Elucidating Binding Sites and Affinities of ERα Agonists and Antagonists to Human Alpha-Fetoprotein by In Silico Modeling and Point Mutagenesis

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
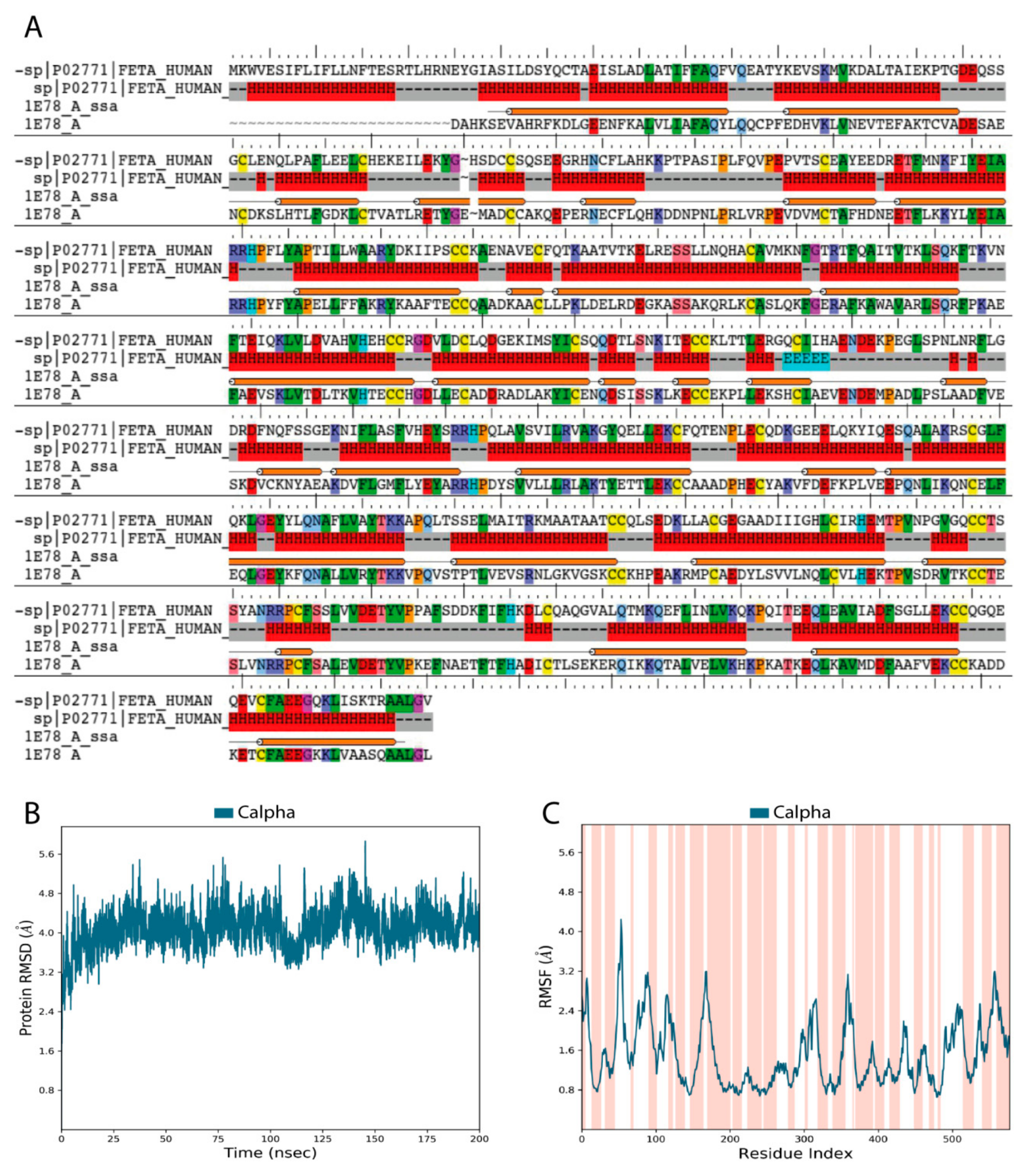
2.1. The Overall Architecture and Quality of Constructed HAFP 3D Model
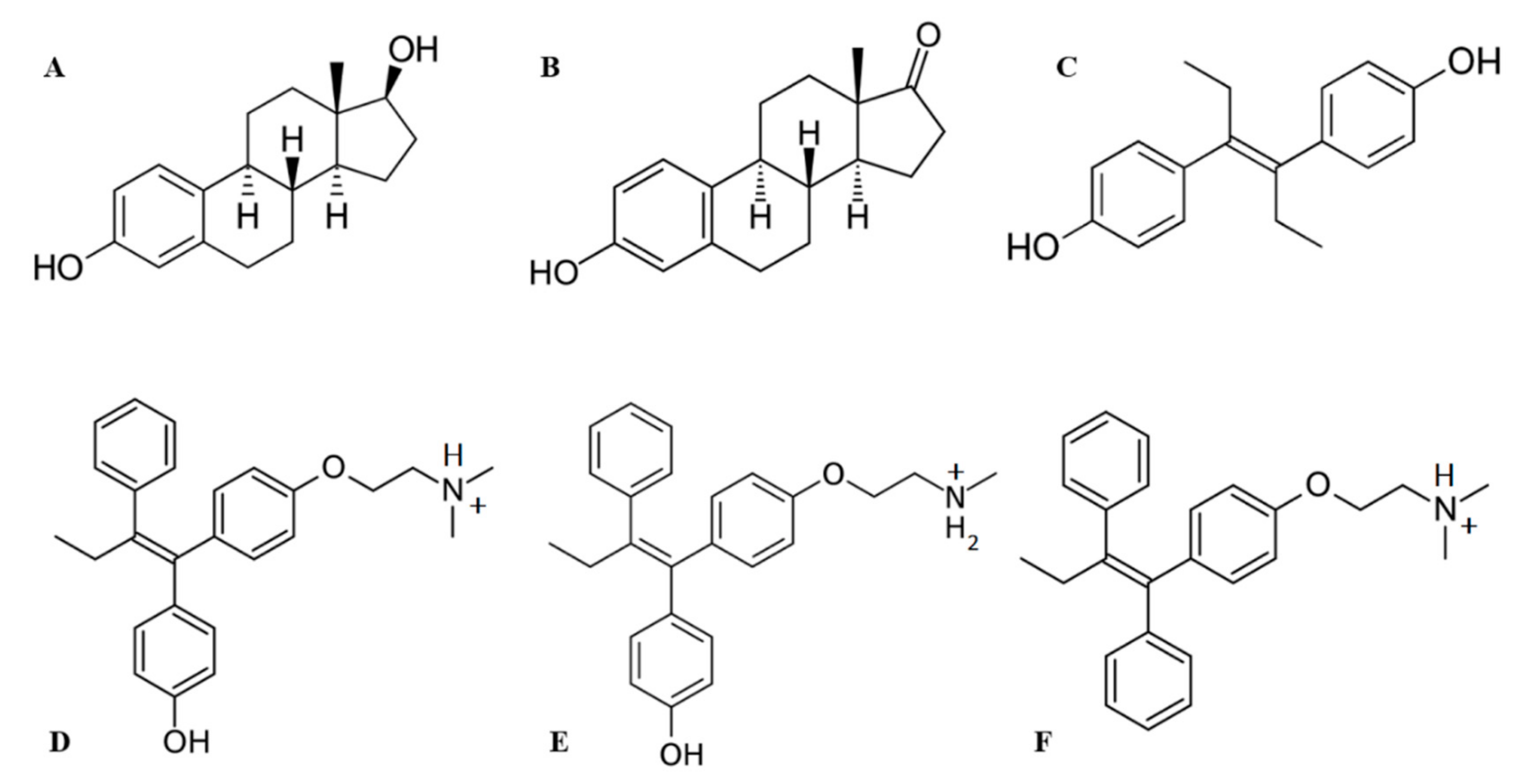
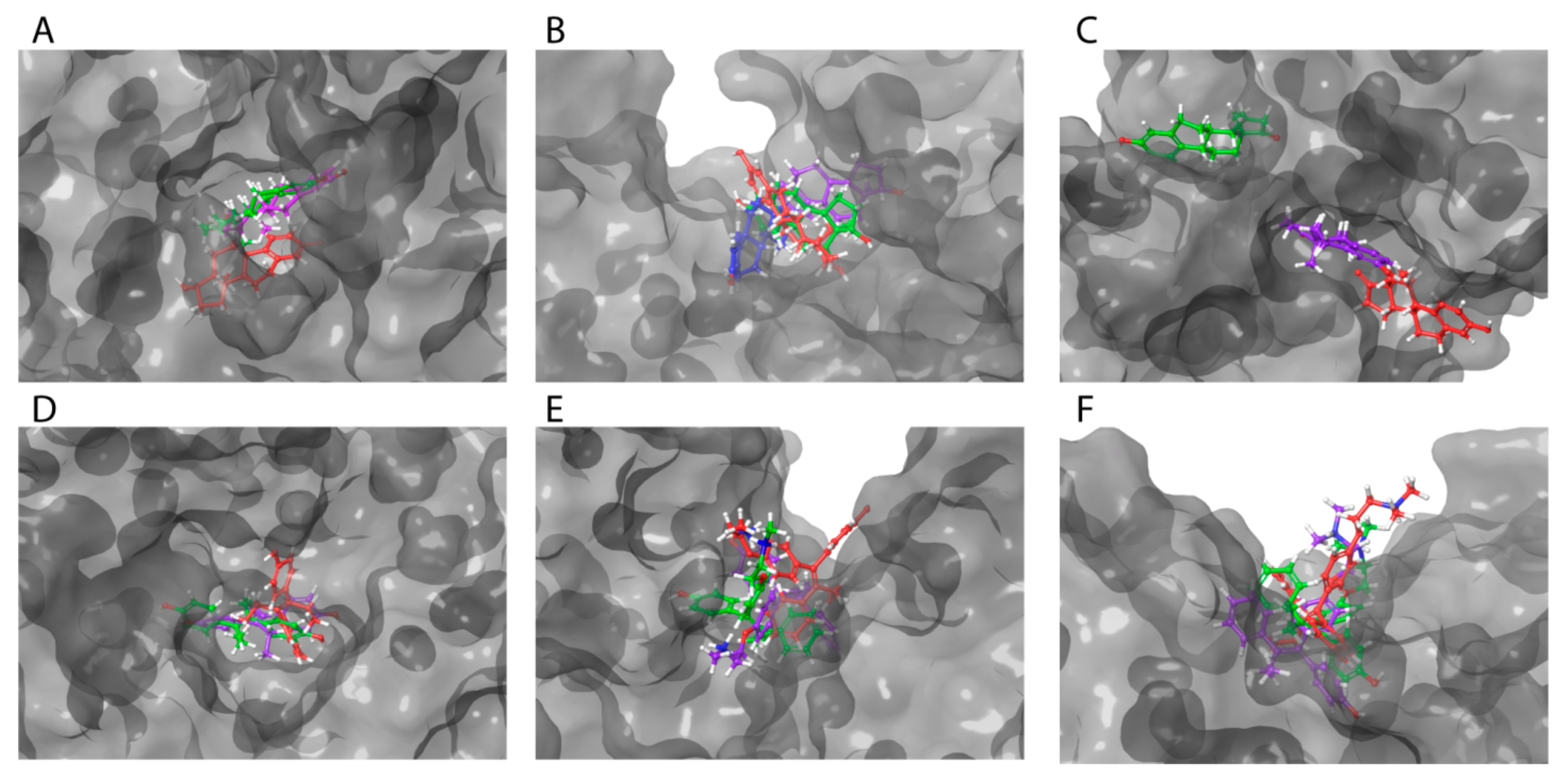
2.2. HAFP–Ligand-Docked Poses
2.3. Binding of Ligands to HAFP Studied by MD Simulation
2.3.1. Stability of HAFP–Ligand Complexes
2.3.2. Ligand Binding Affinities
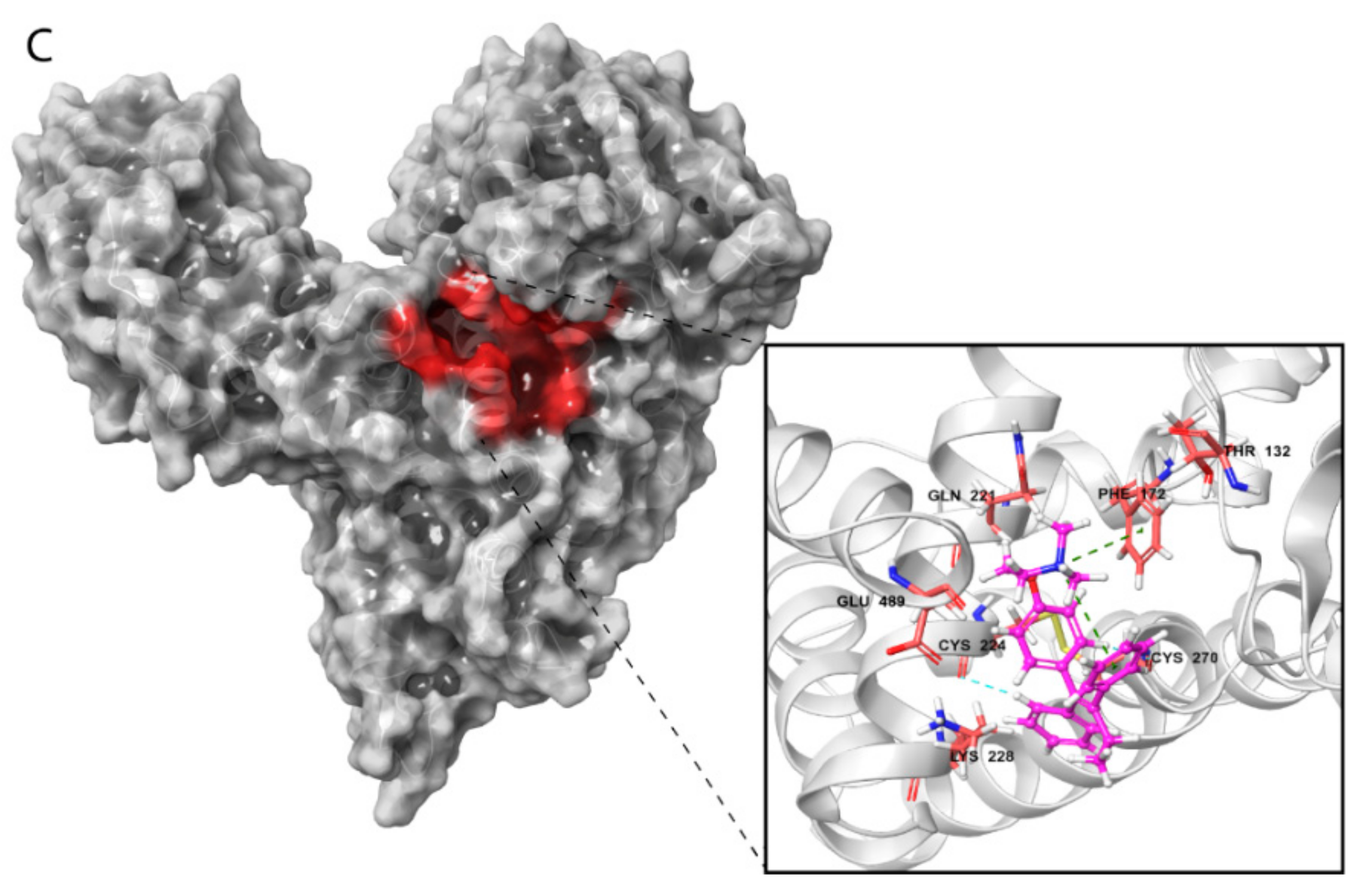
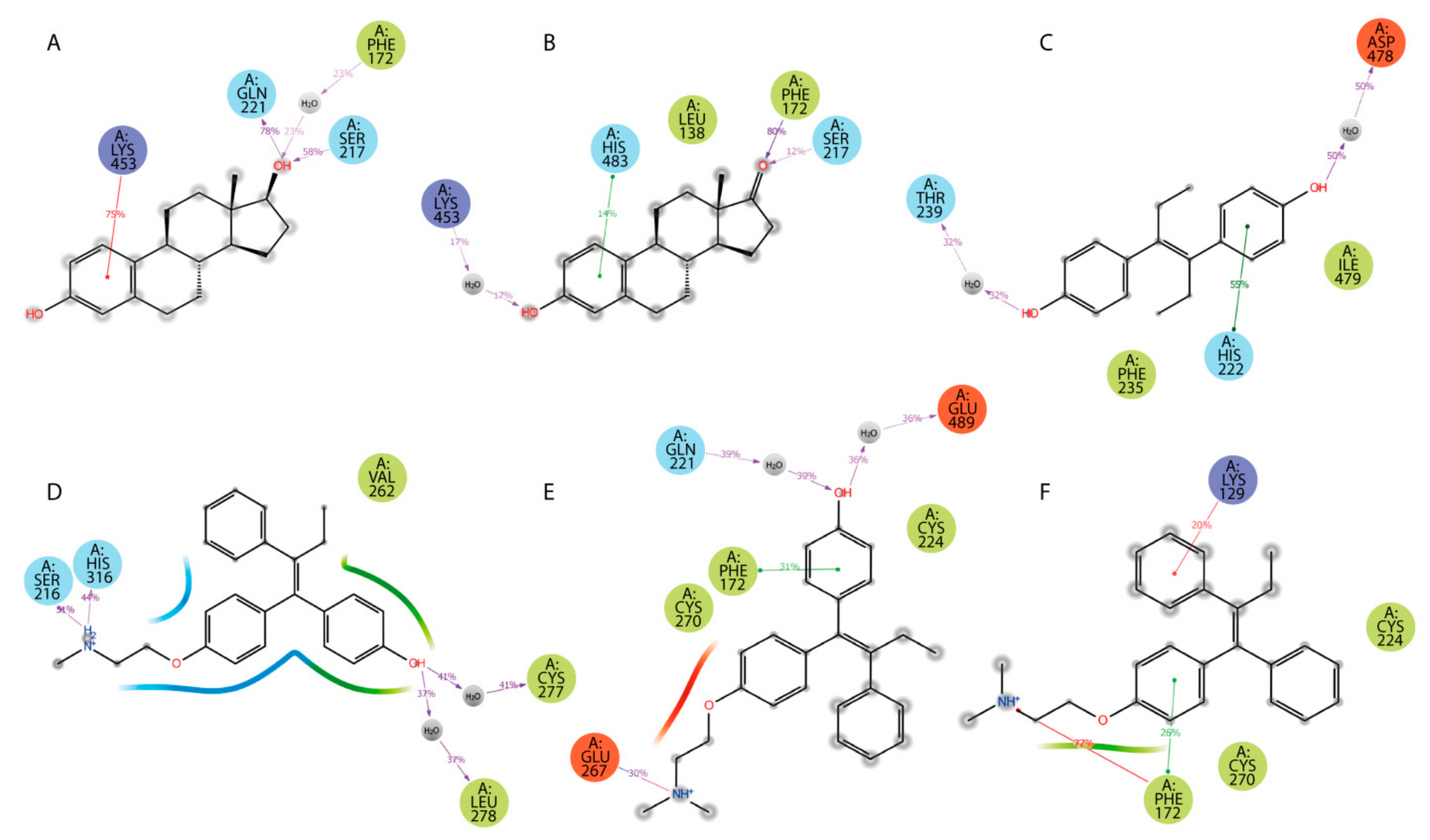
2.3.3. HAFP–Ligand Interaction Modes
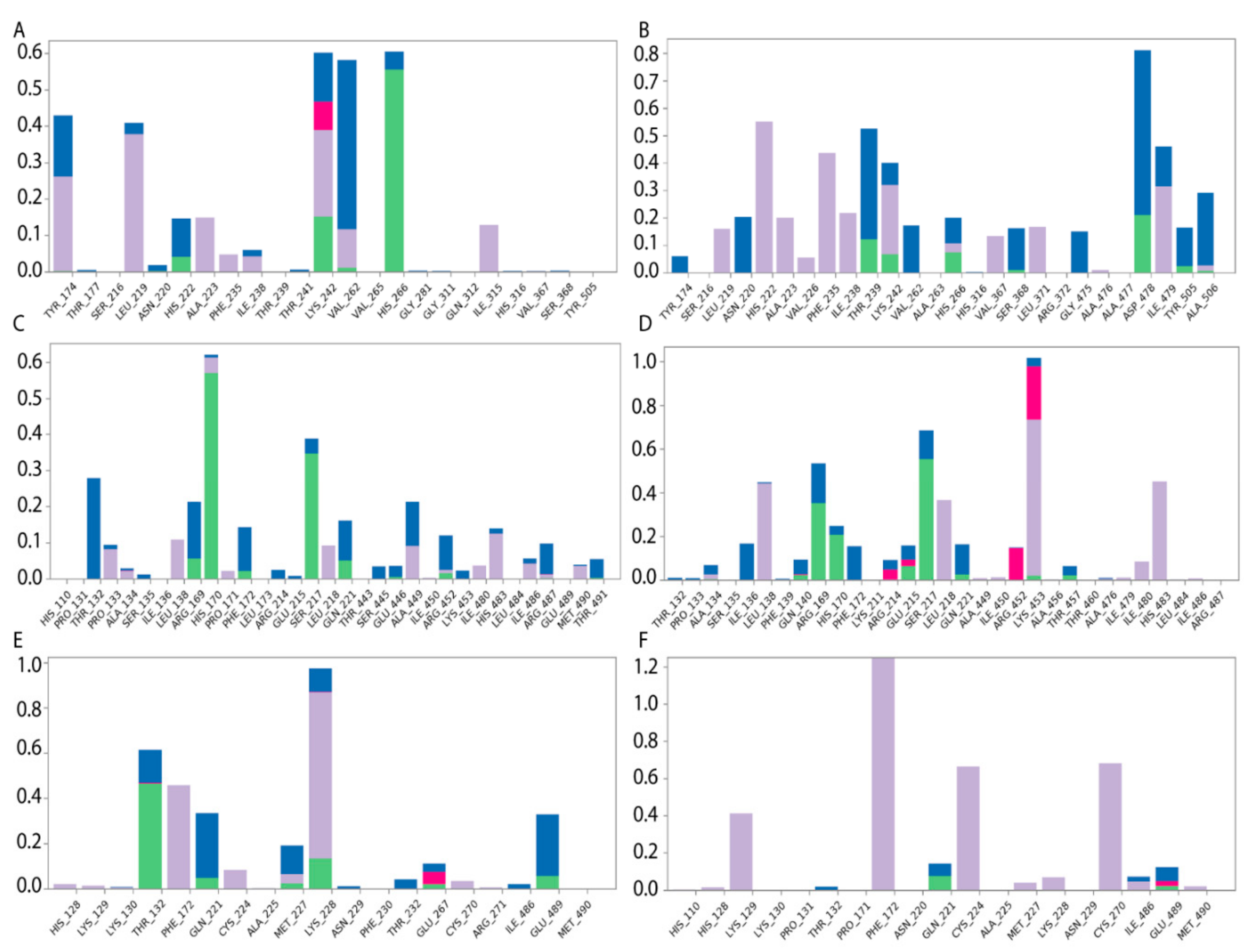
2.4. Effects of In Silico Point Amino Acid Substitutions
3. Discussion
4. Materials and Methods
4.1. HAFP Homology-Based Modeling
4.1.1. Template Identification
4.1.2. Model Generation and Validation
4.1.3. Model Relaxation and Optimization
4.2. Molecular Docking and Scoring
4.2.1. Ligand Preparation
4.2.2. Identification of Protein–Ligand Binding Sites and Grid Generation
4.2.3. Docking Protocol
4.2.4. Scoring Functions
4.3. MD Simulation of Protein–Ligand Complexes
4.4. RMSD and RMSF Calculations for Protein–Ligand Complexes
4.5. In Silico Point Amino Acid Substitutions
4.6. Binding Free Energy Calculations by MM/GBSA Rescoring Method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HAFP | human α-fetoprotein |
| HSA | human serum albumin |
| E2 | 17β-estradiol |
| DES | diethylstilbestrol |
References
- Liu, C.; Xiao, G.Q.; Yan, L.N.; Li, B.; Jiang, L.; Wen, T.F.; Wang, W.T.; Xu, M.Q.; Yang, J.Y. Value of α-fetoprotein in association with clinicopathological features of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Sauzay, C.; Petit, A.; Bourgeois, A.M.; Barbare, J.C.; Chauffert, B.; Galmiche, A.; Houessimon, A. Alpha-foetoprotein (AFP): A multi-purpose marker in hepatocellular carcinoma. Clin. Chim. Acta. 2016, 463, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, R.; Wei, J.; Ye, J.; He, Q.; ChunlingYuan, Y.J.; Li, Y.; Liu, Z.; Kin, Y. Identification of candidate biomarkers in malignant ascites from patients with hepatocellular carcinoma by iTRAQ-based quantitative proteomic analysis. Biomed. Res. Int. 2018, 2018, 5484976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizejewski, G.J. Biological role of alpha-fetoprotein in cancer: Prospects for anticancer therapy. Expert Rev. Anticancer Ther. 2002, 2, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Terentiev, A.A.; Moldogazieva, N.T. Alpha-fetoprotein: A renaissance. Tumor Biol. 2013, 34, 2075–2091. [Google Scholar] [CrossRef]
- Li, M.S.; Li, P.F.; Yang, F.Y.; He, S.P.; Du, G.G.; Li, G. The intracellular mechanism of alpha-fetoprotein promoting the proliferation of NIH 3T3 cells. Cell Res. 2002, 11, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atemezem, A.; Mbemba, E.; Marfaing, R.; Vaysse, J.; Pontet, M.; Saffar, L.; Charnaux, N.; Gattegno, L. Human alpha-fetoprotein binds to primary macrophages. Biochem. Biophys. Res. Commun. 2002, 296, 507–514. [Google Scholar] [CrossRef]
- Matsuura, E.; Kang, Y.; Katikawa, H.; Ogata, P.; Kotani, T.; Ohtaki, S.; Nishi, S. Modulation of T-cell function by alpha-fetoprotein. An in vivo study on porcine thyroid peroxidase induced experimental autoimmune thyroiditis in transgenic mice producing human alpha-fetoprotein. Tumor Biol. 1999, 20, 162–171. [Google Scholar] [CrossRef]
- Milligan, S.R.; Khan, O.; Nash, M. Competitive binding of xenobiotic oestrogens to rat alpha-fetoprotein and sex steroid binding proteins in human and rainbow trout (Oncorhynchus mykiss) plasma. J. Comp. Endocrinol. 1998, 112, 89–95. [Google Scholar] [CrossRef]
- Terent’ev, A.A.; Moldogazieva, N.T.; Tatarinov, I.S. Competitive affinity chromatography of human alpha-fetoprotein on immobilized diethylstilbestrol. Biull. Eksp. Biol. Med. 1990, 109, 438–440. [Google Scholar] [CrossRef]
- Aussel, C.; Masseyeff, R. Human alpha-fetoprotein-fatty acid interaction. Biochem. Biophys. Res. Commun. 1983, 115, 38–45. [Google Scholar] [CrossRef]
- Bennett, J.A.; Zhu, S.; Pagano-Mirarchi, A.; Kellom, T.A.; Jacobson, H.A. Alpha-fetoprotein derived from a human hepatoma prevents growth of estrogen-dependent human breast cancer xenografts. Clin. Cancer Res. 1998, 4, 2877–2884. [Google Scholar] [PubMed]
- Jacobson, H.A.; Bennett, J.A.; Mizejewski, G.J. Inhibition of estrogen-dependent breast cancer growth by a reaction product of alpha-fetoprotein and estradiol. Cancer Res. 1990, 50, 415–420. [Google Scholar] [PubMed]
- Mizejewski, G.J. Mechanism of cancer growth suppression of alpha-fetoprotein derived growth inhibitory peptides (GIP): Comparison of GIP-34 versus GIP-8 (AFPep). Updates and prospects. Cancers 2011, 3, 2709–2733. [Google Scholar] [CrossRef] [Green Version]
- Mizejewski, G.J. Biological roles of alpha-fetoprotein during pregnancy and perinatal development. Exp. Biol. Med. 2004, 229, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Makela, S.; Trenter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef] [PubMed]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, S.; Gustafsson, J.A. Nuclear receptors: Recent drug discovery for cancer therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Blair, R.M.; Fang, H.; Branham, W.S.; Hass, B.S.; Dial, S.L.; Moland, C.L.; Tong, W.; Shi, L.; Perkins, R.; Sheehan, D.M. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: Structural diversity of ligands. Toxicol. Sci. 2000, 54, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Mauvais-Jarvis, F.; Clegg, D.J.; Henever, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 2017, 8, 33. [Google Scholar] [CrossRef]
- Hong, H.; Branham, W.S.; Ng, H.W.; Moland, C.L.; Dial, S.L.; Fang, H.; Perkins, R.; Sheehan, D.; Tong, W. Human sex hormone-binding globulin binding affinities of 125 structurally diverse chemicals and comparison with their binding to androgen receptor, estrogen receptor, and alpha-fetoprotein. Toxicol. Sci. 2015, 143, 333–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.L.; Khosla, S. Modulators of androgen and estrogen receptor activity. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Cornil, C.A.; Ball, G.F.; Balthazart, J. The dual action of estrogen hypothesis. Trends Neurosci. 2015, 38, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojetin, D.J.; Burris, T.P.; Jensen, E.V.; Khan, S.A. Implications of the binding of tamoxifen to the coactivator recognition site of the estrogen receptor. Endocr. Relat. Cancer. 2008, 15, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Chalmers, M.J.; Bruning, J.; Bramlett, K.S.; Osborne, H.E.; Montrose-Rafizadeh, C.; Barr, R.J.; Wang, Y.; Wang, M.; Burris, T.P.; et al. Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc. Natl. Acad. Sci. USA 2008, 105, 7171–7176. [Google Scholar] [CrossRef] [Green Version]
- Terentiev, A.A.; Moldogazieva, N.T. Structural and functional mapping of alpha-fetoprotein. Biochemistry 2006, 71, 120–132. [Google Scholar]
- Tatarinov, Y.S.; Terentiev, A.A.; Moldogazieva, N.T.; Tagirova, A.K. Human alpha-fetoprotein and its purification by chromatography on immobilized estrogens. Tumor Biol. 1991, 12, 125–130. [Google Scholar] [CrossRef]
- Nishi, S.; Matsue, H.; Yoshida, H.; Yamamoto, R.; Sakai, M. Localization of the estrogen-binding site of alpha-fetoprotein in the chimeric human-rat proteins. Proc. Natl. Acad. Sci. USA. 1991, 88, 3102–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazzadeh, D. Estrogen binding activities of recombinant alpha-fetoproteins expressed in yeast. Hokkaido Igaku Zasshi. 1995, 70, 473–483. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, H.; Dugaiszyk, A. Complete structure of the human alpha-albumin gene, a new member of the serum albumin multigene family. Proc. Natl. Acad. Sci. USA 1996, 93, 7557–7561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terentiev, A.A.; Moldogazieva, N.T.; Levtsova, O.V.; Maximenko, D.M.; Borozdenko, D.A.; Shaitan, K.V. Modeling of three dimensional structure of human alpha-fetoprotein complexed with diethylstilbestrol: Docking and molecular dynamics simulation study. J. Bioinform. Comput. Biol. 2012, 10, 1241012. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, W.; Fang, H.; Perkins, R.; Tong, W.; Hong, H. Homology modeling, molecular docking, and molecular dynamics simulations elucidated α-fetoprotein binding modes. BMC Biol. 2013, 14 (Suppl. 14), 56. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Branham, W.S.; Dial, S.L.; Moland, C.L.; Fang, H.; Shen, J.; Perkins, R.; Sheehan, D.; Tong, W. Rat α-fetoprotein binding affinities to a large set of structurally diverse chemicals elucidated the relationships between structures and binding affinities. Chem. Res. Toxicol. 2012, 25, 2553–2566. [Google Scholar] [CrossRef]
- Luft, A.J.; Lorscheider, F.L. Structural analysis of human and bovine α-fetoprotein by electron microscopy, image processing and circular dichroism. Biochemistry 1983, 22, 5971–5978. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Aussel, C.; Masseyeff, R. Interaction of retinoids and bilirubin with the binding of arachidonic acid to human alpha-fetoprotein. Biochem. Biophys. Res. Commun. 1984, 119, 1122–1127. [Google Scholar] [CrossRef]
- Aussel, C. Presence of three different binding sites for retinoids, bilirubin and estrogen or arachidonic acid on rat alpha-fetoprotein. Tumour Biol. 1985, 6, 179–193. [Google Scholar] [PubMed]
- Uriel, J.; Bouillon, D.; Aussel, C.; Dupiers, M. Alpha-fetoprotein: The major high-affinity estrogen binder in rat uterine cytosols. Proc. Natl. Acad. Sci. USA 1976, 73, 1452–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keel, B.A.; Abney, T.O. The kinetics of estrogen binding to rat alpha-fetoprotein. Experientia 1984, 40, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Versee, V.; Barel, A.O. Binding specificity of estrogens and norandrogens to rat alpha-fetoprotein (AFP). FEBS Lett. 1978, 96, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Savu, L.; Benassayag, C.; Vallette, G.; Nunez, E.A. Ligand properties of diethylstilbestrol: Studies with purified native and fatty acid-free rat alpha 1-fetoprotein and albumin. Steroids 1979, 34, 737–748. [Google Scholar] [CrossRef]
- Savu, L.; Benassayag, C.; Vallette, G.; Christeff, N.; Nunez, E.A. Mouse α-fetoprotein and albumin. A comparison of their binding properties with estrogen and fatty acid ligands. J. Biol. Chem. 1981, 256, 9414–9418. [Google Scholar] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Feyfant, E.; Sali, A.; Fiser, A. Modeling mutations in protein structures. Protein Sci. 2007, 16, 2030–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldogazieva, N.T.; Terentiev, A.A.; Shaitan, K.V. Relationship between structure and function of alpha-fetoprotein: Conformational changes and biological activity. Biomed. Khim. 2005, 51, 127–151. [Google Scholar]
- Chen, C.; Huang, H.; Wu, C.H. Protein bioinformatics databases and resources. Methods Mol. Biol. 2017, 1558, 3–39. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-2. Prime; Schrödinger, LLC: New-York, NY, USA, 2018.
- Schrödinger, Release 2018-2. Maestro; Schrödinger, LLC: New York, NY, USA, 2018.
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to heck the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-2. Desmond Molecular Dynamics System; Schrödinger, LLC: New York, NY, USA, 2018.
- Hünenberger, H. Thermostat algorithms for molecular dynamics simulations. In Advanced Computer Simulation; Holm, C., Kremer, K., Eds.; Springer: New York, NY, USA, 2005; pp. 105–149. [Google Scholar]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A. PubChem substance and compound databases. Nucleic Acid Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-2. LigPrep. Schrödinger, LLC: New York, NY, USA, 2018.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar]
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631–1639. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docked Ligand | Amount of Rotational Bonds | Binding Site | Glide Gscore | Glide Emodel |
|---|---|---|---|---|
| 17β-estradiol | 2 | A | −5.420 | −49.732 |
| B | −4.807 | −46.737 | ||
| C | −5.384 | −35.722 | ||
| Estrone | 1 | A | −4.109 | −45.332 |
| B | −4.054 | −45.389 | ||
| C | −4.408 | −38.067 | ||
| DES | 6 | A | −4.934 | −34.058 |
| B | −3.126 | −33.573 | ||
| C | −3.440 | −37.538 | ||
| Afimoxifene | 9 | A | −5.932 | −57.375 |
| B | −3.840 | −55.881 | ||
| C | −5.308 | −54.272 | ||
| Endoxifen | 9 | A | −6.322 | −61.786 |
| B | −4.247 | −50.983 | ||
| C | −5.768 | −59.989 | ||
| Tamoxifen | 8 | A | −4.805 | −53.899 |
| B | −2.976 | −43.853 | ||
| C | −4.169 | −51.536 |
| Complex | MM/GBSA ΔGbind Docked Poses | MM/GBSA ΔGbind Optimized Complexes | ΔGCoulomb Optimized | ΔGvdW Optimized | ΔGGB Optimized | ΔGlipo Optimized | Ligand Efficiency |
|---|---|---|---|---|---|---|---|
| Site A | |||||||
| HAFP–17β-estradiol | −35.03 | −38.29 | −9.55 | −33.20 | 20.50 | −15.99 | −9.58 |
| HAFP–DES | −49.14 | −50.99 | −10.68 | −39.69 | 20.06 | −21.11 | −12.76 |
| HAFP–endoxifen | −64.50 | −64.18 | −42.01 | −47.54 | 56.66 | −28.06 | −14.81 |
| HAFP–afimoxifene | −69.34 | −57.23 | −36.93 | −47.05 | 49.76 | −25.18 | −13.10 |
| HAFP–tamoxifen | −30.53 | −48.09 | −32.86 | −42.11 | 45.60 | −19.74 | −11.10 |
| Site B | |||||||
| HAFP–17β-estradiol | −42.85 | −44.22 | −15.23 | −29.95 | 15.18 | −13.00 | −11.05 |
| HAFP–estrone | −42.51 | −50.31 | −10.81 | −36.08 | 13.86 | −16.32 | −12.59 |
| HAFP–endoxifen | −55.15 | −59.61 | −41.92 | −43.93 | 43.95 | −18.22 | −13.76 |
| Site C | |||||||
| HAFP–endoxifen | −59.13 | −45.38 | −39.41 | −39.13 | 45.95 | −15.91 | −10.47 |
| HAFP–afimoxifene | −53.80 | −61.32 | −39.94 | −47.06 | 46.92 | −22.23 | −14.04 |
| HAFP–tamoxifen | −52.68 | −58.74 | −33.62 | −45.91 | 41.10 | −23.04 | −13.56 |
| HAFP–DES | −44.47 | −36.33 | −9.55 | −22.88 | 14.52 | −17.71 | −9.09 |
| Complex | Binding Site | Substitution | MM/GBSA ΔGbind a | ΔGCoulomb | ΔGvdW | ΔGGB | ΔGlipo | Ligand Efficiency |
|---|---|---|---|---|---|---|---|---|
| HAFP– 17β-estradiol | A | Lys242Leu | −39.3628 (−38.2924) | −7.2468 | −34.8330 | 22.0165 | −19.3103 | −9.8512 |
| His266 Leu | −42.5281 (−38.2924) | −7.8309 | −35.1273 | 19.5618 | −19.4308 | −10.6434 | ||
| B | Ser217Ala | −45.6250 (−44.2185) | −16.5316 | −30.8540 | 16.4249 | −13.7538 | −11.3184 | |
| Gln221Val | −41.9187 (−44.2185) | −11.0667 | −31.4626 | 16.5076 | −14.9278 | −10.4909 | ||
| Lys453Leu | −51.1980 (−44.2185) | −20.9175 | −30.2452 | 17.0518 | −16.4198 | −12.8132 | ||
| HAFP– estrone | B | Leu138 Ser | −47.0881 (−50.31) | −11.0476 | −37.6467 | 16.8381 | −15.1892 | −11.7846 |
| Phe172Ala | −32.3050 (−50.31) | −7.8493 | −30.9881 | 14.3777 | −8.2677 | −8.0849 | ||
| His170 Leu | −40.3051 (−43.88) | −8.1035 | −33.2855 | 15.8254 | −14.8531 | −10.0870 | ||
| HAFP–DES | A | Asp478Ala | −36.5685 (−51.00) | −11.7190 | −30.5551 | 20.7434 | −15.9298 | −9.1519 |
| His222Leu | −48.0213 (−51.00) | −11.2453 | −34.2183 | 16.6168 | −20.4277 | −12.0181 | ||
| HAFP– endoxifen | A | Cys277Val | −54.0787 (−64.18) | −36.8418 | −45.6549 | 55.1793 | −25.6210 | −12.4830 |
| His316Leu | −54.1489 (−64.18) | −38.4019 | −45.9868 | 54.6261 | −23.2189 | −12.4991 | ||
| B | Lys453 Glu | −50.4113 (-59.61) | −36.0847 | −40.3290 | 45.4848 | −19.6389 | −11.6364 | |
| Ser217Ala | −50.5624 (−59.61) | −43.0608 | −41.9079 | 51.3839 | −17.3874 | −11.6713 | ||
| Lys228Glu | −57.2781 (−59.61) | −42.2583 | −41.4879 | 43.1563 | −17.1490 | −13.2215 | ||
| HAFP– afimoxifene | C | Lys228Leu | −66.8559 (−50.07) | −42.6339 | −48.0607 | 51.2632 | −27.7603 | −15.3083 |
| Thr132 Val | −44.5842 (−50.07) | −34.4384 | −41.7174 | 45.6890 | −16.7658 | −10.2087 | ||
| Cys224Ala | −49.2394 (−61.32) | −40.4615 | −39.3824 | 44.3091 | −16.9701 | −11.2746 | ||
| Phe172Ala | −51.4201 (−61.32) | −41.2409 | −45.8675 | 49.0546 | −16.9381 | −11.7739 | ||
| HAFP– tamoxifen | C | Phe172Leu | −51.2034 (−58.74) | −32.8152 | −45.6450 | 43.1294 | −17.2336 | −11.8192 |
| Cys224Val | −41.0369 (−58.74) | −27.3946 | −36.2815 | 36.6834 | −16.0279 | −9.4725 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldogazieva, N.T.; Ostroverkhova, D.S.; Kuzmich, N.N.; Kadochnikov, V.V.; Terentiev, A.A.; Porozov, Y.B. Elucidating Binding Sites and Affinities of ERα Agonists and Antagonists to Human Alpha-Fetoprotein by In Silico Modeling and Point Mutagenesis. Int. J. Mol. Sci. 2020, 21, 893. https://doi.org/10.3390/ijms21030893
Moldogazieva NT, Ostroverkhova DS, Kuzmich NN, Kadochnikov VV, Terentiev AA, Porozov YB. Elucidating Binding Sites and Affinities of ERα Agonists and Antagonists to Human Alpha-Fetoprotein by In Silico Modeling and Point Mutagenesis. International Journal of Molecular Sciences. 2020; 21(3):893. https://doi.org/10.3390/ijms21030893
Chicago/Turabian StyleMoldogazieva, Nurbubu T., Daria S. Ostroverkhova, Nikolai N. Kuzmich, Vladimir V. Kadochnikov, Alexander A. Terentiev, and Yuri B. Porozov. 2020. "Elucidating Binding Sites and Affinities of ERα Agonists and Antagonists to Human Alpha-Fetoprotein by In Silico Modeling and Point Mutagenesis" International Journal of Molecular Sciences 21, no. 3: 893. https://doi.org/10.3390/ijms21030893