STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
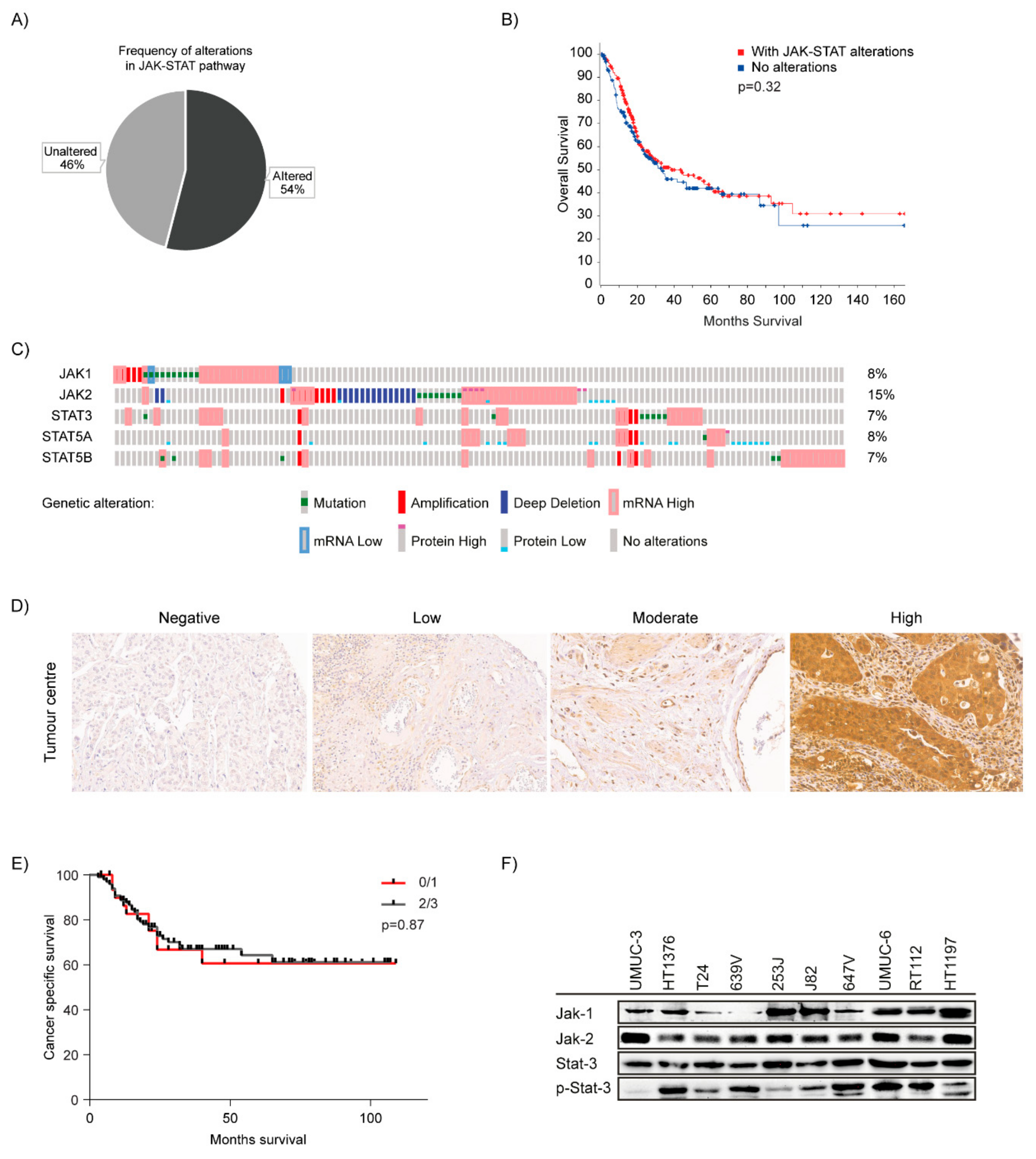
2.1. JAK-STAT Pathway is Dysregulated in Bladder Cancer
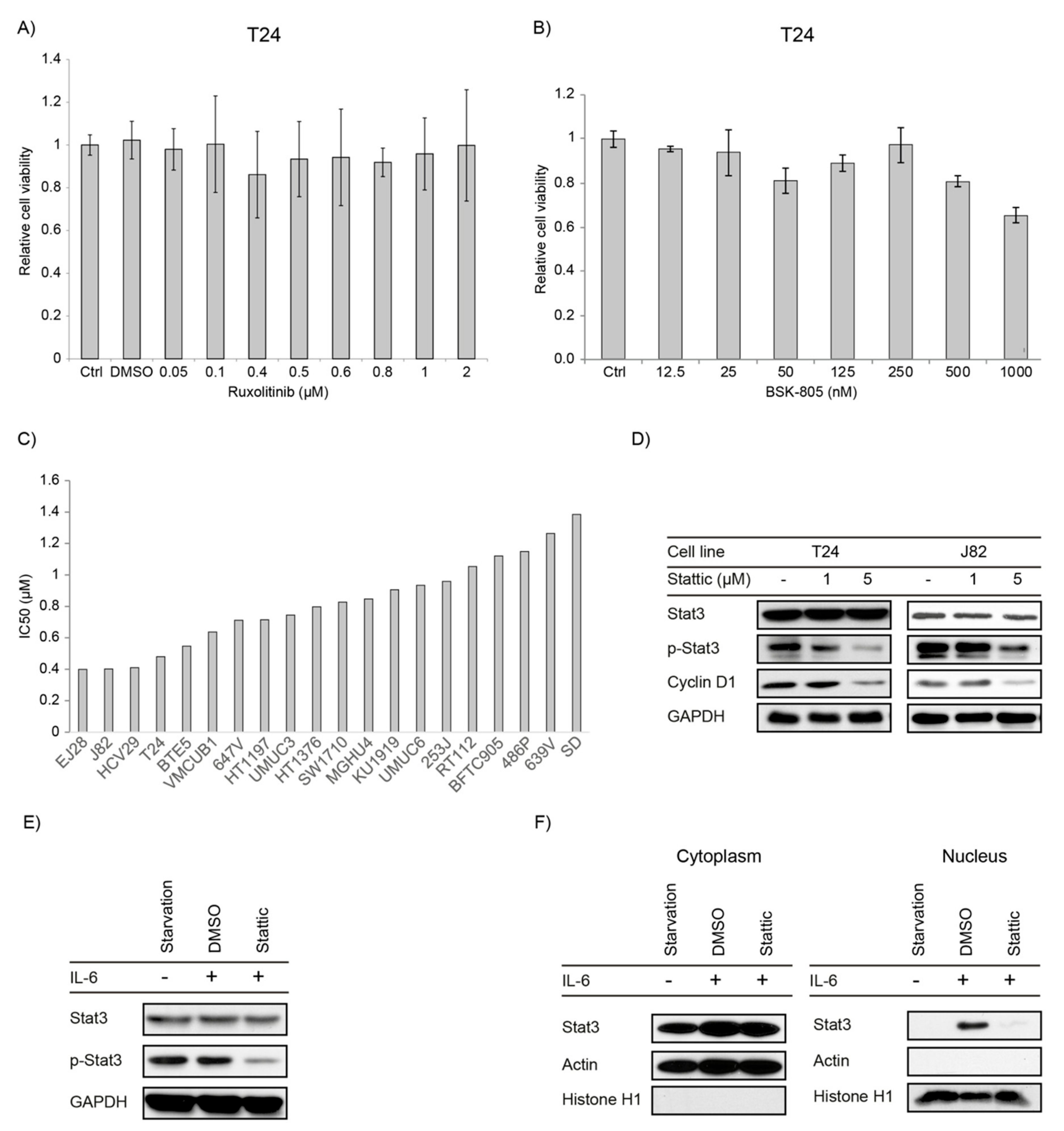
2.2. JAK1/2 Inhibitors Have No Effect on Proliferation in Bladder Cancer Cell Lines
2.3. STAT3/5 Inhibitors Reduced Proliferation and Downstream Signaling in Bladder Cancer Cell Lines
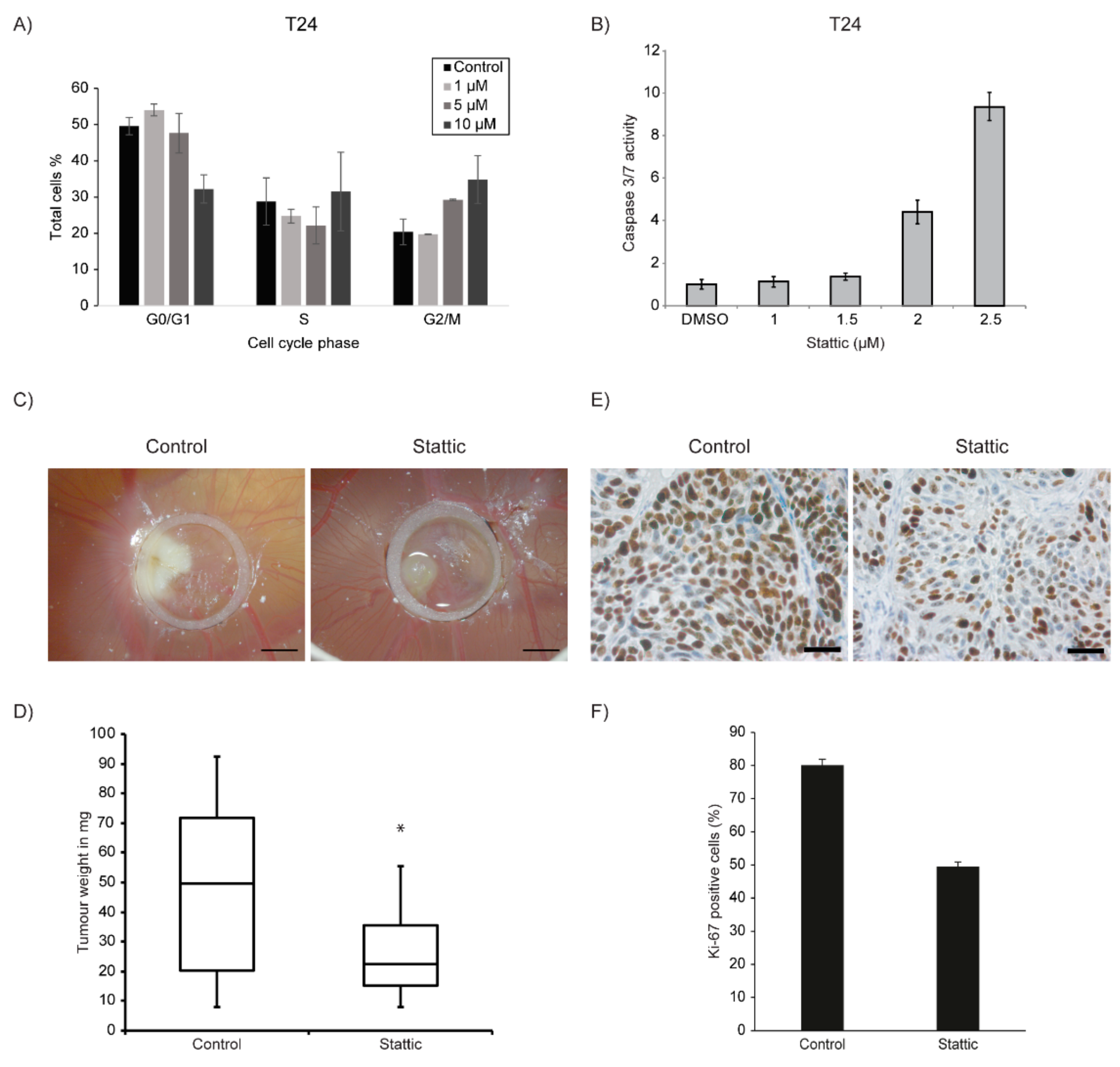
2.4. Stattic Induced G2/M Arrest and Apoptosis in Bladder Cancer Cell Lines
2.5. Stattic Reduced Tumour Growth in 3-Dimensional Xenografts
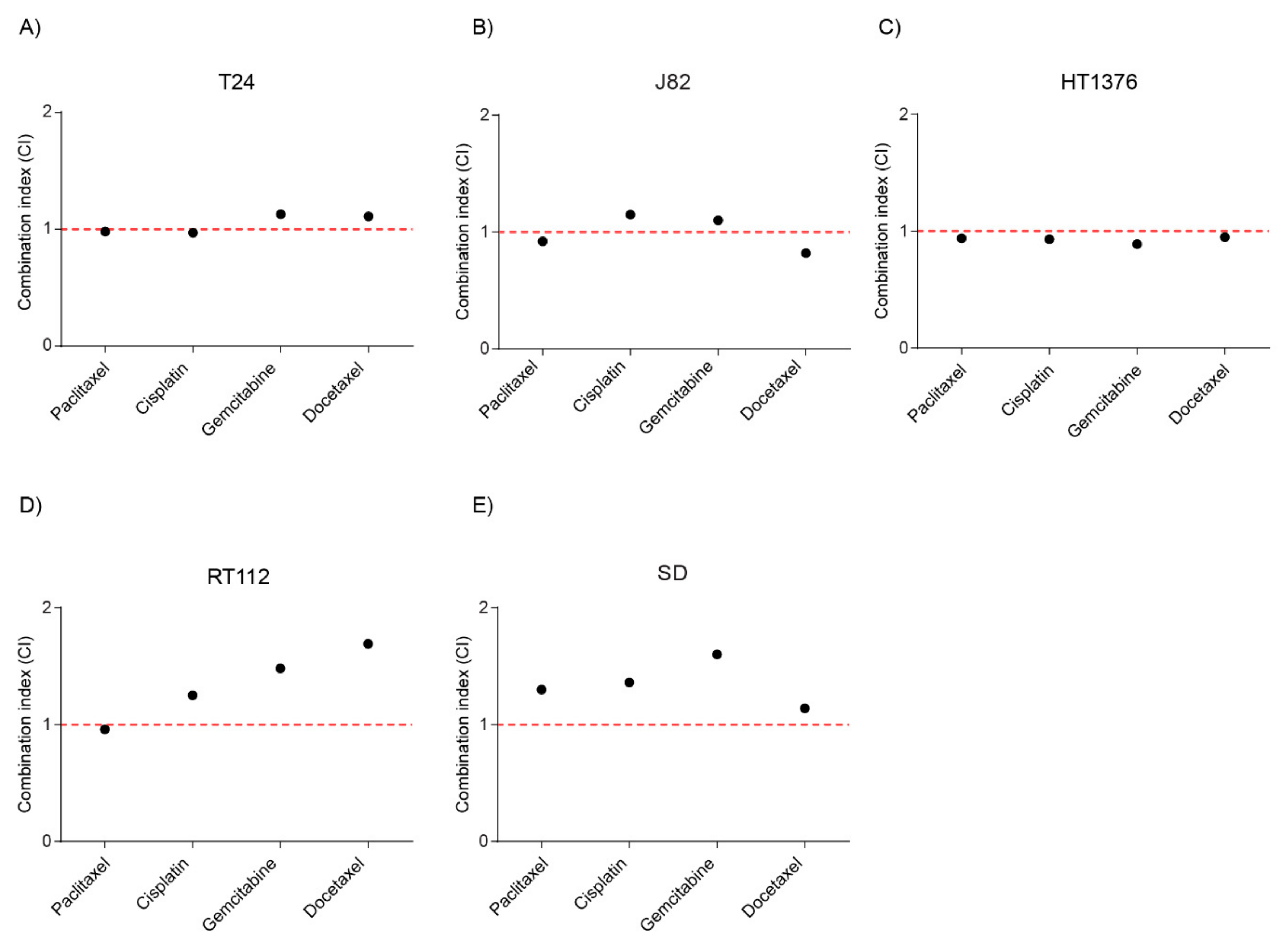
2.6. Combination of Stattic and Chemotherapeutic Agents Showed Additivity
2.7. Combination of Stattic and CDK4/6 inhibitor Palbociclib Showed Additivity
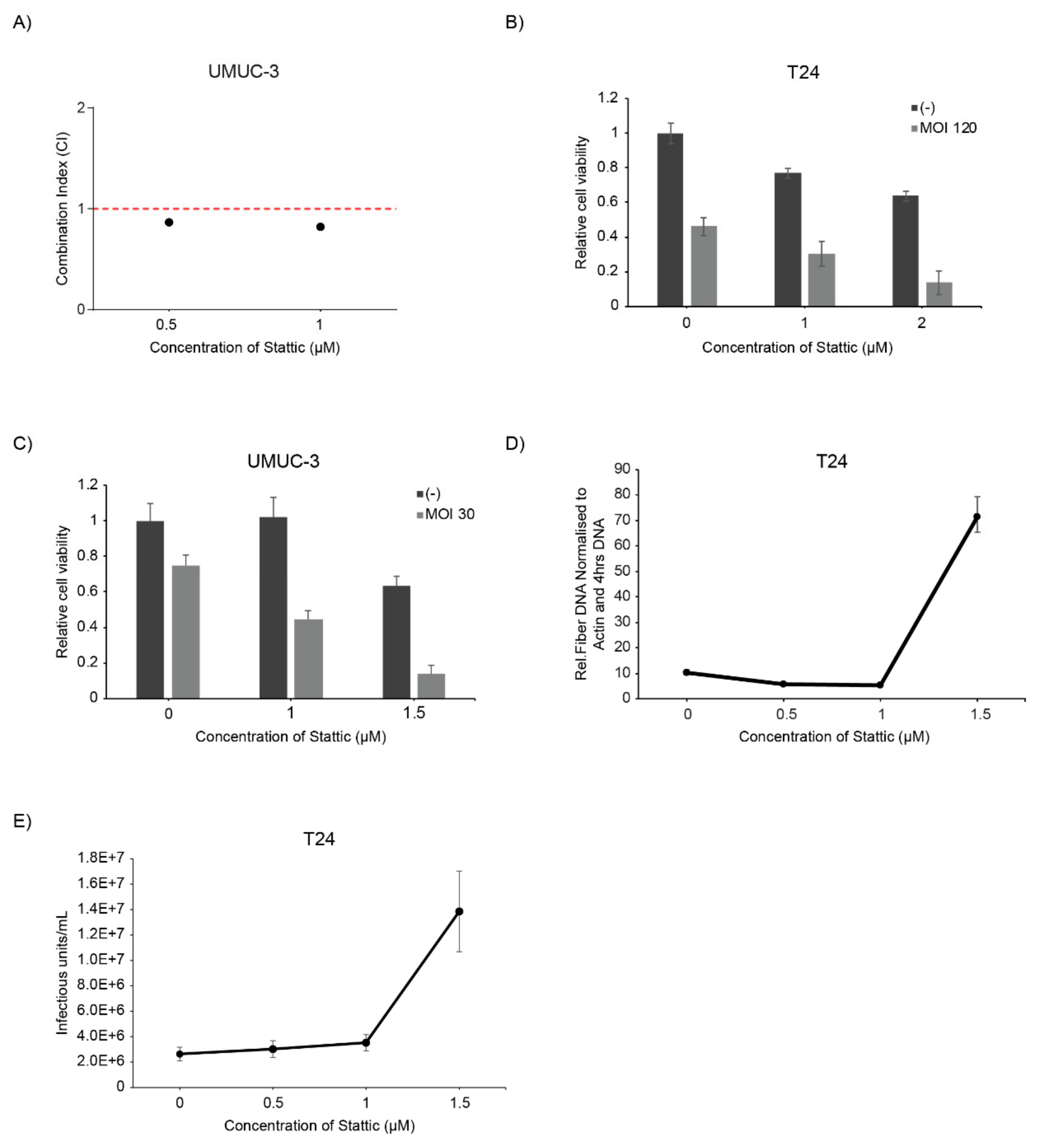
2.8. Stattic Enhanced Oncolytic Virotherapy of the Oncolytic Adenovirus XVir-N-31
3. Discussion
4. Materials and Methods
4.1. Patient Material, Tissue Microarray and Immunohistochemistry
4.2. Cell Lines and Adenovirus
4.3. Small Molecule Inhibitors and Chemotherapeutics
4.4. Cell Viability, Cell Cycle Analysis and Apoptosis Assays
4.5. Combination Index Analysis and Bladder Cancer Molecular Alteration Analysis
4.6. Immunoblot Analysis
4.7. CAM Assay and Immunohistochemistry (IHC)
4.8. Viral Replication and Particle Formation
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAM | Chorioallantoic Membrane |
| FGFR | Fibroblast Growth Factor Receptor |
| JAK | Janus Kinase |
| STAT | Signal Transducer and Activator of Transcription |
| TCGA | The Cancer Genome Atlas |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apolo, A.B. Determining superior first-line therapy in metastatic bladder cancer. Nat. Rev. Urol. 2019, 16, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Boegemann, M.; Aydin, A.M.; Bagrodia, A.; Krabbe, L.M. Prospects and progress of immunotherapy for bladder cancer. Expert Opin. Biol. Ther. 2017, 17, 1417–1431. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, V.S.; Grivas, P. Emerging Role of Immunotherapy in Advanced Urothelial Carcinoma. Curr. Oncol. Rep. 2018, 20, 48. [Google Scholar] [CrossRef]
- Duplisea, J.J.; Dinney, C.P.N. Should chemotherapy still be used to treat all muscle invasive bladder cancer in the “era of immunotherapy”? Expert Rev. Anticancer Ther. 2019, 19, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Grivas, P.; Yu, E.Y. Role of Targeted Therapies in Management of Metastatic Urothelial Cancer in the Era of Immunotherapy. Curr. Treat Options Oncol. 2019, 20, 67. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Da Costa, J.B.; Gibb, E.A.; Nykopp, T.K.; Mannas, M.; Wyatt, A.W.; Black, P.C. Molecular tumor heterogeneity in muscle invasive bladder cancer: Biomarkers, subtypes, and implications for therapy. Urol. Oncol. 2018, 1–8. [Google Scholar] [CrossRef]
- Hammaren, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Guo, C.; Yang, G.; Khun, K.; Kong, X.; Levy, D.; Lee, P.; Melamed, J. Activation of Stat3 in renal tumors. Am. J. Transl. Res. 2009, 1, 283. [Google Scholar]
- Tong, M.; Wang, J.; Jiang, N.; Pan, H.; Li, D. Correlation between p-STAT3 overexpression and prognosis in lung cancer: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, S.; Ushijima, K.; Kawano, K.; Yamaguchi, T.; Terada, A.; Fujiyoshi, N.; Nishio, S.; Tsuda, N.; Ijichi, M.; Kakuma, T.; et al. Expression of activated signal transducer and activator of transcription-3 predicts poor prognosis in cervical squamous-cell carcinoma. Br. J. Cancer 2009, 101, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Cen, L.; Kohout, J.; Hutzen, B.; Chan, C.; Hsieh, F.C.; Loy, A.; Huang, V.; Cheng, G.; Lin, J. Signal transducer and activator of transcription 3 activation is associated with bladder cancer cell growth and survival. Mol. Cancer 2008, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degoricija, M.; Situm, M.; Korac, J.; Miljkovic, A.; Matic, K.; Paradzik, M.; Marinovic Terzic, I.; Jeroncic, A.; Tomic, S.; Terzic, J. High NF-kappaB and STAT3 activity in human urothelial carcinoma: a pilot study. World J. Urol. 2014, 32, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Gu, Z.Q.; Jian, K.; Qi, J. CXCR4-mediated Stat3 activation is essential for CXCL12-induced cell invasion in bladder cancer. Tumor Biol. 2013, 34, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ou, L.; Tang, M.; Wang, Y.; Wang, X.; Chen, E.; Diao, J.; Wu, X.; Luo, C. Knockdown of PLCepsilon inhibits inflammatory cytokine release via STAT3 phosphorylation in human bladder cancer cells. Tumor Biol. 2015, 36, 9723–9732. [Google Scholar] [CrossRef]
- Gatta, L.B.; Melocchi, L.; Bugatti, M.; Missale, F.; Lonardi, S.; Zanetti, B.; Cristinelli, L.; Belotti, S.; Simeone, C.; Ronca, R.; et al. Hyper-Activation of STAT3 Sustains Progression of Non-Papillary Basal-Type Bladder Cancer via FOSL1 Regulome. Cancers 2019, 11, 1219. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.L.; Lay, E.J.; Jian, W.; Parra, D.; Chan, K.S. Stat3 activation in urothelial stem cells leads to direct progression to invasive bladder cancer. Cancer Res. 2012, 72, 3135–3142. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.P.; Pagliarulo, V.; Yang, D.; Waldman, F.M.; Datar, R.H.; Skinner, D.G.; Groshen, S.; Cote, R.J. Generation of a concise gene panel for outcome prediction in urinary bladder cancer. J. Clin. Oncol. 2009, 27, 3929–3937. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cheng, M.K.; RL Griffiths, T.; Kilian Mellon, J.; Kai, B.; Kriajevska, M.; M Manson, M. Inhibition of STAT Signalling in Bladder Cancer by Diindolylmethane-Relevance to Cell Adhesion, Migration and Proliferation. Curr. Cancer Drug Targets 2013, 13, 57–68. [Google Scholar] [CrossRef]
- Vlahou, A.; Gromova, I.; Svensson, S.; Gromov, P.; Moreira, J.M. Identification of BLCAP as a novel STAT3 interaction partner in bladder cancer. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kontzias, A.; Kotlyar, A.; Laurence, A.; Changelian, P.; O’Shea, J.J. Jakinibs: A new class of kinase inhibitors in cancer and autoimmune disease. Curr. Opin. Pharmacol. 2012, 12, 464–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef]
- Zhao, C.; Li, H.; Lin, H.J.; Yang, S.; Lin, J.; Liang, G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol. Sci. 2016, 37, 47–61. [Google Scholar] [CrossRef]
- Shao, D.; Ma, J.; Zhou, C.; Zhao, J.N.; Li, L.L.; Zhao, T.J.; Ai, X.L.; Jiao, P. STAT3 down-regulation induces mitochondria-dependent G2/M cell cycle arrest and apoptosis in oesophageal carcinoma cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 413–420. [Google Scholar] [CrossRef]
- Zhou, C.; Ma, J.; Su, M.; Shao, D.; Zhao, J.; Zhao, T.; Song, Z.; Meng, Y.; Jiao, P. Down-regulation of STAT3 induces the apoptosis and G1 cell cycle arrest in esophageal carcinoma ECA109 cells. Cancer Cell Int. 2018, 18, 53. [Google Scholar] [CrossRef] [Green Version]
- Sathe, A.; Koshy, N.; Schmid, S.C.; Thalgott, M.; Schwarzenbock, S.M.; Krause, B.J.; Holm, P.S.; Gschwend, J.E.; Retz, M.; Nawroth, R. CDK4/6 Inhibition Controls Proliferation of Bladder Cancer and Transcription of RB1. J. Urol. 2016, 195, 771–779. [Google Scholar] [CrossRef]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Sathe, A.; Ebner, B.; Qi, P.; Veltkamp, C.; Gschwend, J.E.; Holm, P.S.; Nawroth, R. Functional genomics identifies predictive markers and clinically actionable resistance mechanisms to CDK4/6 inhibition in bladder cancer. J. Exp. Clin. Cancer Res. 2019, 38, 322. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Karube, H.; Aruga, A. A Comparative Safety Profile Assessment of Oncolytic Virus Therapy Based on Clinical Trials. Ther. Innov. Regul. Sci. 2018, 52, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, E.; Koll, F.; Haas, H.; Mantwill, K.; Janssen, K.P.; Laschinger, M.; Gschwend, J.; Steiger, K.; Black, P.C.; Moskalev, I.; et al. The Oncolytic Adenovirus XVir-N-31 as a Novel Therapy in Muscle-Invasive Bladder Cancer. Hum. Gene Ther. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Taguchi, S.; Fukuhara, H.; Homma, Y.; Todo, T. Current status of clinical trials assessing oncolytic virus therapy for urological cancers. Int. J. Urol. 2017, 24, 342–351. [Google Scholar] [CrossRef] [Green Version]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gomez-Manzano, C.; Rivera-Molina, Y.; Lang, F.F.; Conrad, C.A.; Fueyo, J. Oncolytic adenovirus research evolution: from cell-cycle checkpoints to immune checkpoints. Curr. Opin. Virol. 2015, 13, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.R.; Dash, A.; Jacobson, B.A.; Ji, Y.; Baumann, D.; Ismail, K.; Kratzke, R.A. JAK/STAT inhibition with ruxolitinib enhances oncolytic virotherapy in non-small cell lung cancer models. Cancer Gene Ther. 2019, 26, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Cassady, K.A. Combination Therapy Using Ruxolitinib and Oncolytic HSV Renders Resistant MPNSTs Susceptible to Virotherapy. Cancer Immunol. Res. 2018, 6, 1499–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.T.; Yang, S.F.; Wu, C.C.; Chen, W.T.; Huang, Y.C.; Su, Y.C.; Chai, C.Y. Expression of signal transducer and activator of transcription 3 and suppressor of cytokine signaling in urothelial carcinoma. Kaohsiung J. Med. Sci. 2009, 25, 640–646. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J., Jr.; Chang, H.Y.; van de Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556. [Google Scholar] [CrossRef]
- Horn, T.; Laus, J.; Seitz, A.K.; Maurer, T.; Schmid, S.C.; Wolf, P.; Haller, B.; Winkler, M.; Retz, M.; Nawroth, R.; et al. The prognostic effect of tumour-infiltrating lymphocytic subpopulations in bladder cancer. World J. Urol. 2016, 34, 181–187. [Google Scholar] [CrossRef]
- Horiguchi, A.; Oya, M.; Shimada, T.; Uchida, A.; Marumo, K.; Murai, M. Activation of signal transducer and activator of transcription 3 in renal cell carcinoma—A study of incidence and its association with pathological features and clinical outcome. J. Urol. 2002, 168, 762–765. [Google Scholar] [CrossRef]
- Igelmann, S.; Neubauer, H.A.; Ferbeyre, G. STAT3 and STAT5 Activation in Solid Cancers. Cancers 2019, 11, 1428. [Google Scholar] [CrossRef] [Green Version]
- Fukada, T.; Ohtani, T.; Yoshida, Y.; Shirogane, T.; Nishida, K.; Nakajima, K.; Hibi, M.; Hirano, T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998, 17, 6670–6677. [Google Scholar] [CrossRef] [Green Version]
- Skowron, M.A.; Sathe, A.; Romano, A.; Hoffmann, M.J.; Schulz, W.A.; van Koeveringe, G.A.; Albers, P.; Nawroth, R.; Niegisch, G. Applying the chicken embryo chorioallantoic membrane assay to study treatment approaches in urothelial carcinoma. Urol. Oncol. 2017, 35, 544.e11–544.e23. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, Y.H.; Na, Y.M.; Yoo, Y.B.; Darvin, P.; Sp, N.; Kang, D.Y.; Kim, S.Y.; Kim, H.S.; Choi, Y.H.; Lee, H.K.; et al. Combination of AG490, a Jak2 inhibitor, and methylsulfonylmethane synergistically suppresses bladder tumor growth via the Jak2/STAT3 pathway. Int. J. Oncol. 2014, 44, 883–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintás-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Baffert, F.; Régnier, C.H.; De Pover, A.; Pissot-Soldermann, C.; Tavares, G.A.; Blasco, F.; Brueggen, J.; Chène, P.; Drueckes, P.; Erdmann, D.; et al. Potent and Selective Inhibition of Polycythemia by the Quinoxaline JAK2 Inhibitor NVP-BSK805. Mol. Cancer Ther. 2010, 9, 1945–1955. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haftchenary, S.; Luchman, H.A.; Jouk, A.O.; Veloso, A.J.; Page, B.D.; Cheng, X.R.; Dawson, S.S.; Grinshtein, N.; Shahani, V.M.; Kerman, K.; et al. Potent Targeting of the STAT3 Protein in Brain Cancer Stem Cells: A Promising Route for Treating Glioblastoma. ACS Med. Chem. Lett. 2013, 4, 1102–1107. [Google Scholar] [CrossRef] [Green Version]
- Nelson, E.A.; Walker, S.R.; Kepich, A.; Gashin, L.B.; Hideshima, T.; Ikeda, H.; Chauhan, D.; Anderson, K.C.; Frank, D.A. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 2008, 112, 5095–5102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ye, Y.L.; Li, M.X.; Ye, S.B.; Huang, W.R.; Cai, T.T.; He, J.; Peng, J.Y.; Duan, T.H.; Cui, J.; et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2016, 36, 2095–2104. [Google Scholar] [CrossRef]
- Tsujita, Y.; Horiguchi, A.; Tasaki, S.; Isono, M.; Asano, T.; Ito, K.; Asano, T.; Mayumi, Y.; Kushibiki, T. STAT3 inhibition by WP1066 suppresses the growth and invasiveness of bladder cancer cells. Oncol. Rep. 2017, 38, 2197–2204. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Ren, Y.; Liu, A.; Jin, R.; Jiang, Q.; Huang, Y.; Kong, L.; Wang, X.; Zhang, L. WP1066 sensitizes oral squamous cell carcinoma cells to cisplatin by targeting STAT3/miR-21 axis. Sci. Rep. 2014, 4, 7461. [Google Scholar] [CrossRef] [Green Version]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, J.S.; Qi, J.; Flint, S.J. The human adenovirus type 5 E1B 55 kDa protein obstructs inhibition of viral replication by type I interferon in normal human cells. PLoS Pathog. 2012, 8, e1002853. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Stamminger, T.; Hearing, P. E2F/Rb Family Proteins Mediate Interferon Induced Repression of Adenovirus Immediate Early Transcription to Promote Persistent Viral Infection. PLoS Pathog. 2016, 12, e1005415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, K.; Lee, S.R.; Ying, B.; Spencer, J.F.; Tollefson, A.E.; Sagartz, J.E.; Kong, I.K.; Wang, Z.; Wold, W.S. STAT2 Knockout Syrian Hamsters Support Enhanced Replication and Pathogenicity of Human Adenovirus, Revealing an Important Role of Type I Interferon Response in Viral Control. PLoS Pathog. 2015, 11, e1005084. [Google Scholar] [CrossRef] [Green Version]
- Steinwaerder, D.S.; Carlson, C.A.; Lieber, A. DNA Replication of First-Generation Adenovirus Vectors in Tumor Cells. Hum. Gene Ther. 2000, 11, 1933–1948. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hindupur, S.V.; Schmid, S.C.; Koch, J.A.; Youssef, A.; Baur, E.-M.; Wang, D.; Horn, T.; Slotta-Huspenina, J.; Gschwend, J.E.; Holm, P.S.; et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 1106. https://doi.org/10.3390/ijms21031106
Hindupur SV, Schmid SC, Koch JA, Youssef A, Baur E-M, Wang D, Horn T, Slotta-Huspenina J, Gschwend JE, Holm PS, et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. International Journal of Molecular Sciences. 2020; 21(3):1106. https://doi.org/10.3390/ijms21031106
Chicago/Turabian StyleHindupur, Sruthi V., Sebastian C. Schmid, Jana Annika Koch, Ahmed Youssef, Eva-Maria Baur, Dongbiao Wang, Thomas Horn, Julia Slotta-Huspenina, Juergen E. Gschwend, Per Sonne Holm, and et al. 2020. "STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy" International Journal of Molecular Sciences 21, no. 3: 1106. https://doi.org/10.3390/ijms21031106