 2018, 2018, 4561571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derré, L.; Rivals, J.P.; Jandus, C.; Pastor, S.; Rimoldi, D.; Romero, P.; Michielin, O.; Olive, D.; Speiser, D.E. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J. Clin. Investig. 2010, 120, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, N.; Kamimura, Y.; Hashiguchi, M.; Azuma, M. Expression and function of the B and T lymphocyte attenuator (BTLA/CD272) on human T cells. Biochem. Biophys. Res. Commun. 2006, 344, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Gavrieli, M.; Sedy, J.R.; Yang, J.; Fallarino, F.; Loftin, S.K.; Hurchla, M.A.; Zimmerman, N.; Sim, J.; Zang, X.; et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 2003, 4, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Gavrieli, M.; Watanabe, N.; Loftin, S.K.; Murphy, T.L.; Murphy, K.M. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem. Biophys. Res. Commun. 2003, 312, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.S.; Tan, K.B.; Ni, J.; Oh, K.H.; Lee, Z.H.; Kim, K.K.; Kim, Y.J.; Wang, S.; Gentz, R.; Yu, G.L.; et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 1997, 272, 14272–14276. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.; Solovyev, I.; Colombero, A.; Elliott, R.; Kelley, M.; Boyle, W.J. ATAR, a Novel Tumor Family Member, Signals through TRAF2 and TRAF5. J. Biol. Chem. 1997, 272, 13471–13475. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Anumanthan, A.; Brown, J.A.; Greenfield, E.A.; Zhu, B.; Freeman, G.J. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol. 2008, 9, 176–185. [Google Scholar] [CrossRef]
- Kuncewicz, K.; Spodzieja, M.; Sieradzan, A.; Karczyńska, A.; Dąbrowska, K.; Dadlez, M.; Speiser, D.E.; Derre, L. A structural model of the immune checkpoint CD160 – HVEM complex derived from HDX-mass spectrometry and molecular modeling. Oncotarget 2019, 10, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Compaan, D.M.; Gonzalez, L.C.; Tom, I.; Loyet, K.M.; Eaton, D.; Hymowitz, S.G. Attenuating lymphocyte activity: The crystal structure of the BTLA-HVEM complex. J. Biol. Chem. 2005, 280, 39553–39561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Subudhi, S.K.; Anders, R.A.; Lo, J.; Sun, Y.; Blink, S.; Wang, Y.; Wang, J.; Liu, X.; Mink, K.; et al. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J. Clin. Investig. 2005, 115, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Skalniak, L.; Magiera, K.; Dubin, G.; Holak, T.A.; Zak, K.M.; Musielak, B.; Grudnik, P.; Guzik, K.; Törner, R.; Dömling, A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar]
- Shaabani, S.; Huizinga, H.P.S.; Butera, R.; Kouchi, A.; Guzik, K.; Magiera-Mularz, K.; Holak, T.A.; Dömling, A. A patent review on PD-1/PD-L1 antagonists: small molecules, peptides, and macrocycles (2015-2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Milan, D.; Peal, D. (12) Patent Application Publication. U.S. Patent US 2002/0187020 A1, 2013. [Google Scholar]
- Nevola, L.; Giralt, E. Modulating protein-protein interactions: The potential of peptides. Chem. Commun. 2015, 51, 3302–3315. [Google Scholar] [CrossRef]
- Stiles, K.M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Herpes Simplex Virus Glycoprotein D Interferes with Binding of Herpesvirus Entry Mediator to Its Ligands through Downregulation and Direct Competition. J. Virol. 2010, 84, 11646–11660. [Google Scholar] [CrossRef] [Green Version]
- Spodzieja, M.; Lach, S.; Iwaszkiewicz, J.; Cesson, V.; Kalejta, K.; Olive, D.; Michielin, O.; Speiser, D.E.; Zoete, V.; Derré, L.; et al. Design of short peptides to block BTLA/HVEM interactions for promoting anticancer T-cell responses. PLoS One 2017, 12, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chemie Int. Ed. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 22 January 2019).
- Cheung, T.C.; Humphreys, I.R.; Potter, K.G.; Norris, P.S.; Shumway, H.M.; Tran, B.R.; Patterson, G.; Jean-Jacques, R.; Yoon, M.; Spear, P.G.; et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc. Natl. Acad. Sci. USA. 2005, 102, 13218–13223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumwalde, N.A.; Kenney, S.C.; Jenny, E.; Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Gumperz, J.E. Adoptively transferred V g 9V d 2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model Find the latest version : Adoptively transferred V γ 9V δ 2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis. JCI Insight. 2017, 122, 922–932. [Google Scholar]
- Sieradzan, A.K.; Krupa, P.; Scheraga, H.A.; Liwo, A.; Czaplewski, C. Physics-based potentials for the coupling between backbone- and side-chain-local conformational states in the United Residue (UNRES) force field for protein simulations. J. Chem. Theory Comput. 2015, 11, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieradzan, A.K. Introduction of periodic boundary conditions into UNRES force field. J. Comput. Chem. 2015, 36, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Karczyńska, A.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Bojarski, K.K.; Zaborowski, B.; Liwo, A.; Ślusarz, R.; Ślusarz, M.; Lee, J.; et al. Use of the UNRES force field in template-assisted prediction of protein structures and the refinement of server models: Test with CASP12 targets. J. Mol. Graph. Model. 2018, 83, 92–99. [Google Scholar] [CrossRef]
- Khalili, M.; Liwo, A.; Jagielska, A.; Scheraga, H.A. Molecular dynamics with the united-residue model of polypeptide chains. II. Langevin and Berendsen-bath dynamics and tests on model alpha-helical systems. J. Phys. Chem. B 2005, 109, 13798–13810. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Khalili, M.; Scheraga, H.A. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc. Natl. Acad. Sci. 2005, 102, 2362–2367. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [Green Version]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: Hoboken, HJ, USA, 1986; ISBN 9780471828938. [Google Scholar]
- Güntert, P.; Mumenthaler, C.; Wüthrich, K. Torsion angle dynamics for protein structure calculations with a new program, DYANA. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14. Available online: https://ambermd.org/doc12/Amber14.pdf (accessed on 18 January 2020).
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | System | Method | k1 | k−1 | KD |
|---|---|---|---|---|---|
| 1 | BTLA/HVEM | SPR | 620,000 | 0.12 | 2.5 × 10−7 |
| 2 | BTLA/HVEM (14–39) | SPR | 76,400 | 0.00332 | 1.02 × 10−7 |
| 3 | BTLA/HVEM crystal-based structure | Molecular dynamics | 16.895 | 0.0190 | 1.1 × 10−3 |
| 4 | BTLA/HVEM (14–39) crystal-based structure | Molecular dynamics | 5.594 | 0.00105 | 1.87 × 10−4 |
| 5 | BTLA/HVEM (14–39) NMR-based structure | Molecular dynamics | 68.49 | 0.0116 | 1.69 × 10−4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spodzieja, M.; Kuncewicz, K.; Sieradzan, A.; Karczyńska, A.; Iwaszkiewicz, J.; Cesson, V.; Węgrzyn, K.; Zhukov, I.; Maszota-Zieleniak, M.; Michielin, O.; et al. Disulfide-Linked Peptides for Blocking BTLA/HVEM Binding. Int. J. Mol. Sci. 2020, 21, 636. https://doi.org/10.3390/ijms21020636
Spodzieja M, Kuncewicz K, Sieradzan A, Karczyńska A, Iwaszkiewicz J, Cesson V, Węgrzyn K, Zhukov I, Maszota-Zieleniak M, Michielin O, et al. Disulfide-Linked Peptides for Blocking BTLA/HVEM Binding. International Journal of Molecular Sciences. 2020; 21(2):636. https://doi.org/10.3390/ijms21020636
Chicago/Turabian StyleSpodzieja, Marta, Katarzyna Kuncewicz, Adam Sieradzan, Agnieszka Karczyńska, Justyna Iwaszkiewicz, Valérie Cesson, Katarzyna Węgrzyn, Igor Zhukov, Martyna Maszota-Zieleniak, Olivier Michielin, and et al. 2020. "Disulfide-Linked Peptides for Blocking BTLA/HVEM Binding" International Journal of Molecular Sciences 21, no. 2: 636. https://doi.org/10.3390/ijms21020636