P53: A Guardian of Immunity Becomes Its Saboteur through Mutation
by
 , and
, and
Arjelle Decasa Agupitan
1, 
Paul Neeson
2,3,
Scott Williams
4,
Jason Howitt
5,6,
Sue Haupt
1,2 and
Ygal Haupt
1,2,7,8,* 1
Tumour Suppression Laboratory, Peter MacCallum Cancer Centre, 305 Grattan St, Melbourne 3000, Victoria, Australia
2
Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville 3010, Victoria, Australia
3
Cancer Immunology Research, Peter MacCallum Cancer Centre, Melbourne 3000, Victoria, Australia
4
Division of Radiation Oncology and Cancer Imaging, Peter MacCallum Cancer Centre, Melbourne 3000, Victoria, Australia
5
School of Health Sciences, Swinburne University, Melbourne 3122, Victoria, Australia
6
Florey Institute of Neuroscience and Mental Health, University of Melbourne, Parkville 3010, Victoria, Australia
7
Department of Clinical Pathology, University of Melbourne, Parkville 3010, Victoria, Australia
8
Department of Biochemistry and Molecular Biology, Monash University, Melbourne 3800, Victoria, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(10), 3452; https://doi.org/10.3390/ijms21103452
Submission received: 27 April 2020
/
Revised: 6 May 2020
/
Accepted: 11 May 2020
/
Published: 13 May 2020
(This article belongs to the Special Issue p53 in Cancer and beyond—40 Years after Its Discovery)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Awareness of the importance of immunity in controlling cancer development triggered research into the impact of its key oncogenic drivers on the immune response, as well as their value as targets for immunotherapy. At the heart of tumour suppression is p53, which was discovered in the context of viral infection and now emerges as a significant player in normal and cancer immunity. Wild-type p53 (wt p53) plays fundamental roles in cancer immunity and inflammation. Mutations in p53 not only cripple wt p53 immune functions but also sinisterly subvert the immune function through its neomorphic gain-of-functions (GOFs). The prevalence of mutant p53 across different types of human cancers, which are associated with inflammatory and immune dysfunction, further implicates mutant p53 in modulating cancer immunity, thereby promoting tumorigenesis, metastasis and invasion. In this review, we discuss several mutant p53 immune GOFs in the context of the established roles of wt p53 in regulating and responding to tumour-associated inflammation, and regulating innate and adaptive immunity. We discuss the capacity of mutant p53 to alter the tumour milieu to support immune dysfunction, modulate toll-like receptor (TLR) signalling pathways to disrupt innate immunity and subvert cell-mediated immunity in favour of immune privilege and survival. Furthermore, we expose the potential and challenges associated with mutant p53 as a cancer immunotherapy target and underscore existing therapies that may benefit from inquiry into cancer p53 status.
1. An Immunological Precedent for p53 Function
An interest in the involvement of p53 in cancer immunology is emerging. Beyond its key role in maintaining genomic integrity [1,2], extrinsic (non-cell) autonomous p53 functions affect the surrounding tumour microenvironment (TME) through the induction of senescence, inflammation and immunomodulatory effects [3,4,5]. While overshadowed by the role of p53 in tumour suppression, local immune regulation has long been linked to p53 dysfunction, with viral infections such as Simian virus 40 (SV40), where Large T antigen complexes with and inactivates p53, or Human Papilloma Virus (HPV), where the viral E6 proteins mediates p53 proteasomal destruction [6,7,8], showing reduced immune responses. Furthermore, the impact of mutant p53 on the immune response has been inferred from studies focusing on wild-type p53 (wt p53) functions, rather than being studied directly.
Recent studies have identified several p53 target genes and regulators that play fundamental functions in immune signalling pathways that coordinate the response to cytokine production and inflammatory response, and in innate and cell-mediated immunity. In this review, we discuss the recent finding on the role of mutant p53 in cancer-related inflammation and immunity. Moreover, we highlight the relevance of mutant p53 not only in disabling the inherent wt p53-mediated tumour immunosurveillance, but also in enhancing tumour-associated immune dysfunction as a means to promote tumorigenesis, metastasis, and invasion.
1.1. p53 Influences the Immune and Inflammatory Tumour Microenvironment
A fine homeostatic balance exists between the role of p53 as a tumour suppressor and its impact on immune regulation. This is exemplified by the intrinsic role of wt p53 in facilitating immunity, countered by its contribution to chronic inflammation in cancer development [9,10]. In this section, we discuss how mutant p53 impinges on components of the TME to shape a pro-tumorigenic immune landscape.
Molecular and cellular components of emerging tumours may be hostile to their microenvironment and create a tumouricidal inflammatory niche. With cancer progression, the TME can develop into an immune-suppressive nest (Figure 1). Specifically, stromal cells (blood, cancer-associated fibroblasts, vascular and lymphatic endothelial cells) and infiltrating immune cells (lymphocytes and myeloid cells) cooperate to determine the functional outcome of the TME. An inherent back-and-forth of signalling between the developing tumour and its environment can ultimately maintain and promote an inflammatory, yet immunosuppressive, environment for the tumour, in response to the aberrant secretion of signalling molecules [11,12]. The collective tumour microenvironment is thus one that hijacks inflammatory signals to remodel its surroundings, recruits tumour-associated immune cells, expresses pro-tumorigenic chemokines and cytokines, and promotes neo-angiogenesis [13,14]. Cancer is able to counter normal immune functions that are deleterious to its progression by disrupting common immune effector cell function through a reduction in FAS (Fas cell surface death, CD95) receptor surface expression, and increased expression of FAS-mediated apoptosis inhibitors [15], or by disrupting cytotoxic immune signalling [16]. Moreover, immunotolerant and immune-suppressive signals, such as the checkpoint molecule PD-L1, are utilized, or immunosuppressive cell infiltrate is recruited [17,18,19]. Cellular senescence is induced in stressed and damaged cells as a strategy to maintain tissue and cellular integrity, and it can impact on tumour development. Senescence can trigger several changes in chemokine and cytokine signalling, which are collectively referred to as senescence-associated secretory phenotype (SASP). While SASP serves initially to curb pre-malignant lesions to promote damage repair and cell clearance, the prolonged secretion of inflammatory factors can foster tumour-friendly TME development and angiogenesis [20]. These subversions of the normal immune response promote cancer to flourish in the host environment.
1.2. The Heated Interplay between p53 and Inflammation
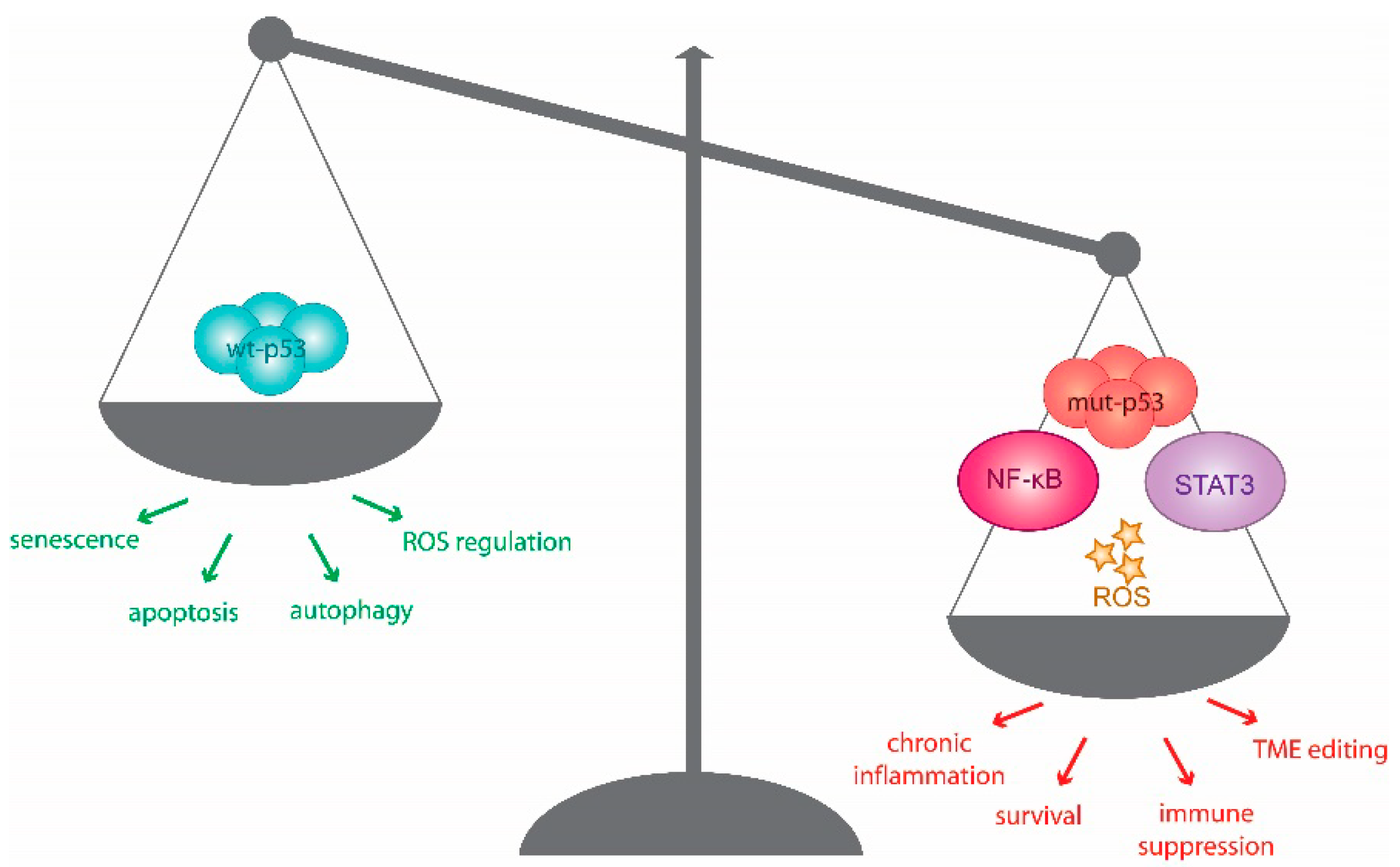
Chronic inflammation stokes environmental and genetic stress, which fuels tumorigenesis [21]. Cancer development frequently coincides with a shift from acute inflammatory tumouricidal mediators, including IL-12 and IFN-γ, to those that are chronically immunosuppressive, such as IL-8, IL-17, IL-23, and IL-13. This modification of the cytokine landscape requires the activation of NF-κB and Signal-Transducer and -Activator of Transcription (STAT) pathways, not only in cancer cells, but also among the TME components. The resulting environment is rich in reactive oxygen species (ROS) generated from increased receptor signalling, metabolic activity, mitochondrial dysfunction and infiltrating immune cells, which ultimately prompts chronic stress and genotoxic signalling [22]. The SASP, which also contributes to and fuels such shifts in inflammatory signalling, is also regulated by p53, its isoforms, and protein family members, and co-opts several of the aforementioned inflammatory triggers [23,24]. The effects of SASP on cancer is mediated primarily through the inflammatory phenotype and the consequent changes in cellular signalling it produces, which have been extensively reviewed elsewhere [25,26]. Below, we discuss studies that suggest that mutant p53 may take centre stage and orchestrate this immunological shift to promote a tumour-promoting TME (Figure 1).
1.2.1. The Inflammatory Crosstalk between NF-κB and p53
The NF-κB and wt p53 pathways are often considered antagonistic transcriptional networks. While the canonical role of wt p53 is growth restrictive in nature, that of NF-κB promotes cell survival and inflammation. The downstream activity of both pathways may reciprocally regulate the expression of the other. Specifically, constitutive activation of NF-κB is commonly observed in chronically inflamed and malignant tissues with the capacity to repress normal apoptotic and senescence-inducing p53 activity [27,28,29,30]. If wt p53 function is lost, aberrant inflammation can enhance tumour development [31,32]. Consistent with this, wt p53 can directly suppress the transcriptional activity of NF-κB [33,34].
Intriguingly, NF-κB and wt p53 activities can also converge to promote common outcomes. NF-κB can participate in wt p53-mediated apoptosis [35] and cooperate with wt p53 to promote senescence in IMR-90 cells but not human BJ fibroblasts [36]. NF-κB is also activated downstream of p53-inducible death-domain-containing protein in response to DNA damage [37]. Pertinently, NF-κB is essential for wt p53-dependent regulation of several pro-inflammatory genes in macrophages and monocytes to amplify responses to damage signals in tissues and inflammatory environments [38].
The balance of inflammatory signalling between p53 and NF-κB signalling is maintained by the regulatory, yet cooperative, push-and-pull from both transcriptional networks. On the other hand, mutant p53 introduces a kink in this regulatory axis. Loss-of-function p53 mutations disable the measured molecular responses in this reciprocal relationship, while gain-of-function (GOF) p53 mutations can augment pro-inflammatory and survival activities of NF-κB target genes [39,40,41]. A study by Cooks et al. demonstrated that mutant p53, in cooperation with tumour necrosis factor-α (TNFα), prolongs NF-κB activation and results in a chronic-inflammatory phenotype and development of colon carcinoma in mutant p53 mouse models [39]. These observations echo the correlation of accumulated missense p53 mutants and NF-κB activation in human colitis-associated cancer [39]. Di Minin et al. uncovered a novel cytoplasmic mutant p53 GOF in human breast cancer cells, and RAS-transformed mouse embryonic fibroblasts (MEFs), capable of altering the TNF-dependent activation of NF-κB and (c-Jun N-terminal kinase) JNK pathways through the inhibition of RasGAP Disabled 2 Interacting Protein (DAB2IP). This GOF demonstrated in R280K p53 mutant, and is predicted to occur in other hotspot mutants, which are nuclear-excluded, and hence can shift TNF-induced transcription toward products that promote cell migration and lymphocyte recruitment, thereby enabling an invasive phenotype that is resistant to TNF-induced apoptosis [40]. Mutant p53 may also interact directly with NF-κB, influencing enhancer binding in response to chronic TNFα-signalling in colon carcinomas. The simultaneous binding of NF-κB and of the R273H p53 mutant and other detected mutant forms of p53 in colorectal carcinomas regulates RNA polymerase II recruitment to these enhancers and facilitates mRNA synthesis and the activation of tumour-promoting genes like MMP9 and CCL2 [41]. Mutant p53 in synergy with NF-κB can thus shape the inflammatory TME, coercing both epithelial and non-epithelial cells to favour cancer-promoting gene expression [4,38,39].
Consequently, opposing the pro-tumorigenic arms of the NF-κB-p53 axis is an appealing target for cancer therapy [42]. Indeed, NF-κB inhibition to restore wt p53 function is a rational approach that has previously been demonstrated using derivatives of 9-aminoacridine in renal cell carcinomas [43], and small molecule curaxins in several cancer cell lines and mouse tumour xenografts [44]. In a mutant p53 context, wt p53 reactivation strategies could thus supplement current NF-κB-dependent treatments [45,46].
1.2.2. The Reciprocal Relationship of STAT and p53 in Response to Inflammatory Signalling
STAT pathways transcriptionally regulate biological responses to cytokines, chemokines and growth factor signals alongside NF-κB [47]. Like NF-κB, STAT3 is often constitutively activated in malignant tumour cells and immune cells. In fact, STAT3 interacts with NF-κB in context-dependent manners to promote several cancer hallmark characteristics including: the inhibition of cell death, increased proliferation, survival, and inflammation [48]. STAT3, and, in some cases, STAT5 and STAT6, affect the TME by promoting immunosuppressive TMEs and inhibiting anti-tumour immunity [49,50].
Pertinently, STATs can channel the inflammatory TME to impinge upon p53 activity. Like NF-κB, STAT3 impedes p53 expression, limiting its canonical tumour suppressive function [51,52,53]. In contrast, alternative phosphorylated forms of STAT3 can upregulate p53 expression through promoter binding [54]. In a manner suggestive of a feed-back loop, wt p53 is able to reduce tyrosine phosphorylation, and thus prevent STAT3 DNA-binding activity, as demonstrated in breast [55] and prostate cancer cells [56]. This reciprocal negative regulation of the phosphorylated forms of STAT3 does not occur when p53 is mutated. Indeed, the capacity of phosphorylated or alternatively spliced STAT3 to promote p53 expression may be an anticipated cancer risk when p53 is mutated. Therefore, constitutive activation of STAT3 may be selectively present in cancer cells that harbour inactivating mutation or deletion of the p53 gene, which may enable cancer cells to escape inhibition by wt p53 pathway, particularly after DNA damage. This hypothesis is supported by the status of STAT3 and p53 in prostate (DU145 and Tsu), breast (MDA-MB-468 and SK-BR-3) and ovarian (MDAH 2774, SKOV-3 and Caov-3) cancer cell lines, which express constitutively active STAT3 and either express mutant p53 or are p53 null [56]. A recent study has also shown that the R248Q p53 mutant mediates hyperactive STAT3/Jak signalling, and ablation of this mutant is sufficient to inhibit growth and invasion of colorectal cancer cell lines [57]. Although unexplored, this study likely demonstrates the ability of mutant p53 to exert novel GOFs in cancer through the differential regulation of the STAT3 pathway in inflammatory microenvironments.
1.2.3. ROS Fuels the Pro-Tumourigenic Activity of Mutant p53 in Inflammatory Environments
DNA damage-induced ROS stimulates several immune pathways, including the NF-κB and STAT pathways. Wt p53 and ROS dynamically engage in maintaining the balance of these pathways, with wt p53 monitoring and maintaining ROS at permissible homeostatic threshold levels. If exceeded, as frequently occur in chronic inflammation, elevated levels of stress-associated ROS trigger apoptotic machinery [58,59,60,61,62].
Cox-2 is induced by pro-inflammatory cytokine signatures and ROS accumulation and is overexpressed in several cancers, modulating cancer cell proliferation and apoptosis [63,64]. In response to ROS activation, Cox-2 is upregulated by and interacts with p53. The consequent interaction interferes with p53 transcription and hinders stress-induced apoptosis, while augmenting cell proliferation and hepatocyte-like stem cell differentiation [65,66,67]. Specific mutations in p53 are associated with Cox-2 overexpression and in response to ROS, favour pro-tumorigenic environmental stimuli [68]. Thus, contexts of chronic inflammatory ROS, Cox-2 inhibitors like NS-398 together with an activator of p53-dependent apoptosis, doxorubicin, present an attractive therapeutic combination, as demonstrated in wt p53 expressing normal human cells [66]. In the absence of inflammatory ROS stress, Cox-2 may positively regulate wt p53 levels. The accumulation of wt p53 in response to doxorubicin or etoposide is lower in Cox-2 knockout MEFs due to the inhibition of ROS-mediated JNK activation [69]. Thus, Cox-2 inhibitors can antagonize the cytotoxicity of therapeutic agents, as exemplified by the suppression of doxorubicin-induced cytotoxicity and p53 accumulation by NS-398 in U2OS and MCF-7 cell lines. Moreover, it is specifically the Cox-2 and ROS-associated accumulation of wt p53 that is attenuated, while the accumulation of mutant p53 in HT-29 and MDA-MB-231 cells persists when NS-398 is used with doxorubicin [69].
Mutant p53 may also impinge on endogenous antioxidant systems by differentially regulating components of the NRF2 transcriptional program responsible for the expression of antioxidant response element-dependent genes [70]. The overall effect of this interaction promotes a pro-survival ROS response in a highly inflammatory tumour environment [71,72]. By and large, the integrity of the p53 pathway is important in managing oxidative stress associated with chronic inflammatory TMEs.
1.3. Mutant p53 Supports and Alters Components of the Tumour Milieu
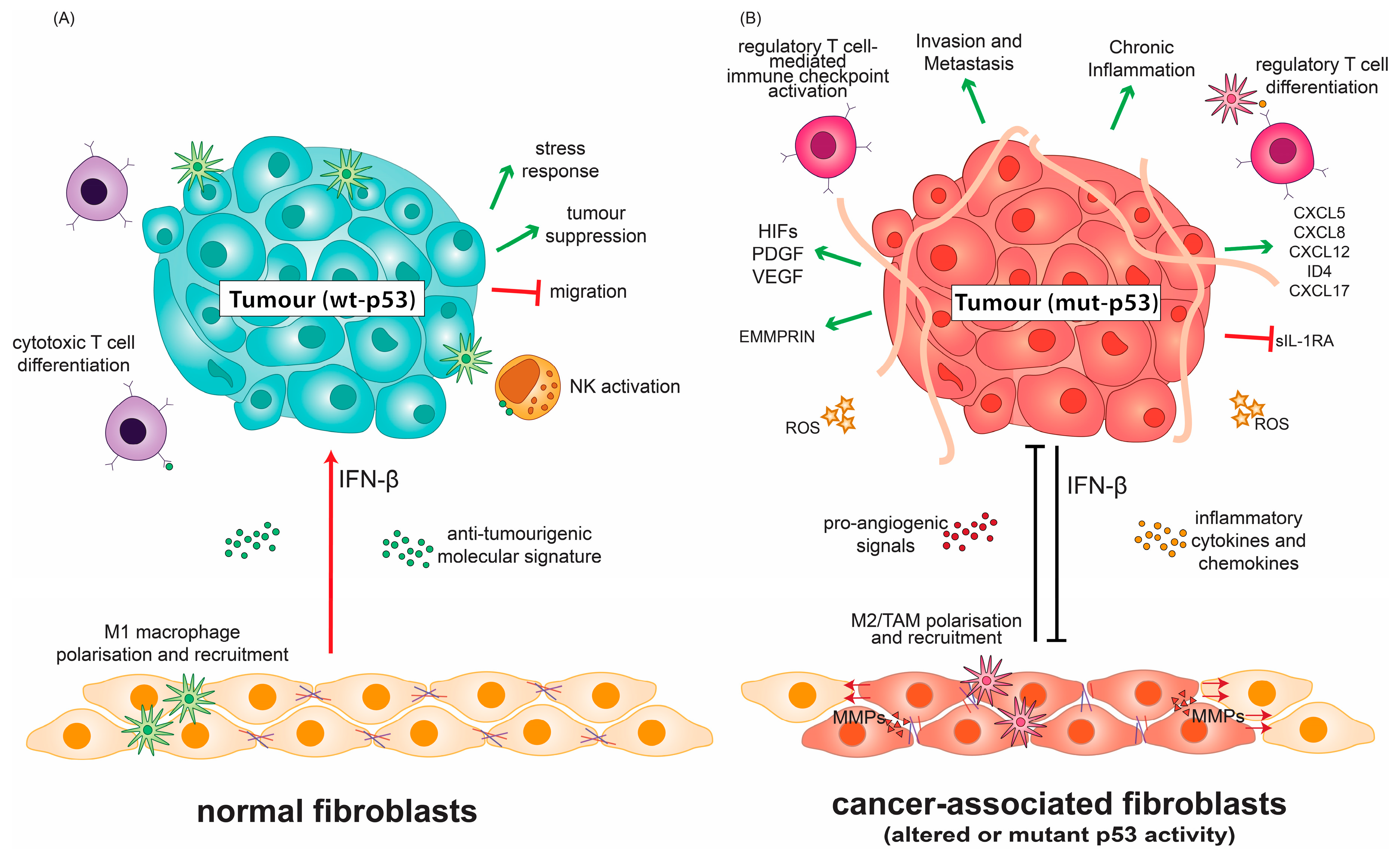
In the contexts discussed thus far, mutant p53 ultimately harnesses growth factor, chemokine and cytokine production as an instrument of pro-inflammatory, yet immunosuppressive molecular signalling. Mutant p53 has been reported to induce CXCL5, CXCL8, and CXCL12, which are pro-angiogenic and pro-invasive chemokines, which are implicated in cancer and several inflammatory diseases [73,74]. Mutant p53 (R175H, R273H, R280K) also upregulates ID4, a post-transcriptional regulator of several pro-angiogenic and tumour-supporting cytokines [75]. The mutant p53-mediated suppression of anti-inflammatory signals can also promote the tumour’s pro-inflammatory effects. For example, the suppression of the anti-inflammatory cytokine sIL-1RA by mutant p53 results in chronic inflammation associated with tumour progression [76]. Overall, mutant p53 exposed to inflammatory TMEs can form an inflammatory feed-forward loop that affects not only its encompassing cancer cell population and those surrounding it, but also the tumour stroma, extracellular matrix (ECM) and associated immune cells infiltrate (Figure 2) [5,77].
1.3.1. Cancer-Associated Fibroblasts Work with Altered and Mutant p53 to Promote Inflammation and Tumorigenesis
A complex role for p53 in cancer immunity and inflammation is emerging from studies of its alteration in tumour stroma. Cancer-associated fibroblasts (CAFs) are an integral part of the TME and are heavily involved in receiving and conveying signals for inflammation and leukocyte recruitment [78,79,80,81]. CAFs in contact with cancer cells can undergo the activation of their IFN-β pathway, which cooperates with wt p53 in fibroblasts to suppress tumour growth, respond to stress and prevent cancer cell migration (Figure 2A) [82,83].
Altered p53 status in CAFs consequently affects the tumour inflammatory milieu. Cancer cells are able to suppress wt p53 activity, or rewire intact p53 pathways to mutant p53-like cancer-promoting states in associated stroma [84,85,86,87]. The resulting p53 dysfunction in CAFs alters the molecular crosstalk between the tumour stroma and cancer cells through the enhanced production of CXCL12, IL-6, and SDF-1 to promote inflammation and oncogenic signals (Figure 2B) [3,88,89,90]. The loss of wt p53 activity in CAFs can further exert selective pressure to promote transformation in neighbouring epithelial cells through ablation of p53-dependent senescence programs and skewed macrophage polarisation (Figure 2B) [91]. Less phosphorylated forms of wt p53 in lung-derived CAFs have been shown to support CAF-like properties such as migration when compared to normal fibroblasts, in part due to altered mutant-like conformation despite being genetically wild-type. Wt p53 in these CAFs also supports an altered secretome, which facilitates ECM degradation, migration and invasion [92]. Cancer cells can “educate” normal fibroblasts gradually to adopt CAF-like properties [93]. In this re-education process of normal fibroblasts, the wt p53 transcriptional program is altered upon co-culture with H460 or H1299 cancer cell lines [92]. The activity of this mutant-like wt p53 population in CAFs has profound ramifications that limit the use of treatments, which depend on canonical wt p53 activities.
While relatively uncommon, p53 mutation may occur in CAFs in hereditary cancers [94,95], in which case the surrounding microenvironment is primed for tumour formation [96]. In sporadic cancers, p53 mutation in fibroblasts occurs independently of that in tumours and is potentially clonal in origin. These mutant stromal cells potentially provide a favourable microenvironment for tumour spread [94]. In a study of bladder cancer, these stromal mutations act as neoplastic seeds for urothelial carcinoma [97]. The loss of wt p53 and alterations in its expression in CAFs further promotes ROS accumulation and alters the properties of the tumour ECM [92,98,99], potentially affecting the surrounding tumour milieu.
In contrast to its wild-type counterpart, mutant p53 in cancer cells modulates and prevents the tumour-suppressive response to IFN-β, by inhibiting STAT1 phosphorylation and downstream targets of IFN-β. In turn, IFN-β secreted by CAFs can reduce mutant p53 RNA levels in tumours (Figure 2B). This regulatory network employed by CAFs constitutes a molecular standstill that both limits and promotes the tumorigenic effects of mutant p53 in cancer cells, the balance of which can be tipped by the inflammatory microenvironment [100]. The mutational status of p53 is thus paramount to directing IFN-β-related therapies, and the reactivation of wt p53 activity may constitute a synergistic opportunity for these therapies [100].
1.3.2. Mutant p53 Favours Neo-Angiogenesis and Extracellular Matrix Remodelling
In the TME, the ECM is altered to favour the infiltration of specific subsets of tumour-supportive immune cells and the formation of neo-vasculature, thus participating in what can be considered figurative inflammatory terraforming (Figure 2) [101]. Previous studies have demonstrated that the ECM can exert a regulatory effect on the p53 activity of cultured cells through pro-survival signals that suppress its apoptotic functions [102,103]. The reciprocal role of p53 in regulating the ECM has been fleshed out in recent years particularly in hypoxic contexts [104,105]. Hypoxic tumour conditions go hand-in-hand with ROS accumulation and inflammation [106]. Transcription factors that are activated in such environments play a large role in shaping the TME. In hypoxic conditions, hypoxia-inducible factors (HIFs) can induce the expression of pro-angiogenic factors including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) [107]. Furthermore, HIFs are also overexpressed in supportive cells of the TME and regulate their activity [108].
Wt p53 is able to promote the degradation of HIF-1α through MDM-2-mediated ubiquitination. Consequently, in the absence of p53, HIF-1 levels and its downstream transcriptional targets increase [109]. It has recently been demonstrated that R273H and R246I p53 mutants cooperate with HIF-1 in non-small cell lung cancer cells to transcriptionally regulate ECM components, favouring aggressive invasion and poor clinical prognosis (Figure 2B) [110]. This activity of HIFs is important for the expression of chemo-attractants to recruit supportive cells and illustrates how mutant p53 regulates these aspects of the TME [111].
Wt p53 also negatively regulates extracellular matrix metalloproteinase inducer (EMMPRIN), which is known to increase the production of several matrix metalloproteinases (MMPs) responsible for ECM remodelling, angiogenesis, and mediating tumour cell–macrophage interactions [112,113,114,115,116,117]. The down-regulation of EMMPRIN by wt p53 is not transcriptionally-dependent but is sensitive to the inhibition of the endosomal pathway by chloroquine [115]. As a corollary, p53 dysfunction can lead to EMMPRIN upregulation, and thus ECM remodelling in highly invasive cancers. Indeed, there is a correlation between mutant p53 and EMMPRIN expression in intestinal and diffuse-type gastric carcinoma, which hint at a potential GOF for mutant p53 in this context [118]. Human melanoma cells expressing mutant p53 overexpress MMP2, which is ablated upon introduction of wt p53 [119]. MMP2, in turn impinges on anti-tumour immunity [120]. The tumour ECM affects the recruitment of various cellular components of the TME, which can promote tumour cell growth as discussed below.
1.3.3. P53 Status Influences the Tumour Immune Cell Infiltrate
The abundance and composition of the immune cell compartment of the TME vary among different tumour types and can contribute to the rate of disease progression and prognosis [121,122]. The immune landscape of the TME is modulated by the crosstalk between macrophages, dendritic cells, myeloid-derived suppressor cells (MDSCs), T cells, mast cells and natural killer (NK) cells (Figure 2A) [18,123]. Through sustained NF-κB and STAT3 signalling, the impairment of the epithelial barrier, and transcriptional activation of CXCL17, the loss of wt p53 activity can increase macrophage, neutrophil, monocyte and CD4+ T cell infiltration, while limiting the infiltration of potent anti-cancer CD8+ T cells (Figure 2B) [124,125,126,127,128].
In addition, mutant p53 can support myeloid cell infiltration through NF-κB-dependent inflammatory cytokine signatures [39]. The R280K p53 mutant has also been demonstrated to act in accordance with TNFα to elicit a chemokine signature that modulates the immune cell infiltrate [40]. Lymphocyte metagene analysis of tumours harbouring this p53 mutant identified a higher expression of cytotoxic T cell lymphocytes, NK cells, and Th1 genes characteristic of a pro-inflammatory immune cell signature [122]. Despite promoting cancer motility and survival, high levels of pro-inflammatory immune infiltrate correlate with better disease-free survival in patients with mutant p53 basal breast cancers [40]. This illustrates how p53 status in cancer cells can affect the clinical prognosis of cancer, depending on the nature of the immune cell landscape [127,129].
2. Mutant p53 Disrupts Innate Tumour Immunity
Innate immunity is the first line of defence to engage immediate short-term immune operations against pathogens without establishing immunological memory. This similarly applies to the host’s initial response to the danger signals from a developing tumour and cancer-associated inflammation [130]. Key players in innate immunity include cells of the myeloid lineage that mount effector responses in addition to priming further adaptive immune responses [131,132]. Changes in the composition of cancer cell surface proteins, and, in some cases, secreted tumour antigens, enable the activation of both arms of the innate immunity: the complement system and the toll-like receptor (TLR) system [133]. Furthermore, crosstalk between the complement and TLR pathways makes innate immunity a dynamic and robust network for first-line pathogen response [134,135].
The innate immune system is an active participant in immunosurveillance against cancer cells. Abnormal cells may, however, undergo immunoediting in response to selective pressure exerted by innate immune cells like natural killer (NK) cells, which, in concert with adaptive immune cells, kill immunogenic clones selectively. These abnormal cells evolve to evade eradication, escaping immune control and eventually developing into clinical tumours and malignancy. Identifying pathways driving the evasion of the innate immune system in the tumour context offers scope for fine-tuning therapies to reprime this intrinsic cancer defence system. Several cellular components of the inflammatory TME modulate tumour innate immunity [136]. In this section, we discuss how mutant p53 influences innate immunosurveillance in cancer.
2.1. P53 Mediates the Genotoxic Stress Response of the Toll-Like Receptor Pathway
Several studies hint that wt p53 acts through an expansive integrated network that mediates host intrinsic tumour immunity. Moreover, several studies demonstrate that the role wt p53 plays in cancer inflammation is not exclusive to tumour contexts and instead may be a general regulatory mechanism that aids in host responses to infection. Wt p53 expression is induced in response to viral infection as part of the host immune defence, and its role in host antiviral response has been extensively reviewed [137,138,139,140,141]. P53 is inactivated by viruses, regardless of their tumourigenic potential, to prevent host cell-mediated apoptosis [142,143]. Interestingly, the activation of wt p53 in response to cancer echoes the mechanisms employed in antiviral response, and, as such, a role for wt p53 in innate tumour immunity has been explored and developed over the years [82].
Wt p53 and the Toll-like receptor (TLR) pathways are directly linked in humans [144]. Wt p53 regulates the expression of seven out of the 10 TLRs [145]. TLRs are endosomal and plasma-membrane associated receptors that are expressed not only on immune cells, but also on cells of the TME [146]. TLRs recognize pathogen- and damage-associated molecular patterns to promote a protein kinase cascade that induces the expression of inflammatory cytokines and interferons. Additionally, TLR signalling can activate and increase natural killer (NK) cell cytotoxicity, as well as extrinsic and intrinsic apoptotic pathways in tumours [147]. Several pro-angiogenic and growth-promoting factors lie downstream of TLR signalling pathways that function in normal acute immune responses to boost the activity of antigen-presenting cells (APCs) and effector T cell responses [148,149]. Depending on the subset of immune cells activated by these factors, their response can be modulated to promote cancer [146,150,151,152].
TLR activity is canonically modulated, not by the increased expression of its member receptor proteins, but rather by the upstream stimulation of relevant receptors [153]. Though the transcriptional control of TLRs has been described, the regulation of the differential expression of TLRs in cells is commonly epigenetic in nature, and largely an indirect effect of external environmental stresses [148,154,155,156]. Thus, the discovery that several TLRs are transcriptionally regulated by a central stress-response factor, p53, constitutes an important direct link between common cellular stresses and induction of the TLR innate immune response [139,144,157].
Wt p53 regulates the expression of the TLR gene family in T-lymphocytes, and to a lesser extent in macrophages, in a manner dependent on the genetic stress and the host genetic context [144,158,159]. In particular, polymorphisms in p53 response elements in the promoters of TLR genes confer differential sensitivity to genetic stress and infection [157].
The anti-tumour benefits of such TLR induction are apparent when considering the merit of APC reactivation in the tumour microenvironment, whereby activated TLR pathways increase immune detection and activity against tumour-antigen bearing cells [160]. However, TLR expression in tumour cells and surrounding cells can prove pro-tumorigenic [161,162,163,164]. TLR4 is expressed in several human cancer cell lines, such as MDA-MB-231, MCF7, A549 and H1299. Moreover, TLR4 is functional in A549 and H1299, where it activates the p38 MAPK and NF-κB signalling pathways upon exposure to LPS treatment. This activation promotes tumour immune escape and resistance to apoptosis through production of immunosuppressive cytokines like VEGF, TGF-β and IL-8 [165]. The activation of MAPK and NF-κB are common threads in TLR-4-expressing colorectal cancers, increasing proliferative potential, apoptotic resistance and metastatic potential [166]. Moreover, TLR-4 expression in breast cancer correlates with poor survival rates and invasiveness [167,168].
Additionally, the upregulation of TLR1-10, NF-κB and p53 expression is observed in oral lichenoid disease, an auto-immune disease associated with chronic inflammation and malignant potential owing to atypical lichenoid lesions in cases of greater chronic inflammatory response [169,170]. The downstream generation of ROS and activation of NF-κB can indeed amplify the innate immune response, but may also act to generate abnormal patterns of inflammation, which, when chronically activated, prove to be pro-tumorigenic, as previously discussed [171,172]. This has a profound implication on genotoxic and TLR agonist strategies for targeting tumours, whereby enhancing TLR-signalling may instead foster a highly inflammatory yet immunosuppressive TME.
Notably, the p53-TLR regulatory axis exists only in primates and humans [144]. This evolutionary distinction is of prime consideration when evaluating TLR-mediated cancer therapies, as mouse models do not recapitulate the regulatory axis that exists in humans [162].
Mutant p53 Hijacks TLR Signalling in Cancer
The direct transcriptional regulation of TLR proteins by wt p53 does not necessarily reflect a linear increase in the respective ligand-dependent cytokine responses. This demonstrates the highly context-dependent nature of p53 immune signalling. Downstream TLR activity is also not solely transcriptionally dependent on p53. Target genes that crosstalk between the two pathways can cooperate to mediate and control downstream immune signalling [159]. Polymorphisms on the p53 gene and its TLR targets may contribute to the variability in downstream TLR responses observed. Indeed, single nucleotide polymorphisms in wt p53 response elements on the promoter of TLR8 modulate the wt p53 regulation of downstream immune response. Some p53 mutants retain functionality in regulating TLR expression, while several others demonstrate altered TLR regulatory spectrums. Not surprisingly, transcriptionally inactive p53 mutants, which constitute the majority of p53 mutations, are unable to mediate the upregulation of TLRs in response to genetic stress [144,158,159], supporting a role for the p53-TLR axis in the cellular response to genotoxic stress.
Notably, the clinical relevance of TLR-4 in breast cancer has been shown to be p53-dependent. In the presence of mutant p53, patients with a low expression of TLR4 correlate with better survival than those with high expression levels, whereas the inverse is true in patients with wt p53 tumours. This clinical distinction is attributed to IFN-γ secretion in wt p53 breast cancer cells that mediates growth inhibition by TLR-4. Strikingly, functional TLR-4 retention correlates with a broad spectrum of cancers with functional p53 loss, including serous ovarian, head and neck, and bladder cancers. This indicates a selective advantage to retaining TLR-4 signalling in the absence of wt p53. Inversely, tumours that retain wt p53 function correlate with a higher incidence of TLR-4 alterations, as exemplified in lung cancers [173,174].
P53 mutation not only leads to the differential expression of the TLR genes, but also impose altered downstream effects. These impact the sensitivity and responsiveness of TLR3 to its known ligands, affecting the downstream type I interferon response and genotoxic-stress induced apoptosis. This modulation of TLR3 responsiveness is directly attributed to the presence of transcriptionally active or TLR3-enhancing p53 mutants like P151H and R337H, while other mutants may conversely dampen the TLR3-mediated immune response [145,175,176]. Knowledge of TLR responsiveness to specific p53 mutants prior to the application of TLR agonists may be appropriate for enhancing the efficacy of its anti-tumour therapy. In the same vein, the restoration of p53 activity proves valuable in combination with TLR agonists to rescue TLR activation and responsiveness effects [159,161].
Mutations in p53 may also have indirect effects on TLR signalling owing to GOF effects. Arf6 and its downstream effectors are major targets for induction by p53 mutations in epithelial cells, pancreatic ductal adenocarcinoma, and mevalonate pathway-driven malignancies [177,178,179]. Strikingly, Arf6 is crucial for downstream TLR signalling and its overexpression and activity has been associated with increased proliferation, invasion and metastasis [180,181,182,183,184]. Arf6 lies in the crossroad of these two pathways through its roles in ROS production that affect both p53 and TLR pathways [58,185]. Moreover, Arf6 activation is facilitated by the platelet-derived growth factor (PDGF), a product of TLR4 signalling, and its β receptor (PDGFRβ), a direct target of mutant p53 [178,186,187]. The pro-tumorigenic effects of Arf6 in cancer have been largely attributed to its roles in receptor recycling, matrix disruption, manipulation of cellular adhesion, aberrant growth-factor signalling, and metabolic dysfunction [180]. Arf6 may further stand as a functional link between mutant p53 and the regulation of innate tumour immunity.
2.2. Mutant p53 Favours a Pro-Tumour Macrophage Signature
Macrophages are one of the most abundant immune cell populations in the TME [188]. When macrophage activation is driven by lipopolysaccharide and IFN-γ, they tend toward an M1-like phenotype, which is pro-inflammatory, inducing cytotoxic responses against cancers and pathogens. In contrast, IL-4 or IL-13-mediated macrophage activation tends to elicit an M2-like anti-inflammatory phenotype, associated with wound healing and tumour promotion [189,190]. The activity of both macrophage responses is integral to several steps of tumour progression. More specifically, M1-like polarized tumour-associated macrophage (TAM) responses mediate an inflammatory environment that imposes selective pressure against highly immunogenic cancer cells driving their elimination. If the tumour escapes immune detection, the TME induces an M2-like polarization of TAMs, which favours immunosuppression and pro-tumorigenic activity [190,191].
It is generally accepted that both M1-like and M2-like polarisation are associated with increased levels of p53 expression to varying degrees. High-levels of p53 activity are elicited as a brake in M1-like macrophage polarisation to inhibit detrimental prolonged activation of the inflammatory NF-κB and STAT1 pathways, resulting in diminished M1-like gene expression over time [192]. For example, in response to iron overload, increased ROS levels enable prime p53 acetylation, while driving M1-like polarisation [193]. The elicited activity of wt p53, present only in low levels in M2-like polarized cells, permits M2-like gene expression and phenotype. The inactivation or mutation of p53 in M2-like macrophages can result in increased M2-like gene expression [194].
Wt p53 not only regulates macrophage polarisation but also impacts on the resulting inflammatory gene expression of macrophages. In macrophages, the usual antagonistic relationship of NF-κB and wt p53 is reoriented, instead forming a cooperative relationship to promote inflammatory response to genotoxic stress [38]. Demonstrating this, the Nutlin-3-mediated stabilisation of wt p53 can enhance NF-κB function in macrophage populations, amplifying early pro-inflammatory macrophage signalling. This is in stark contrast to the anti-tumorigenic effects Nutlin-3 has upon the stabilisation of wt p53 in epithelial cells, further highlighting programmatic differences in the p53-NF-κB pathway in macrophage populations [34,195,196]. Much like the TLR/p53 axis, this distinct regulatory program is absent in rodents, which, aside from having evolutionary implications, could affect the evaluation of its biological consequences using mouse models [38]. Stabilised wt p53 in macrophages potentially activates pro-inflammatory gene expression, but may also allow macrophages to adopt a senescence secretory phenotype to ensure continuous turnover in the TME through the p53 pathway [197].
Mutant p53-expressing cancers can exert similar non-cell-intrinsic reprogramming of macrophages into TAM-like M2 phenotypes through the exosomal transfer of microRNA [198]. Exosomal transfer of microRNA is functionally relevant in several cancers and may thus constitute an additional mutant-specific GOF of p53 [198,199,200]. Strikingly, exosomes from R248W and R273H mutant p53-expressing colon cancer cells are enriched for miR-1246, a microRNA found to promote invasiveness and stemness [201,202,203,204]. Following treatment of M2-like macrophages with these exosomes, genes characteristic of the tumour-supportive TAM phenotype were upregulated, demonstrating a mechanism by which mutant p53 cancers can alter innate immune cell function to promote tumorigenesis [198].
3. Mutant p53 Alters Cell-Mediated Immunity in Cancer
Cancer immunoediting relies on three component phases: elimination, equilibrium, and escape. The activities of cells of the adaptive immune system are also central in cancer immune editing [136]. Immunotherapy directed to tumour specific antigens relies on the integrity of APCs [130]. However, these immunogenic APCs are scantly detected in tumours, and, if are present, they display impaired or dysfunctional antigen-presenting activity. While p53 mutations are less common in immune cells, p53 can influence cell-mediated immunity through specific molecular signatures brought about by the tumour or stromal cells, which in turn affect recruitment and activation of immune cells [4]. It can also regulate the expression of class one major histocompatibility complex and associated downstream immune effects [205].
P53 has recently been shown to influence the differentiation of monocytes from already abundant myeloid precursor cells in the periphery of tumours [206]. P53 also modulates NKG2D-mediated NK cell activity through pathways in senescence. The mutation or loss of p53 in these contexts impairs NK cell-mediated immunity [138,207].
Interestingly, the role of wt p53 in inducing regulatory T cells to suppress autoimmunity hints at a mechanism by which aberrant activation of these pathways can be misappropriated in cancer [208]. Nutlin-3a-induced wt p53 reactivation in a TME rich with tumour-infiltrating leukocytes, such as in EL4 tumours, is sufficient to induce antitumor immunity, in contrast to B16 with low infiltration. This is due to an increase in the activated dendritic cell population that elicits expansion of CD8+ cytotoxic T cells and induces an immunogenic cell death. Indeed, the efficacy of the reactivation is p53-dependent, not only in the tumour but also in the leukocyte population within the TME [46]. Consistently, mutant p53 fails to trigger and can inhibit the tumour antigenicity that is normally caused by genomic instability, or by the prolonged activation and accumulation of wt p53, as demonstrated in gastric cancer [209]. Lymphocyte invasion, particularly that of cytotoxic T cells, is also impaired when wt p53 pathways are compromised in ER-negative breast cancer and basal-like breast tumours. Both the loss of heterozygosity and p53 mutation show lower rates of T cell infiltration and correlate with poor prognosis [210]. The loss of p53 in several genetically engineered mouse breast cancer models resulted in increased inflammatory Wnt signalling in tumour-associated macrophages, prompting systemic neutrophilia and ultimately metastasis [211]. A study using a murine melanoma model has also highlighted the requirement of p53 stabilisation for the assembly of endosomal sorting complexes that mediate immunosurveillance within the metastatic niche [212]. These studies highlight a role for wt p53 in facilitating productive tumour immunity that is compromised when p53 is lost or mutated.
More recently, connections with wt p53 and immune checkpoints have been uncovered. P53 transactivates programmed death-ligand 1 (PD-L1) and its receptor programmed death-1 (PD-1) in cancer cells, and in normal T cells in response to stress [213,214]. Physiologically, the interaction between PD-L1 on tissue and PD-1 on T cells suppresses activation signals generated following the T cell receptor recognition of antigen; this immune checkpoint controls inflammation. The overexpression of PD-L1 on tumours, however, takes advantage of this immune checkpoint pathway to suppress tumour recognition and induce immune tolerance [215]. Similarly, FOXP3 is induced by wt p53 in breast and colon cancer cell lines in response to DNA damage. Ectopic FOXP3 expression in turn converts normal T cells to T regulatory cells, which promote an immunosuppressive tumour milieu [216]. A study using h3T T cell receptor mice with human tyrosinase epitope-reactive T cells showed that p53 knockout T cells actually served to augment anti-tumour functions. While this increased robustness was associated with decreased ROS production, which increased T cell longevity, the authors imply a role for p53-dependent FOXP3 in the observed phenotype [217]. These examples illustrate cases wherein intact wt p53 pathways are subverted by cancers to develop tumour tolerance to adaptive immunity.
More recently, connections with wt p53 and immune checkpoints have been uncovered. P53 transactivates programmed death-ligand 1 (PD-L1) and its receptor programmed death-1 (PD-1) in cancer cells, and in normal T cells in response to stress [213,214]. Physiologically, the interaction between PD-L1 on tissue and PD-1 on T cells suppresses activation signals generated following the T cell receptor recognition of antigen; this immune checkpoint controls inflammation. The overexpression of PD-L1 on tumours, however, takes advantage of this immune checkpoint pathway to suppress tumour recognition and induce immune tolerance [215]. Similarly, FOXP3 is induced by wt p53 in breast and colon cancer cell lines in response to DNA damage. Ectopic FOXP3 expression in turn converts normal T cells to T regulatory cells, which promote an immunosuppressive tumour milieu [216]. A study using h3T T cell receptor mice with human tyrosinase epitope-reactive T cells showed that p53 knockout T cells actually served to augment anti-tumour functions. While this increased robustness was associated with decreased ROS production, which increased T cell longevity, the authors imply a role for p53-dependent FOXP3 in the observed phenotype [217]. These examples illustrate cases wherein intact wt p53 pathways are subverted by cancers to develop tumour tolerance to adaptive immunity.
In some contexts, mutant p53 can mediate increased immunogenic activity. In mutant p53-expressing breast cancer, tumours displayed a higher enrichment of immunogenic activity than those expressing wt p53. Moreover, these observations have been recapitulated in vitro, where mutant p53 regulation of cell cycle, apoptosis, Wnt, JAK-STAT, NOD-like receptor and glycolysis pathways were found to promote immunogenicity of cultured breast cancer cells. Thus, in some contexts, mutant p53 could potentially be a useful biomarker for immunotherapy responsiveness and can be associated with better survival prognosis owing to unique immunogenic signatures [218].
4. Mutant p53 as a Tumour Antigen
Mutations in tumour suppressor gene products can lead to the formation of novel epitopes not normally presented in self-tissues, and can thus be considered tumour antigens [219]. Mutant p53 that accumulates in cancers belongs to this class of tumour antigens. Antibodies against p53 can be detected in patient sera across several cancer types and these are strongly correlated with p53 alteration and over-expression [220,221,222,223]. The presence of p53 antibodies has been used as an early marker for diagnosing pre-malignant disease and early stages of cancer; however, the prognostic and diagnostic values of mutant p53-associated expression of p53 antibodies in sera is limited and differs depending on the type of cancer [220,224]. Mutant p53 has been considered as an appealing tumour-specific antigen target for immunotherapy, but limited success has been achieved in this field due to the inefficient presentation of mutant p53 antigen on cells for recognition [225,226,227,228].
Mutant p53 can bind to human leukocyte antigen (HLA) class I molecules to enable cytotoxic targeting by CD8+ T cells [229,230,231,232,233], but can also bind to HLA class II molecules [234]. The latter has been demonstrated to skew the CD4+ T cell response to adapt a pro-tumour Th2 phenotype in head and neck cancers [235]. Despite this, there is a low correlation with accumulated levels of p53 and their recognition by targeted T cells. In fact, the mutational status of p53 has been shown to affect its detection by human T cells. Consequently, p53-bearing destabilizing mutations like R175H and Y220C favour antigen presentation to T cells, eliciting distinct immune reactivity independent of expression levels [236]. Indeed, several studies explore the potential development of mutant p53 cancer vaccines that take advantage of this immunogenicity [237,238,239].
Recently, a screen for T cell responses against the naturally processed neoantigens of several hotspot p53 mutants (R175H, Y220C, G245S, G245D, R248L, R248Q, R248W, R249S, R273C, R273H, R273L, and R282W) revealed broad immunogenic responses that varied across patients with different metastatic epithelial cancer types and different p53 mutational status [240]. Moreover, the HLA alleles involved in the presentation of different mutant p53 antigens varied from patient to patient, highlighting a context-dependent threshold for mutant p53 antigen presentation. Despite this, HLA alleles that were capable of eliciting mutant p53 immunogenic response were frequent across several racial patient demographics, indicating a broad potential benefit for p53-neoantigen therapy. A similar screen was conducted on mutant p53 neoantigens in ovarian cancers [241]. While not all hotspot p53 mutations were immunogenic, those that were (G245S and Y220C) elicited mutant-specific T cell infiltration of ovarian cancer metastasis, further emphasizing the potential for mutant p53 as a target for T cell immune and gene therapy. Interestingly, neoantigens that arose from random, somatic, non-synonymous mutations were also recognized by T cells and were unique to each patient. Elevated levels of p53 associated with its mutation are linked to the generation of anti-p53 auto-antibodies, underpinning the potential role of p53 in mediating tumour antigenicity [220,221,228,235,242]. Consequently, fragments of p53 proteins have since been used as tumour-associated antigens for the generation of therapeutic vaccines [243,244,245,246].
Despite the promise that mutant p53 holds in the field of immunotherapy, the selective pressure that T cells exert could lead to eventual therapeutic resistance. Recent studies have employed a fusion of mutant p53 transduced dendritic cells and antigen-expressing tumour cells to generate a broad-acting vaccine, obtaining results that could potentially overcome such a pitfall [247]. The development of targeted immunotherapies against mutant p53 has proven a challenging feat, but one that potentially can reap great outcome considering the multifaceted nature of mutant p53 tumorigenic activity and its prevalence in human cancers.
5. Wild-Type p53 in Post-Apoptotic Cell Clearance
The post-mortem fate of cells targeted by effective anti-tumour activity also constitutes an intriguing aspect of tumour immunity and inflammation. The normal turnover of cells during the resolution of injury and infection is key to preventing aberrant inflammation and triggering the immune recognition of dead cell antigens [248]. Wt p53 further extends its role in immunity to mediating macrophage clearance of dead cells by inducing the expression of Death Domain 1α (DD1α, also named VISTA), a gene implicated in triggering the immune checkpoint [249]. The presence of DD1α on the surface of dying cells specifically facilitates their recognition and subsequent phagocytosis by macrophages. Inflammation due to the failure to recognize and resolve dead cell clearance may explain the resulting autoimmune phenotype associated with DD1α disruption. Similar interactions can occur between macrophage DD1α and T cell DD1α, rendering them susceptible to immunosuppression and preventing the further recognition of tumour antigens [213,250]. The role of p53 in modulating DD1α expression highlights both its tumour suppressive function and its role in modulating tumour surveillance. While this pathway is wholly reliant on intact wt p53 function, the prospect of this pathway failing, or being hyperactive in mutant p53 contexts may reinforce targeting DD1α as an effective cancer therapy.
6. Conclusions
The body of knowledge implicating mutant p53 in disruption of immunity that normally limits cancer control is ever increasing. A deeper understanding of the intrinsic roles of wt p53 in the TME illuminates the subversive activities of its mutant counterparts. It is now established that, aside from functional loss of p53, mutant p53 GOF contributes extensively to promoting immune and inflammatory hallmarks of cancer by (1) responding to and fuelling inflammatory signalling in the TME, (2) shaping the tumour milieu to facilitate cancer progression, (3) disrupting innate tumour immunity, (4) modulating the activity of infiltrating immune cells, and (5) potentially impinging on post-apoptotic cell clearance (Figure 3).
These proposed roles for mutant p53 predict its relevance as a target for immunotherapy as a means to limit tumour growth and metastasis. Pertinently through, the activity of mutant p53 in tumour immunity is largely context dependent. The nature of its genetic mutations, the cellular compartment in which the mutations function, as well as the molecular and cellular conditions of the surrounding TME are all significant variables able to impact the influence mutant p53 exerts over immune responses.
Efforts to target mutant p53 hold promise for intervening in its immune-regulatory function. These include the use of mutant p53 specific vaccines and autoantibodies, the reactivation of wt p53 in cellular components of the TME, and disrupting regulatory axes involving mutant p53. Overall, the complexity of mutant p53 interactions in the tumour niche convolute and limit the application of these proposed therapies, and further understanding of mutant p53 GOF is needed to fully appreciate the therapeutic capacity of targeting mutant p53 immune and inflammatory pathways.
Funding
The work in the author’s lab is supported by grants from National Health and Medical Research Council (NHMRC 1123057), Sister Institute Network Fund (MD Anderson Cancer Centre/Peter MacCallum Cancer Centre), and the Peter MacCallum Cancer Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, K.; Xu, J.; Zairis, S.; Resnick-Silverman, L.; Abate, F.; Steinbach, N.; Ozturk, S.; Saal, L.H.; Su, T.; Cheung, P.; et al. p53 Maintains Baseline Expression of Multiple Tumor Suppressor Genes. Mol. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskovits, N.; Kalinkovich, A.; Bar, J.; Lapidot, T.; Oren, M. p53 attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Guo, G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef] [Green Version]
- Stein, Y.; Aloni-grinstein, R.; Rotter, V. Mutant p53—A potential player in shaping the tumor–stroma crosstalk. J. Mol. Cell Biol. 2019, 11, 600–604. [Google Scholar] [CrossRef]
- Miciak, J.; Bunz, F. Long story short: p53 mediates innate immunity. BBA Rev. Cancer 2016. [Google Scholar] [CrossRef] [Green Version]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual role of p53 in innate antiviral immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the viral connection: Back into the future. Cancers 2018, 10, 178. [Google Scholar] [CrossRef] [Green Version]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53: A tale of two stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the crossfire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Richmond, A. Chemokins modulate immune surveillance in tumorignesis, metastatsis, and response to immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.R.; Ho, P.C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015, 22, 549–559. [Google Scholar] [CrossRef]
- Nicolini, A.; Ferrari, P.; Diodati, L.; Carpi, A. Alterations of signaling pathways related to the immune system in breast cancer: New perspectives in patient management. Int. J. Mol. Sci. 2018, 19, 2733. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 distribution and perspective for cancer immunotherapy—Blockade, knockdown, or inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Irish J. Med. Sci. (1971 -) 2017, 186, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Munn, L. Cancer and Inflammation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Chen, X. Senescence regulation by the p53 protein family. Methods Mol. Biol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K. P53 isoforms in cellular senescence-and ageing-associated biological and physiological functions. Int. J. Mol. Sci. 2019, 20, 6023. [Google Scholar] [CrossRef] [Green Version]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef]
- Webster, G.A.; Perkins, N.D. Transcriptional Cross Talk between NF-κB and p53. Mol. Cell. Biol. 1999. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; Von Werder, A.; Schmid, R.M.; et al. Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 2010. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, V.; Komarov, A.P.; Ippolito, T.; Bonneau, K.; Chenchik, A.A.; Gudkov, A.V. Peptides genetically selected for NF-κB activation cooperate with oncogene Ras and model carcinogenic role of inflammation. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [Green Version]
- Son, D.S.; Kabir, S.M.; Dong, Y.L.; Lee, E.; Adunyah, S.E. Inhibitory Effect of Tumor Suppressor p53 on Proinflammatory Chemokine Expression in Ovarian Cancer Cells by Reducing Proteasomal Degradation of IκB. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Uehara, I.; Tanaka, N. Role of p53 in the regulation of the inflammatory tumor microenvironment and tumor suppression. Cancers 2018, 10, 219. [Google Scholar] [CrossRef] [Green Version]
- Gudkov, A.V.; Komarova, E.A. p53 and the carcinogenicity of chronic inflammation. Cold Spring Harb. Perspect. Med. 2016. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 induces NF-κB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res. Commun. 2008. [Google Scholar] [CrossRef]
- Liu, G.; Park, Y.-J.; Tsuruta, Y.; Lorne, E.; Abraham, E. p53 Attenuates Lipopolysaccharide-Induced NF-κB Activation and Acute Lung Injury. J. Immunol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.M.; Ernst, M.K.; Rice, N.R.; Vousden, K.H. Role of NF-κB in p53-mediated programmed cell death. Nature 2000. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011. [Google Scholar] [CrossRef] [Green Version]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD Mediates NF-κB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. P53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014. [Google Scholar] [CrossRef] [Green Version]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rozenfeld, N.; et al. Mutant p53 Prolongs NF-κB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Di Minin, G.; Bellazzo, A.; DalFerro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 Reprograms TNF Signaling in Cancer Cells through Interaction with the Tumor Suppressor DAB2IP. Mol. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Rahnamoun, H.; Lu, H.; Duttke, S.H.; Benner, C.; Glass, C.K.; Lauberth, S.M. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Dey, A.; Tergaonkar, V.; Lane, D.P. Double-edged swords as cancer therapeutics: Simultaneously targeting p53 and NF-κB pathways. Nat. Rev. Drug Discov. 2008, 7, 1031–1040. [Google Scholar] [CrossRef]
- Gurova, K.V.; Hill, J.E.; Guo, C.; Prokvolit, A.; Burdelya, L.G.; Samoylova, E.; Khodyakova, A.V.; Ganapathi, R.; Ganapathi, M.; Tararova, N.D.; et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-κB-dependent mechanism of p53 suppression in tumors. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef] [Green Version]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compounds that simultaneously suppress NF-κB and activate p53 by targeting FACT. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef]
- Binayke, A.; Mishra, S.; Suman, P.; Das, S.; Chander, H. Awakening the “guardian of genome”: Reactivation of mutant p53. Cancer Chemother. Pharmacol. 2019, 83, 1–15. [Google Scholar] [CrossRef]
- Guo, G.; Yu, M.; Xiao, W.; Celis, E.; Cui, Y. Local Activation of p53 in the Tumor Microenvironment Overcomes Immune Suppression and Enhances Antitumor Immunity. Cancer Res. 2017, 77, 2292–2306. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Li, N.; Grivennikov, S.I.; Karin, M. The Unholy Trinity: Inflammation, Cytokines, and STAT3 Shape The Cancer Microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in Regulating p53 Expression and Function. Mol. Cell. Biol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Yue, X.; Zhao, Y.; Li, X.; Wu, L.; Zhang, C.; Liu, Z.; Lin, K.; Xu-Monette, Z.Y.; Young, K.H.; et al. LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Sainz-Perez, A.; Gary-Gouy, H.; Gaudin, F.; Maarof, G.; Marfaing-Koka, A.; de Revel, T.; Dalloul, A. IL-24 Induces Apoptosis of Chronic Lymphocytic Leukemia B Cells Engaged into the Cell Cycle through Dephosphorylation of STAT3 and Stabilization of p53 Expression. J. Immunol. 2008. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, J.; Liu, J.; Liang, X.; Jin, X.; Xie, J.; Zhang, L.; Chen, D.; Fiskesund, R.; Tang, K.; et al. STAT3/p53 pathway activation disrupts IFN-β-induced dormancy in tumor-repopulating cells. J. Clin. Invest. 2018. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Jin, X.; Rothman, K.; Lin, H.J.; Tang, H.; Burke, W. Modulation of signal transducer and activator of transcription 3 activities by p53 tumor suppressor in breast cancer cells. Cancer Res. 2002. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Tang, H.; Jin, X.; Jia, G.; Hsieh, J.T. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene 2002. [Google Scholar] [CrossRef] [Green Version]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Chen, Y.; St. Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Simon, H.U. A novel link between p53 and ROS. Cell Cycle 2013, 12, 201. [Google Scholar] [CrossRef]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreou, K.; Rajendran, R.; Krstic-Demonacos, M.; Demonacos, C. Regulation of CXCR4 gene expression in breast cancer cells under diverse stress conditions. Int. J. Oncol. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [Green Version]
- Diederich, M.; Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int. J. Cell Biol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, C.A.; He, Q.; Huang, Y.; Saeed Sheikh, M. Cyclooxygenase-2 interacts with p53 and interferes with p53-dependent transcription and apoptosis. Oncogene 2005. [Google Scholar] [CrossRef] [Green Version]
- Han, J.A.; Kim, J.I.; Ongusaha, P.P.; Hwang, D.H.; Ballou, L.R.; Mahale, A.; Aaronson, S.A.; Lee, S.W. p53-mediated induction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. EMBO J. 2002. [Google Scholar] [CrossRef] [Green Version]
- Khajeniazi, S.; Allameh, A.; Soleimani, M.; Mortaz, E. Changes in COX-2 and oxidative damage factors during differentiation of human mesenchymal stem cells to hepatocyte-like cells is associated with downregulation of P53 gene. Biol. Chem. 2013. [Google Scholar] [CrossRef]
- Niki, T.; Kohno, T.; Iba, S.; Moriya, Y.; Takahashi, Y.; Saito, M.; Maeshima, A.; Yamada, T.; Matsuno, Y.; Fukayama, M.; et al. Frequent co-localization of cox-2 and laminin-5 γ2 chain at the invasive front of early-stage lung adenocarcinomas. Am. J. Pathol. 2002. [Google Scholar] [CrossRef]
- Kim, J.; Shim, M. COX-2 inhibitor NS-398 suppresses doxorubicin-induced p53 accumulation through inhibition of ROS-mediated Jnk activation. Mol. Carcinog. 2016. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.N.; Jin, H.O.; Park, J.A.; Kim, J.H.; Kim, J.Y.; Kim, B.; Kim, W.; Hong, S.E.; Lee, Y.H.; Chang, Y.H.; et al. Heme oxygenase-1 determines the differential response of breast cancer and normal cells to piperlongumine. Mol. Cells 2015. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543. [Google Scholar] [CrossRef]
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012, 33, 442–451. [Google Scholar] [CrossRef]
- Fontemaggi, G.; Dell’Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009. [Google Scholar] [CrossRef]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nisticò, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015. [Google Scholar] [CrossRef]
- Cordani, M.; Pacchiana, R.; Butera, G.; D’Orazi, G.; Scarpa, A.; Donadelli, M. Mutant p53 proteins alter cancer cell secretome and SS: Involvement in cancer invasion and metastasis. Cancer Lett. 2016, 376, 303–309. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiantore, M.V.; Vannucchi, S.; Accardi, R.; Tommasino, M.; Percario, Z.A.; Vaccari, G.; Affabris, E.; Fiorucci, G.; Romeo, G. Interferon-β induces cellular senescence in cutaneous human papilloma virus-transformed human keratinocytes by affecting p53 transactivating activity. PLoS ONE 2012, 7, e36909. [Google Scholar] [CrossRef]
- Bar, J.; Feniger-Barish, R.; Lukashchuk, N.; Shaham, H.; Moskovits, N.; Goldfinger, N.; Simansky, D.; Perlman, M.; Papa, M.; Yosepovich, A.; et al. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, A.C.; Shih, S.C.; Cliffe, A.R.; Hida, K.; Klagsbrun, M. Attenuated p53 activation in tumour-associated stromal cells accompanies decreased sensitivity to etoposide and vincristine. Br. J. Cancer 2008. [Google Scholar] [CrossRef]
- Schmid, J.O.; Dong, M.; Haubeiss, S.; Friedel, G.; Bode, S.; Grabner, A.; Ott, G.; Mürdter, T.E.; Oren, M.; Aulitzky, W.E.; et al. Cancer cells cue the p53 response of cancer-associated fibroblasts to cisplatin. Cancer Res. 2012. [Google Scholar] [CrossRef] [Green Version]
- Kurose, K.; Gilley, K.; Matsumoto, S.; Watson, P.H.; Zhou, X.P.; Eng, C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002. [Google Scholar] [CrossRef]
- Guo, G.; Marrero, L.; Rodriguez, P.; Del Valle, L.; Ochoa, A.; Cui, Y. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid-like stromal network. Cancer Res. 2013, 73, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005. [Google Scholar] [CrossRef]
- Addadi, Y.; Moskovits, N.; Granot, D.; Lozano, G.; Carmi, Y.; Apte, R.N.; Neeman, M.; Oren, M. p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 2010. [Google Scholar] [CrossRef] [Green Version]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; Van Der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P. Primed for cancer: Li Fraumeni Syndrome and the pre-cancerous niche. Ecancermedicalscience 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, D.; Malkin, D. p53 and hereditary cancer. Subcell. Biochem. 2014. [Google Scholar] [CrossRef]
- Paterson, R.F.; Ulbright, T.M.; MacLennan, G.T.; Zhang, S.; Pan, C.X.; Sweeney, C.J.; Moore, C.R.; Foster, R.S.; Koch, M.O.; Eble, J.N.; et al. Molecular Genetic Alterations in the Laser-Capture-Microdissected Stroma Adjacent to Bladder Carcinoma. Cancer 2003. [Google Scholar] [CrossRef]
- Trachootham, D.; Chen, G.; Zhang, W.; Lu, W.; Zhang, H.; Liu, J.; Huang, P. Loss of p53 in stromal fibroblasts promotes epithelial cell invasion through redox-mediated ICAM1 signal. Free Radic. Biol. Med. 2013. [Google Scholar] [CrossRef] [Green Version]
- Alexandrova, A.; Ivanov, A.; Chumakov, P.; Kopnin, B.; Vasiliev, J. Changes in p53 expression in mouse fibroblasts can modify motility and extracellular matrix organization. Oncogene 2000. [Google Scholar] [CrossRef] [Green Version]
- Madar, S.; Harel, E.; Goldstein, I.; Stein, Y.; Kogan-Sakin, I.; Kamer, I.; Solomon, H.; Dekel, E.; Tal, P.; Goldfinger, N.; et al. Mutant p53 Attenuates the Anti-Tumorigenic Activity of Fibroblasts-Secreted Interferon Beta. PLoS ONE 2013. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.N.; Wang, D.R.; Sato, M.; Kojima, N.; Imai, K.; Higashi, N.; Senoo, H. Extracellular matrix-regulated p53 expression and nuclear localization in cultured Detroit 562 cells derived from pharyngeal carcinoma. Arch. Histol. Cytol. 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilić, D.; Almeida, E.A.C.; Schlaepfer, D.D.; Dazin, P.; Aizawa, S.; Damsky, C.H. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 1998. [Google Scholar] [CrossRef] [PubMed]
- Royds, J.A.; Dower, S.K.; Qwarnstrom, E.E.; Lewis, C.E. Response of tumour cells to hypoxia: Role of p53 and NFkB. J. Clin. Pathol. Mol. Pathol. 1998, 51, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell. Longev. 2016, 2016, 3907147. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, D.; Kim, K.; Noha, M.; Teramoto, A. Hypoxia inducible factor 1-α regulates of platelet derived growth actor-B in human glioblastoma cells. J. Neurooncol. 2006. [Google Scholar] [CrossRef]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia- inducible factor 1α. Genes Dev. 2000. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.J.; Chen, C.; Zhou, Z.H.; Gao, S.T.; Tee, T.J.; Yang, L.Q.; Xu, Y.Y.; Pang, T.H.; Xu, X.Y.; Sun, Q.; et al. Hypoxia-inducible factor-1 alpha correlates with tumor-associated macrophages infiltration, influences survival of gastric cancer patients. J. Cancer 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Song, Z.; Zhang, S.; Nanda, A.; Li, G. CD147: A Novel Modulator of Inflammatory and Immune Disorders. Curr. Med. Chem. 2014. [Google Scholar] [CrossRef] [PubMed]
- Caudroy, S.; Polette, M.; Nawrocki-Raby, B.; Cao, J.; Toole, B.P.; Zucker, S.; Birembaut, P. EMMPRIN-mediated MMP regulation in tumor and endothelial cells. Clin. Exp. Metastasis 2002. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hemler, M.E. Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res. 2001, 61, 2276–2281. [Google Scholar]
- Zhu, H.; Evans, B.; O’Neill, P.; Ren, X.; Xu, Z.; Hait, W.N.; Yang, J.M. A role for p53 in the regulation of extracellular matrix metalloproteinase inducer in human cancer cells. Cancer Biol. Ther. 2009. [Google Scholar] [CrossRef] [Green Version]
- Amit-Cohen, B.C.; Rahat, M.M.; Rahat, M.A. Tumor cell-macrophage interactions increase angiogenesis through secretion of EMMPRIN. Front. Physiol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Bougatef, F.; Quemener, C.; Kellouche, S.; Naïmi, B.; Podgorniak, M.P.; Millot, G.; Gabison, E.E.; Calvo, F.; Dosquet, C.; Lebbé, C.; et al. EMMPRIN promotes angiogenesis through hypoxia-inducible factor-2α-mediated regulation of soluble VEGF isoforms and their receptor VEGFR-2. Blood 2009. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Takahashi, H.; Murai, Y.; Cui, Z.; Nomoto, K.; Miwa, S.; Tsuneyama, K.; Takano, Y. Pathobiological characteristics of intestinal and diffuse-type gastric carcinoma in Japan: An immunostaining study on the tissue microarray. J. Clin. Pathol. 2007. [Google Scholar] [CrossRef]
- Toschi, E.; Rota, R.; Antonini, A.; Melillo, G.; Capogrossi, M.C. Wild-type p53 gene transfer inhibits invasion and reduces matrix metalloproteinase-2 levels in p53-mutated human melanoma cells. J. Invest. Dermatol. 2000. [Google Scholar] [CrossRef] [Green Version]
- Godefroy, E.; Bhardwaj, N. Dysregulation of anti-tumor immunity by the matrix metalloproteinase-2. Oncoimmunology 2012, 1, 109–111. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Bindea, G.; Kirilovsky, A.; Angell, H.K.; Obenauf, A.C.; Tosolini, M.; Church, S.E.; Maby, P.; Vasaturo, A.; Angelova, M.; et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-B signalling in a mouse model of lung adenocarcinoma. Nature 2009. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in Enterocytes Generates an Inflammatory Microenvironment Enabling Invasion and Lymph Node Metastasis of Carcinogen-Induced Colorectal Tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2–STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016. [Google Scholar] [CrossRef] [Green Version]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 2018. [Google Scholar] [CrossRef]
- Hacke, K.; Rincon-Orozco, B.; Buchwalter, G.; Siehler, S.Y.; Wasylyk, B.; Wiesmüller, L.; Rösl, F. Regulation of MCP-1 chemokine transcription by p53. Mol. Cancer 2010. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yuan, L.; Lu, Q.; Xu, H.; He, X. Distinctive profiles of tumor-infiltrating immune cells and association with intensity of infiltration in colorectal cancer. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of Tumors by the Innate Immune System and Natural Killer Cells. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2014; Volume 122, ISBN 9780128002674. [Google Scholar]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lambris, J.D. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010, 31, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.C. Crosstalk between Complement and Toll-Like Receptors. Toxicol. Pathol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Anazawa, Y.; Iiizumi, M.; Fukuda, S.; Nakamura, Y.; Arakawa, H. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 2002. [Google Scholar] [CrossRef] [Green Version]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013. [Google Scholar] [CrossRef]
- Taura, M.; Eguma, A.; Suico, M.A.; Shuto, T.; Koga, T.; Komatsu, K.; Komune, T.; Sato, T.; Saya, H.; Li, J.-D.; et al. p53 Regulates Toll-Like Receptor 3 Expression and Function in Human Epithelial Cell Lines. Mol. Cell. Biol. 2008, 28, 6557–6567. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Wei, J.; Deng, X.; Shi, Z.; Zhu, Z.; Shao, D.; Li, B.; Wang, S.; Tong, G.; Ma, Z. Transcriptional analysis of immune-related gene expression in p53-deficient mice with increased susceptibility to influenza A virus infection. BMC Med. Genomics 2015. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. P53 and The Immune Response: 40 Years of Exploration-A Plan for the Future. Int. J. Mol. Sci. 2020, 21, 541. [Google Scholar] [CrossRef] [Green Version]
- Collot-Teixeira, S.; Bass, J.; Denis, F.; Ranger-Rogez, S. Human tumor suppressor p53 and DNA viruses. Rev. Med. Virol. 2004, 14, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.T.; Rebouillat, D.; Ramana, C.V.; Murakami, J.; Hill, J.E.; Gudkov, A.; Silverman, R.H.; Stark, G.R.; Williams, B.R.G. Down-Regulation of p53 by Double-Stranded RNA Modulates the Antiviral Response. J. Virol. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, D.; Lowe, J.M.; Snipe, J.; Resnick, M.A. Ligand dependent restoration of human TLR3 signaling and death in p53 mutant cells. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Reyes, S.; Marín, L.; González, L.; González, L.O.; Del Casar, J.M.; Lamelas, M.L.; González-Quintana, J.M.; Vizoso, F.J. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer 2010. [Google Scholar] [CrossRef] [Green Version]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like receptor’s two-edged sword: When immunity meets apoptosis. Eur. J. Immunol. 2007, 37, 3311–3318. [Google Scholar] [CrossRef]
- Barr, T.A.; Brown, S.; Ryan, G.; Zhao, J.; Gray, D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur. J. Immunol. 2007. [Google Scholar] [CrossRef]
- Rahman, A.H.; Taylor, D.K.; Turka, L.A. The contribution of direct TLR signaling to T cell responses. Immunol. Res. 2009, 45, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016. [Google Scholar] [CrossRef] [Green Version]
- González-Reyes, S.; Fernández, J.M.; González, L.O.; Aguirre, A.; Suárez, A.; González, J.M.; Escaff, S.; Vizoso, F.J. Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol. Immunother. 2011. [Google Scholar] [CrossRef]
- Cen, X.; Liu, S.; Cheng, K. The role of toll-like receptor in inflammation and tumor immunity. Front. Pharmacol. 2018, 9, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mahony, D.S.; Pham, U.; Iyer, R.; Hawn, T.R.; Liles, W.C. Differential constitutive and cytokine-modulated expression of human Toll-like receptors in primary neutrophils, monocytes, and macrophages. Int. J. Med. Sci. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzio, M.; Bosisio, D.; Polentarutti, N.; D’amico, G.; Stoppacciaro, A.; Mancinelli, R.; van’t Veer, C.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; et al. Differential Expression and Regulation of Toll-Like Receptors (TLR) in Human Leukocytes: Selective Expression of TLR3 in Dendritic Cells. J. Immunol. 2000. [Google Scholar] [CrossRef] [Green Version]
- Bell, M.P.; Svingen, P.A.; Rahman, M.K.; Xiong, Y.; Faubion, W.A. Forkhead Box P3 Regulates TLR10 Expression in Human T Regulatory Cells. J. Immunol. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsatsanis, C.; Androulidaki, A.; Alissafi, T.; Charalampopoulos, I.; Dermitzaki, E.; Roger, T.; Gravanis, A.; Margioris, A.N. Corticotropin-Releasing Factor and the Urocortins Induce the Expression of TLR4 in Macrophages via Activation of the Transcription Factors PU.1 and AP-1. J. Immunol. 2006. [Google Scholar] [CrossRef] [Green Version]
- Tomso, D.J.; Inga, A.; Menendez, D.; Pittman, G.S.; Campbell, M.R.; Storici, F.; Bell, D.A.; Resnick, M.A. Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatz, M.; Menendez, D.; Resnick, M.A. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012. [Google Scholar] [CrossRef] [Green Version]
- Shatz, M.; Shats, I.; Menendez, D.; Resnick, M.A. p53 amplifies Toll-like receptor 5 response in human primary and cancer cells through interaction with multiple signal transduction pathways. Oncotarget 2015, 6, 16963–16980. [Google Scholar] [CrossRef] [Green Version]
- So, E.Y.; Ouchi, T. The application of toll like receptors for cancer therapy. Int. J. Biol. Sci. 2010, 6, 675. [Google Scholar] [CrossRef] [Green Version]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [Green Version]
- Pradere, J.P.; Dapito, D.H.; Schwabe, R.F. The Yin and Yang of Toll-like receptors in cancer. Oncogene 2014, 33, 3485–3495. [Google Scholar] [CrossRef] [Green Version]
- Zeromski, J.; Mozer-Lisewska, I.; Kaczmarek, M. Significance of toll-like receptors expression in tumor growth and spreading: A short review. Cancer Microenviron. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korneev, K.V.; Atretkhany, K.S.N.; Drutskaya, M.S.; Grivennikov, S.I.; Kuprash, D.V.; Nedospasov, S.A. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine 2017, 89, 127–135. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, Q.; Wang, L.; Chen, W.; Li, N.; Cao, X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol. Immunol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.L.; Qian, Z.R.; Nakasono, M.; Tanahashi, T.; Yoshimoto, K.; Bando, Y.; Kudo, E.; Shimada, M.; Sano, T. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br. J. Cancer 2010. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhao, F.; Zhang, H.; Zhu, Y.; Wu, K.; Tan, G. Significance of TLR4/MyD88 expression in breast cancer. Int. J. Clin. Exp. Pathol. 2015. [Google Scholar]
- Yang, H.; Wang, B.; Wang, T.; Xu, L.; He, C.; Wen, H.; Yan, J.; Su, H.; Zhu, X. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Cortés-Ramírez, D.A.; Gainza-Cirauqui, M.L.; Echebarria-Goikouria, M.A.; Aguirre-Urizar, J.M. Oral lichenoid disease as a premalignant condition: The controversies and the unknown. Med. Oral Patol. Oral Cir. Bucal 2009, 14, E118–E122. [Google Scholar]
- Rusanen, P.; Marttila, E.; Uittamo, J.; Hagström, J.; Salo, T.; Rautemaa-Richardson, R. TLR1-10, NF-κB and p53 expression is increased in oral lichenoid disease. PLoS ONE 2017. [Google Scholar] [CrossRef] [Green Version]
- Teselkina, Y.O.; Khorevaa, M.V.; Veselovaa, A.V.; Babenkovaa, I.V.; Osipova, A.N.; Gankovskaya, L.V.; Vladimirova, Y.A. No TitleTLR-Mediated Production of Reactive Oxygen Species and Tumor Necrosis Factor Alpha by Human Peripheral Blood Neutrophils. Biophysics (Oxf) 2018, 63, 187–192. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Haricharan, S.; Brown, P. TLR4 has a TP53-dependent dual role in regulating breast cancer cell growth. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.V.; Seiva, F.R.F.; Carniato, A.P.; de Mello Júnior, W.; Duran, N.; Macedo, A.M.; de Oliveira, A.G.; Romih, R.; da Silva Nunes, I.; da Silva Nunes, O.; et al. Increased toll-like receptors and p53 levels regulate apoptosis and angiogenesis in non-muscle invasive bladder cancer: Mechanism of action of P-MAPA biological response modifier. BMC Cancer 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, A.; Starace, D.; Galli, R.; Padula, F.; De Cesaris, P.; Filippini, A.; Ziparo, E.; Riccioli, A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-α-dependent mechanism. Carcinogenesis 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taura, M.; Fukuda, R.; Suico, M.A.; Eguma, A.; Koga, T.; Shuto, T.; Sato, T.; Morino-Koga, S.; Kai, H. TLR3 induction by anticancer drugs potentiates poly I:C-induced tumor cell apoptosis. Cancer Sci. 2010. [Google Scholar] [CrossRef]
- Hashimoto, A.; Oikawa, T.; Hashimoto, S.; Sugino, H.; Yoshikawa, A.; Otsuka, Y.; Handa, H.; Onodera, Y.; Nam, J.M.; Oneyama, C.; et al. P53- and mevalonate pathway-driven malignancies require Arf6 for metastasis and drug resistance. J. Cell Biol. 2016. [Google Scholar] [CrossRef]
- Hashimoto, S.; Furukawa, S.; Hashimoto, A.; Tsutaho, A.; Fukao, A.; Sakamura, Y.; Parajuli, G.; Onodera, Y.; Otsuka, Y.; Handa, H.; et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [Green Version]
- Handa, H.; Hashimoto, A.; Hashimoto, S.; Sugino, H.; Oikawa, T.; Sabe, H. Epithelial-specific histone modification of the miR-96/182 locus targeting AMAP1 mRNA predisposes p53 to suppress cell invasion in epithelial cells. Cell Commun. Signal. 2018. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Peng, C.; Zhang, X.; Wu, Y.; Pan, S.; Xiao, Y. Roles of Arf6 in cancer cell invasion, metastasis and proliferation. Life Sci. 2017, 182, 80–84. [Google Scholar] [CrossRef]
- Yoo, J.H.; Brady, S.W.; Acosta-Alvarez, L.; Rogers, A.; Peng, J.; Sorensen, L.K.; Wolff, R.K.; Mleynek, T.; Shin, D.; Rich, C.P.; et al. The small GTPase ARf6 activates PI3K in melanoma to induce a prometastatic state. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Van Acker, T.; Tavernier, J.; Peelman, F. The small GTPase Arf6: An overview of its mechanisms of action and of its role in host- pathogen interactions and innate immunity. Int. J. Mol. Sci. 2019, 20, 2209. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Liu, T.; Zhang, H.; Tang, S.; Min, W. AIP1 functions as Arf6-GAP to negatively regulate TLR4 signaling. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [Green Version]
- Van Acker, T.; Eyckerman, S.; Vande Walle, L.; Gerlo, S.; Goethals, M.; Lamkanfi, M.; Bovijn, C.; Tavernier, J.; Peelman, F. The small GTPase Arf6 is essential for the Tram/Trif pathway in TLR4 signaling. J. Biol. Chem. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of Reactive Oxygen Species in Toll-Like Receptor 4-Dependent Activation of NF-κB. J. Immunol. 2004. [Google Scholar] [CrossRef] [Green Version]
- Vallance, T.M.; Zeuner, M.T.; Williams, H.F.; Widera, D.; Vaiyapuri, S. Toll-Like Receptor 4 Signalling and Its Impact on Platelet Function, Thrombosis, and Haemostasis. Mediators Inflamm. 2017, 2017, 9605894. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris IV, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399. [Google Scholar] [CrossRef]
- Zheng, S.J.; Lamhamedi-Cherradi, S.E.; Wang, P.; Xu, L.; Chen, Y.H. Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Que, K.T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.P.; Liu, Z.J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ng, D.S.W.; Mah, W.C.; Almeida, F.F.; Rahmat, S.A.; Rao, V.K.; Leow, S.C.; Laudisi, F.; Peh, M.T.; Goh, A.M.; et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Wong, E.T.; Bist, P.; Tergaonkar, V.; Lane, D.P. Nutlin-3 inhibits the NFκB pathway in a p53-dependent manner: Implications in lung cancer therapy. Cell Cycle 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.Y.; Xiang, C.; Zhang, C.X.; Xie, Y.Y.; Chen, L.; Zhang, G.X.; Lu, Y.; Liu, G. P53 in the Myeloid Lineage Modulates an Inflammatory Microenvironment Limiting Initiation and Invasion of Intestinal Tumors. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yu, F.; Ding, H.; Wang, Y.; Li, P.; Wang, K. Emerging Function and Clinical Values of Exosomal MicroRNAs in Cancer. Mol. Ther. Nucleic Acids 2019, 16, 791. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Kim, G.; An, H.J.; Lee, M.J.; Song, J.Y.; Jeong, J.Y.; Lee, J.H.; Jeong, H.C. Hsa-miR-1246 and hsa-miR-1290 are associated with stemness and invasiveness of non-small cell lung cancer. Lung Cancer 2016. [Google Scholar] [CrossRef]
- Shimomura, A.; Shiino, S.; Kawauchi, J.; Takizawa, S.; Sakamoto, H.; Matsuzaki, J.; Ono, M.; Takeshita, F.; Niida, S.; Shimizu, C.; et al. Novel combination of serum microRNA for detecting breast cancer in the early stage. Cancer Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.C.; Chin, T.M.; Yang, H.; Nga, M.E.; Lunny, D.P.; Lim, E.K.H.; Sun, L.L.; Pang, Y.H.; Leow, Y.N.; Malusay, S.R.Y.; et al. Tumour-initiating cell-specific MIR-1246 and MIR-1290 expression converge to promote non-small cell lung cancer progression. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Neerincx, M.; Sie, D.L.S.; Van De Wiel, M.A.; Van Grieken, N.C.T.; Burggraaf, J.D.; Dekker, H.; Eijk, P.P.; Ylstra, B.; Verhoef, C.; Meijer, G.A.; et al. MiR expression profiles of paired primary colorectal cancer and metastases by next-generation sequencing. Oncogenesis 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Niu, D.; Lai, L.; Ren, E.C. P53 increases MHC class i expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Sharma, M.D.; Rodriguez, P.C.; Koehn, B.H.; Baban, B.; Cui, Y.; Guo, G.; Shimoda, M.; Pacholczyk, R.; Shi, H.; Lee, E.J.; et al. Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c+CD103+ Monocytic Antigen-Presenting Cells in Tumors. Immunity 2018. [Google Scholar] [CrossRef] [Green Version]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, H.; Takatori, H.; Suzuki, K.; Iwata, A.; Yokota, M.; Suto, A.; Minamino, T.; Hirose, K.; Nakajima, H. Tumor Suppressor p53 Inhibits Systemic Autoimmune Diseases by Inducing Regulatory T Cells. J. Immunol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Immunogenomics Analysis Reveals that TP53 Mutations Inhibit Tumor Immunity in Gastric Cancer. Transl. Oncol. 2018, 11, 1171–1187. [Google Scholar] [CrossRef]
- Quigley, D.; Silwal-Pandit, L.; Dannenfelser, R.; Langerød, A.; Vollan, H.K.M.; Vaske, C.; Siegel, J.U.; Troyanskaya, O.; Chin, S.F.; Caldas, C.; et al. Lymphocyte invasion in IC10/basal-like breast tumors is associated with wild-type TP53. Mol. Cancer Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; Van Miltenburg, M.H.; Slagter, M.; De Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef]
- Schuldner, M.; Dörsam, B.; Shatnyeva, O.; Reiners, K.S.; Kubarenko, A.; Hansen, H.P.; Finkernagel, F.; Roth, K.; Theurich, S.; Nist, A.; et al. Exosome-dependent immune surveillance at the metastatic niche requires BAG6 and CBP/p300-dependent acetylation of p53. Theranostics 2019. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.W.; Byun, S.; Kwon, E.; Hwang, S.Y.; Chu, K.; Hiraki, M.; Jo, S.H.; Weins, A.; Hakroush, S.; Cebulla, A.; et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiem, A.; Hesbacher, S.; Kneitz, H.; Di Primio, T.; Heppt, M.V.; Hermanns, H.M.; Goebeler, M.; Meierjohann, S.; Houben, R.; Schrama, D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J. Exp. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Jung, D.J.; Jin, D.H.; Hong, S.W.; Kim, J.E.; Shin, J.S.; Kim, D.; Cho, B.J.; Hwang, Y.I.; Kang, J.S.; Lee, W.J. Foxp3 expression in p53-dependent DNA damage responses. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Thyagarajan, K.; Chatterjee, S.; Chakraborty, P.; Kesarwani, P.; Soloshchenko, M.; Al-Hommrani, M.; Andrijauskaite, K.; Moxley, K.; Janakiraman, H.; et al. Lack of p53 augments antitumor functions in cytolytic T cells. Cancer Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Jiang, Z.; Gao, Y.; Wang, L.; Chen, C.; Wang, X. TP53 Mutations Promote Immunogenic Activity in Breast Cancer. J. Oncol. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, N. Human Tumor Antigens and Cancer Immunotherapy. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Lubin, R.; Zalcman, G.; Bouchet, L.; Trédaniel, J.; Legros, Y.; Cazals, D.; Hirsch, A.; Soussi, T. Serum p53 antibodies as early markers of lung cancer. Nat. Med. 1995. [Google Scholar] [CrossRef]
- Garziera, M.; Montico, M.; Bidoli, E.; Scalone, S.; Sorio, R.; Giorda, G.; Lucia, E.; Toffoli, G. Prognostic role of serum antibody immunity to p53 oncogenic protein in ovarian cancer: A systematic review and a meta-analysis. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Labrecque, S.; Naor, N.; Thomson, D.; Matlashewski, G. Analysis of the Anti-p53 Antibody Response in Cancer Patients. Cancer Res. 1993, 53, 3468–3471. [Google Scholar] [PubMed]
- Volkmann, M.; Müller, M.; Hofmann, W.J.; Meyer, M.; Hagelstein, J.; Räth, U.; Kommerell, B.; Zentgraf, H.; Galle, P.R. The humoral immune response to p53 in patients with hepatocellular carcinoma is specific for malignancy and independent of the α-fetoprotein status. Hepatology 1993. [Google Scholar] [CrossRef]
- Soussi, T. p53 Antibodies in the sera of patients with various types of cancer: A review. Cancer Res. 2000, 60, 1777–1788. [Google Scholar] [PubMed]
- Lane, D.P.; Brown, C.J.; Verma, C.; Cheok, C.F. New insights into p53 based therapy. Discov. Med. 2011, 12, 107–117. [Google Scholar] [PubMed]
- Yen, N.; Ioannides, C.G.; Xu, K.; Swisher, S.G.; Lawrence, D.D.; Kemp, B.L.; El-Naggar, A.K.; Cristiano, R.J.; Fang, B.; Glisson, B.S.; et al. Cellular and humoral immune responses to adenovirus and p53 protein antigens in patients following intratumoral injection of an adenovirus vector expressing wild-type p53 (Ad-p53). Cancer Gene Ther. 2000. [Google Scholar] [CrossRef]
- Nijman, H.W.; Lambeck, A.; van der Burg, S.H.; van der Zee, A.G.J.; Daemen, T. Immunologic aspect of ovarian cancer and p53 as tumor antigen. J. Transl. Med. 2005, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suppiah, A.; Greenman, J. Clinical utility of anti-p53 auto-antibody: Systematic review and focus on colorectal cancer. World J. Gastroenterol. 2013, 19, 4651. [Google Scholar] [CrossRef]
- Yanuck, M.; Carbone, D.P.; David Pendleton, C.; Tsukui, T.; Winter, S.F.; Minna, J.D.; Berzofsky, J.A. A Mutant p53 Tumor Suppressor Protein Is a Target for Peptide-induced CD8+ Cytotoxic T-Cells. Cancer Res. 1993, 53, 3257–3261. [Google Scholar]
- Houbiers, J.G.A.; Nijman, H.W.; Van Der Burg, S.H.; Drijfhout, J.W.; Kenemans, P.; Van De Velde, C.J.H.; Brand, A.; Momburg, F.; Kast, W.M.; Melief, C.J.M. In vitro induction of human cytotoxic T lymphocyte responses against peptides of mutant and wild-type p53. Eur. J. Immunol. 1993. [Google Scholar] [CrossRef]
- Röpke, M.; Hald, J.; Guldberg, P.; Zeuthen, J.; Nørgaard, L.; Fugger, L.; Svejgaard, A.; Van Der Burg, S.; Nijmanu, H.W.; Melief, C.J.M.; et al. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53-derived peptide. Proc. Natl. Acad. Sci. USA 1996. [Google Scholar] [CrossRef] [Green Version]
- Sirianni, N.; Ha, P.K.; Oelke, M.; Califano, J.; Gooding, W.; Westra, W.; Whiteside, T.L.; Koch, W.M.; Schneck, J.P.; DeLeo, A.; et al. Effect of human papillomavirus-16 infection on CD8+ T-cell recognition of a wild-type sequence p53264-272 peptide in patients with squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theobald, M.; Biggs, J.; Dittmer, D.; Levine, A.J.; Sherman, L.A. Targeting p53 as a general tumor antigen. Proc. Natl. Acad. Sci. USA 1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Senju, S.; Yokomizo, H.; Saya, H.; Ogawa, M.; Matsushita, S.; Nishimura, Y. Evidence that HLA class II-restricted human CD4+ T cells specific to p53 self peptides respond to p53 proteins of both wild and mutant forms. Eur. J. Immunol. 1998. [Google Scholar] [CrossRef]
- Couch, M.E.; Ferris, R.L.; Brennan, J.A.; Koch, W.M.; Jaffee, E.M.; Leibowitz, M.S.; Nepom, G.T.; Erlich, H.A.; Sidransky, D. Alteration of cellular and humoral immunity by mutant p53 protein and processed mutant peptide in head and neck cancer. Clin. Cancer Res. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamalov, K.; Levy, S.N.; Horovitz-Fried, M.; Cohen, C.J. The mutational status of p53 can influence its recognition by human T-cells. Oncoimmunology 2017. [Google Scholar] [CrossRef] [Green Version]
- Carbone, D.P.; Ciernik, I.F.; Kelley, M.J.; Smith, M.C.; Nadaf, S.; Kavanaugh, D.; Maher, V.E.; Stipanov, M.; Contois, D.; Johnson, B.E.; et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: Immune response and clinical outcome. J. Clin. Oncol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Albers, A.E.; Ferris, R.L.; Kim, G.G.; Chikamatsu, K.; DeLeo, A.B.; Whiteside, T.L. Immune responses to p53 in patients with cancer:enrichment in tetramer+ p53 peptide-specific T cells and regulatory T cells at tumor sites. Cancer Immunol. Immunother. 2005. [Google Scholar] [CrossRef]
- Cohen, C.J.; Zheng, Z.; Bray, R.; Zhao, Y.; Sherman, L.A.; Rosenberg, S.A.; Morgan, R.A. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J. Immunol. 2006. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell responses to TP53 “Hotspot” Mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Crawford, L.V.; Pim, D.C.; Bulbrook, R.D. Detection of antibodies against the cellular protein p53 in sera from patients with breast cancer. Int. J. Cancer 1982. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.B. p53-based immunotherapy of cancer. Adv. Otorhinolaryngol. 2005. [Google Scholar] [CrossRef]
- Lauwen, M.M.; Zwaveling, S.; De Quartel, L.; Ferreira Mota, S.C.; Grashorn, J.A.C.; Melief, C.J.M.; Van Der Burg, S.H.; Offringa, R. Self-tolerance does not restrict the CD4+ T-helper response against the p53 tumor antigen. Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeo, A.B.; Whiteside, T.L. Development of multi-epitope vaccines targeting wild-type-sequence p53 peptides. Expert Rev. Vaccines 2008, 7, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijman, H.W.; Vermeij, R.; Leffers, N.; Van Der Burg, S.H.; Melief, C.J.; Daemen, T. Immunological and clinical effects of vaccines targeting p53-overexpressing malignancies. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef]
- Humar, M.; Azemar, M.; Maurer, M.; Groner, B. Adaptive Resistance to Immunotherapy Directed Against p53 Can be Overcome by Global Expression of Tumor-Antigens in Dendritic Cells. Front. Oncol. 2014. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Kroemer, G. A p53-regulated immune checkpoint relevant to cancer. Science 2015. [Google Scholar] [CrossRef]
Figure 1.
Mutant p53 feeds off and fuels inflammation. Wild-type p53 (Wt p53) contributes to effective tumour immunity through its roles in senescence, apoptosis, autophagy and ROS regulation, as well as established roles in regulating immune response. Mutant p53 is able to subvert these effects through dominant negative effects and novel tumourigenic functions mediated through the inflammatory pathways of NF-κB and signal transducer and activator of transcription 3 (STAT3). The excessive production of reactive oxygen species (ROS) can fuel inflammatory microenvironments without the regulation of wt p53, consequently feeding mutant p53 function and tipping the scales toward tumorigenesis. It is worth noting that innate immunity is not included in this model and is discussed later on.
Figure 1.
Mutant p53 feeds off and fuels inflammation. Wild-type p53 (Wt p53) contributes to effective tumour immunity through its roles in senescence, apoptosis, autophagy and ROS regulation, as well as established roles in regulating immune response. Mutant p53 is able to subvert these effects through dominant negative effects and novel tumourigenic functions mediated through the inflammatory pathways of NF-κB and signal transducer and activator of transcription 3 (STAT3). The excessive production of reactive oxygen species (ROS) can fuel inflammatory microenvironments without the regulation of wt p53, consequently feeding mutant p53 function and tipping the scales toward tumorigenesis. It is worth noting that innate immunity is not included in this model and is discussed later on.

Figure 2.
Mutant p53 alters its immunogenic niche. The tumour microenvironment is comprised of cellular and molecular components which shape the immune niche of the growing tumour. (A) Normal fibroblasts help mediate immunosurveillance by modulating anti-tumour immune cell infiltration and employing the IFN-β/p53 axis of regulation to activate anti-tumour responses. (B) Mutant p53 in the tumour alters the crosstalk between the tumour and its microenvironment, suppressing IFN-β signalling and supporting pro-angiogenic signalling. This altered crosstalk converts normal fibroblasts into tumour-supporting cancer-associated fibroblasts with altered p53 functionality. The altered transcriptional program of cancer-associated fibroblasts (CAFs) supports pro-tumorigenic signalling and extracellular matrix remodelling. The demographic and functionality of infiltrating immune cells is consequently shifted to favour immunosuppression, ultimately leading to a chronically inflamed environment and enhanced migration and invasion.
Figure 2.
Mutant p53 alters its immunogenic niche. The tumour microenvironment is comprised of cellular and molecular components which shape the immune niche of the growing tumour. (A) Normal fibroblasts help mediate immunosurveillance by modulating anti-tumour immune cell infiltration and employing the IFN-β/p53 axis of regulation to activate anti-tumour responses. (B) Mutant p53 in the tumour alters the crosstalk between the tumour and its microenvironment, suppressing IFN-β signalling and supporting pro-angiogenic signalling. This altered crosstalk converts normal fibroblasts into tumour-supporting cancer-associated fibroblasts with altered p53 functionality. The altered transcriptional program of cancer-associated fibroblasts (CAFs) supports pro-tumorigenic signalling and extracellular matrix remodelling. The demographic and functionality of infiltrating immune cells is consequently shifted to favour immunosuppression, ultimately leading to a chronically inflamed environment and enhanced migration and invasion.

Figure 3.
Mutant p53 impacts on the immune and inflammatory hallmarks of cancer. Mutant p53 responds to and contributes to cancer-associated chronic inflammation which facilitates several immune gain-of-functions. These are highlighted by the mutant p53-mediated alteration of the tumour milieu (pro-invasive extracellular matrix structure, cancer-associated fibroblast activity, tumour-tolerant immune cell infiltrate and chemical signatures), disabling of the innate immune response through aberrant toll-like receptor signalling, the inhibition of cell-mediated cancer immunity and potentially disrupting post-apoptotic cell clearance.
Figure 3.
Mutant p53 impacts on the immune and inflammatory hallmarks of cancer. Mutant p53 responds to and contributes to cancer-associated chronic inflammation which facilitates several immune gain-of-functions. These are highlighted by the mutant p53-mediated alteration of the tumour milieu (pro-invasive extracellular matrix structure, cancer-associated fibroblast activity, tumour-tolerant immune cell infiltrate and chemical signatures), disabling of the innate immune response through aberrant toll-like receptor signalling, the inhibition of cell-mediated cancer immunity and potentially disrupting post-apoptotic cell clearance.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Agupitan, A.D.; Neeson, P.; Williams, S.; Howitt, J.; Haupt, S.; Haupt, Y. P53: A Guardian of Immunity Becomes Its Saboteur through Mutation. Int. J. Mol. Sci. 2020, 21, 3452. https://doi.org/10.3390/ijms21103452
AMA Style
Agupitan AD, Neeson P, Williams S, Howitt J, Haupt S, Haupt Y. P53: A Guardian of Immunity Becomes Its Saboteur through Mutation. International Journal of Molecular Sciences. 2020; 21(10):3452. https://doi.org/10.3390/ijms21103452
Chicago/Turabian StyleAgupitan, Arjelle Decasa, Paul Neeson, Scott Williams, Jason Howitt, Sue Haupt, and Ygal Haupt. 2020. "P53: A Guardian of Immunity Becomes Its Saboteur through Mutation" International Journal of Molecular Sciences 21, no. 10: 3452. https://doi.org/10.3390/ijms21103452
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.