Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures


1
Department of Neuroscience, Central Clinical School, Monash University, Melbourne, Victoria 3004, Australia
2
Department of Neurology, Alfred Health, Melbourne, Victoria 3004, Australia
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(10), 3676; https://doi.org/10.3390/ijms21103676
Submission received: 17 April 2020
/
Revised: 20 May 2020
/
Accepted: 20 May 2020
/
Published: 23 May 2020
(This article belongs to the Special Issue Neuronal, Glial, and Immune Changes in Models of Epilepsy and Epileptogenesis)
{kind=link}
{kind=link}
Abstract
:Alzheimer’s disease (AD) is the most common form of dementia. An increasing body of evidence describes an elevated incidence of epilepsy in patients with AD, and many transgenic animal models of AD also exhibit seizures and susceptibility to epilepsy. However, the biological mechanisms that underlie the occurrence of seizure or increased susceptibility to seizures in AD is unknown. Glycogen synthase kinase-3 (GSK-3) is a serine/threonine kinase that regulates various cellular signaling pathways, and plays a crucial role in the pathogenesis of AD. It has been suggested that GSK-3 might be a key factor that drives epileptogenesis in AD by interacting with the pathological hallmarks of AD, amyloid precursor protein (APP) and tau. Furthermore, seizures may also contribute to the progression of AD through GSK-3. In this way, GSK-3 might be involved in initiating a vicious cycle between AD and seizures. This review aims to summarise the possible role of GSK-3 in the link between AD and seizures. Understanding the role of GSK-3 in AD-associated seizures and epilepsy may help researchers develop new therapeutic approach that can manage seizure and epilepsy in AD patients as well as decelerate the progression of AD.
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia and is clinically characterised by cognitive impairment and progressive loss of memory. It has been estimated that one in every 85 individuals worldwide will be living with AD by 2050 [1], imposing a tremendous psychological and financial burden on the patient’s family and the community. There is currently no cure for AD, and treatments are mostly symptomatic [2].
Recent evidence has raised the possibility of a connection between AD and epilepsy, another common CNS disorder in the elderly that is characterised by an enduring predisposition to generate unprovoked seizures. Multiple studies have reported an increased incidence of unprovoked seizures in AD patients [3,4,5]. For example, prospective studies showed that AD patients are 6- to 10-fold more likely to develop seizures than age-matched controls [4,5], while it has been estimated that 10%–17% of AD patients have unprovoked seizures, suggesting that AD itself might be a risk factor of epilepsy [3,6]. Additionally, it is reported that unprovoked seizures might aggravate cognitive deficits of AD patients [7,8], which suggests that recurrent seizures lead to a vicious cycle of cognition exacerbation in AD patients.
In addition to the clinical evidence, animal models harbouring AD-associated mutations provide a useful insight on the possible causation and mechanisms underlying AD-associated epilepsy [9]. There are two main pathological hallmarks in AD, senile plaques and neurofibrillary tangles (NFT), the major molecular constituents of which are the amyloid precursor protein (APP) and microtubule-associated protein tau (MAPT), respectively [10]. Several lines of evidence describe AD-relevant mouse models, particularly those carrying APP or tau mutations, as being more susceptible of developing unprovoked seizures and induced seizures [11,12,13,14]. Others have shown that the increase of seizure susceptibility in APP mouse model was dependent on the expression of tau [15,16], which suggests that intervening the interaction between amyloid and tau might have therapeutic potential for epilepsy management in AD patients. Therefore, it is important to clarify the interaction between APP and tau and their implication in AD-associated seizures.
Glycogen synthase kinase-3 (GSK-3) is a proline-rich serine/threonine kinase which plays a pivotal role in the pathogenesis of AD [17,18,19]. It is suggested that GSK-3 may be a critical connecting factor between amyloid and tau [20,21,22], as amyloid activates GSK-3, which subsequently phosphorylates tau, the process of which is shown in Figure 1. Since the occurrence of seizures might be related to the interaction between amyloid and tau, it is likely that GSK-3 may play a unique role in the development of unprovoked seizures in AD. Therefore, a better understanding of the role of GSK-3 in connecting AD and seizures would be helpful for understanding causal mechanisms and for improving seizure management in AD patients.
In this article, we will review the possible mechanisms of AD-related seizures, present hypothesised pathways of the role of GSK-3 in the development of seizures in AD, and propose future research to clarify the role of GSK-3 in this process. It is worth noting that multiple studies using AD animal models employed models that harbour familial AD mutations and, thus, the neuropathological process in the context of familial AD. However, that does not exclude patients with sporadic AD from the possibility of similar pathological changes.
2. AD-Associated Epilepsy and Epileptic Seizures
There is growing clinical evidence suggestive of an increased risk of epilepsy or epileptic seizures in AD. Higher incidence of unprovoked seizures in AD patients has been reported by numerous studies [3,23,24,25]. It has been shown that seizure-free AD patients are more likely to develop unprovoked seizures compared to age-match controls [4,6]. Furthermore, evidence showed that familial AD was strongly associated with epileptogenesis [25,26], indicating that some AD-related mutations might contribute to abnormal neuronal network functions that lead to the development of epilepsy. However, it remains unclear whether seizure is an integral part of AD. There are a few confounding factors that need to be considered. On the one hand, the causal relationship is far from conclusive as the onset of seizures might merely be concurrent with severe AD or consequential to AD symptoms instead of AD per se. It is reported that about 22% of patients with dementia might conduct self-injurious behaviors [27]. Poor judgement and impaired consciousness can also lead to epilepsy-causing injuries in AD patients. On the other hand, it is also possible that the incidence of seizures is underestimated. AD patients might develop nonconvulsive seizures that can be misdiagnosed as AD symptoms such as altered consciousness or amnestic wandering [8]. Using long-term electroencephalogram (EEG) recording, clinically silent seizures were detected in AD patients [28,29], indicating that subtle seizures might not be documented in some AD patients without routine EEG recording. Therefore, the causal relationship and the underlying mechanism between AD and epilepsy need to be further clarified in prospective clinical studies.
In parallel to the clinical evidence, many transgenic mouse models carrying identified AD mutations also show unprovoked seizures or epileptiform EEG patterns. Although AD mouse models cannot fully recapitulate the pathological features of AD, they serve as controllable models of the disease with one or few genetic modifications that manifest AD-associated symptoms. The most well-studied models are mice that overexpress human mutant APP or specific APP cleavage products such as amyloid intracellular domain (AICD). The process of APP cleavage is shown in Figure 1A. Several APP models with various mutations exhibit spontaneous seizures or epileptiform discharges [13,14,30,31,32], while the genetic suppression of APP can abate epileptiform activities in APP transgenic mice [33], suggesting that APP is involved in the development of seizure. Furthermore, transgenic mice overexpressing AICD, the intracellular domain of APP, also exhibit EEG abnormalities and increased susceptibility to induced seizure [34]. In addition, APP transgenic models with a C-terminus cleavage site mutation which prevents the cleavage of AICD showed a complete reversal of AD phenotypes susceptibility to induced-seizure [34,35,36], suggesting that AICD might be the excitotoxic fragment of APP that contributes to the development of AD phenotypes and seizures in AD models. Overall, a vast literature documents consistent evidence that mutated or overexpressed APP or its cleavage products can result in seizures.
Aside from the potential immediate consequences of epileptic seizures, such as physical injuries, to AD patients, it is found that the occurrence of seizures might also be relevant to the acceleration of cognition impairment, which creates a complex bidirectional association between AD and seizures. The cognitive functions of patients with AD or other dementias may abruptly worsen after the first onset of seizure [37,38], suggesting a short-term impact on cognitive deficit. In addition, it has also been reported that AD patients with seizures had an earlier onset of cognitive decline and worse cognitive outcome compared to AD patients without seizures [30,39,40], suggesting that seizures might also have long-term effects on cognition. Similar to the clinical observation, studies on animal models also showed worsened cognitive conditions in animals with seizures [39]. Further studies suggested that epileptiform activity might disrupt memory consolidation and cause cognitive impairment in animal models [30,40,41]. Strikingly, multiple lines of evidence showed that antiepileptic drugs (AEDs), which can prevent these seizures and/or epileptiform discharges, seem capable of rescuing the cognitive deficit in AD models [42,43,44,45]. These studies highlight the significance of seizures and epilepsy in the development of AD, which also points to a potentially new therapeutic strategy for decelerating the cognitive deficit in AD patients.
3. GSK-3 Is Involved in the Development of Seizures in AD
GSK-3 is a ubiquitously expressed kinase that is involved in various physiological and pathological processes. GSK-3 was originally found as a key kinase that phosphorylated glycogen synthase during glycogen accumulation, a fundamental process of energy storage [46]. There are two genes coding for the GSK-3 protein, from which two homologous forms are expressed, GSK-3α and GSK-3β. Similar to most kinases, the activity of GSK-3 is regulated by phosphorylation at certain sites. The inhibition of GSK-3 is regulated by phosphorylation at serine residue near N-terminus (Ser9 of GSK-3β and Ser21 of GSK-3α) [47]. When GSK-3 is activated, the tyrosine site of GSK-3 will be auto-phosphorylated (Tyr216 of GSK-3β and Tyr279 of GSK-3α) [48]. Although GSK-3α shares 98% sequence identity with GSK-3β [49], most studies have focused on GSK-3β and the functions of GSK-3α are not well understood. It is possible that GSK-3β serves a more fundamental physiological function because the GSK-3β knockout is lethal [50], whereas knockout of GSK-3α is tolerable [51]. GSK-3 is involved in multiple molecular events including cell signalling [52], maintaining cellular structure [53], and modulation of transcription [52]. Other studies suggested that GSK-3 might also be involved in epileptogenesis [54].
The role of GSK-3 in the pathogenesis of AD has been discussed in recent reviews [17,55,56,57]. GSK-3 was initially known as one of the key kinases that was involved in the phosphorylation of tau [46,58,59], which is a biological process that regulates the binding of tau to microtubules, as being shown in Figure 1C [60]. However, GSK-3 is excessively activated in AD, which contributes to the hyper-phosphorylation of tau [60]. Hyper-phosphorylated tau has been identified as one of the critical factors and causal drivers of pathogenesis of AD [61,62]. Furthermore, studies showed that hyper-phosphorylation of tau also plays a role in epileptogenesis and ictogenesis in AD. It is reported that the hyper-phosphorylation of tau is implicated in the development of seizures in AD model [63] while reduction of tau or tau phosphorylation supresses seizure and impedes the development of epilepsy [64,65,66,67]. This body of evidence suggests a mechanistic relationship between tau phosphorylation and development of seizures and epilepsy. It is likely that GSK-3, as one of the main kinases of tau, plays a critical role in this process. Furthermore, studies suggested that GSK-3 might be a downstream regulator of other tau kinases such as cyclin-dependent kinase 5 (CDK5) as well as protein phosphatase 2A (PP2A) [68,69,70], which suggested the potential regulating role of GSK-3 in tau phosphorylation. Therefore, GSK-3 represents an attractive target of intervention in AD-associated seizures, such that inhibiting the activity of GSK-3 might have a therapeutic potential in ameliorating or suppressing epileptic seizures in AD models. Although the therapeutic potential of GSK-3 inhibitor in AD patients has not been clarified, several GSK-3 inhibitors such as AZD1080 and Tideglusib are undergoing clinical trial [71,72].
In addition, studies showed that GSK-3 might be involved in AD-associated seizures through alternate pathways. Multiple studies have shown that GSK-3β can be associated with the activity of Fyn [73,74,75], a tyrosine kinase involved in mediating seizure susceptibility through activation of N-methyl-D-aspartate (NMDA receptors which is an ion channel modulating the influx of calcium [76]. Fyn could phosphorylate a unique site Tyr18 (Y18) of tau [77], and Y18-phosphorylated tau is implicated in tau-related NMDA receptor overactivation and excitotoxicity in vitro [78]. Y18-phosphorylated tau subsequently over-activates NMDA receptors and causes excitotoxicity and eventually seizures. Taken together, these results might lead to another pathway of seizure development that involves GSK-3 in AD (Figure 2). However, it is worth mentioning that evidence in physiological condition is required to prove this hypothesis. Studies in AD animal models are also important to connect the dots among GSK-3, Fyn, and AD.
4. Putative Interaction Between APP and GSK-3
Although the interaction between GSK-3 and tau has been widely studied, the mechanisms which initiate the dysregulation of GSK-3 activity potentially leading to subsequent seizures and epileptiform events in AD remain to be clarified. Multiple results from in vitro and in vivo experiments showed that GSK-3β might be activated by APP or its metabolites [57,79,80,81]. Studies found that the C-terminus fragment of APP, including AICD and its downstream cleavage product C31, increased the expression of GSK-3 [82,83]. Furthermore, other study also found the increase of activated GSK-3 in AICD overexpressing mice [84]. These lines of evidence showed that it might be this intracellular cleavage product instead of the extracellular β-amyloid peptides that activates GSK-3β (Figure 1B). However, further research is needed to clarify specifically which metabolite(s) of the APP protein are relevant.
Interestingly, some evidence showed that the AICD may also be pertinent to the occurrence of spontaneous seizures and increased susceptibility of induced seizures in animal models. It is reported that the cleavage of APP at the C-terminus may be related to neuronal network abnormality [34], while other studies suggested that the AICD from C-terminus cleavage is critical to neuronal network excitability, possibly leading to the increased spontaneous seizures in transgenic mice harbouring the human AICD gene [34,85]. Therefore, these studies established that AICD or the pathway that AICD is involved in may modulate neuronal excitability and epileptogenesis. Furthermore, there are some studies mentioning that the excitotoxic effect of AICD was exerted through GSK-3 [82,83] whereas inhibition of GSK-3 abated AD phenotypes in AICD transgenic mice [86], which suggests that GSK-3 might be part of the AICD pathway involved in AD phenotypes. However, if GKS-3 is involved in an AICD-related pathway, the relationship between GSK-3 and AICD is unclear, as the activation of GSK-3 in AICD transgenic mice does not clarify whether GSK-3 is upstream or downstream of AICD. On the one hand, observations that GSK-3 activation occurs after the expression of AICD in vitro [87] and in vivo [84,86] provide some lines of evidence that GSK-3 might be downstream of AICD. On the other hand, another report showed that GSK-3 acts upstream of AICD by regulating the activity of AICD through phosphorylation at site Thr668 (T668), thereby greatly enhancing binding of AICD and Fe65 [83], an adaptor protein that facilitates the functions of AICD [88]. As results from multiple studies showed that the binding between AICD and its binding partner Fe65 is essential for the pathogenic role of AICD in AD and epilepsy [34,86,88], it is possible that the activity of AICD is regulated by GSK-3. However, more research is required to prove the causal relationship between AICD and GSK-3, as direct evidence is still missing to clarify whether GSK-3 activity is involved in the development of seizures in AICD transgenic mice. It is possible that activation of GSK-3 and the development of seizures are two independent events in AICD transgenic mice. Therefore, further study is necessary to support the role of GSK-3 in epileptogenesis and ictogenesis in AD.
Some other studies suggest that GSK-3 might also be involved in the production of AICD through the cleavage of APP [55,89]. Phiel and colleagues showed that GSK-3 accelerated the cleavage of APP and the production of C-terminus fragments, whereas inhibition of GSK-3 reduces the production of C-terminus fragment [90]. Therefore, as the increased production of AICD is correlated to the development of seizures and epilepsy [34,85], it is possible that GSK-3 is implicated in epilepsy development through increasing the production of AICD. However, in what way GSK-3 is involved in the processing of APP has not been clarified to date. Although it has been reported that suppressing the expression of GSK-3α after birth of mice reduces the production of C-terminus fragments [90], other study showed that knockout of GSK-3α gene before birth does not alter the processing of APP [89]. This discrepancy can be explained by a compensatory mechanism that both GSK-3α and GSK-3β mediate the processing of APP. While inhibiting GSK-3α acutely decreases the cleavage of APP, the functions of GSK-3α might be overtaken by GSK-3β chronically. However, further research is needed to verify this mechanism.
Collectively, accumulating evidence suggests that there might be a GSK-3-amyloid-tau triad that induces the development of seizures and epilepsy in AD. However, the specific interaction between GSK-3 and AICD remains to be clarified. Answering these questions might help us understand the pathophysiology of AD-associated epilepsy as well as develop new therapeutic approaches to manage seizures and epilepsy in AD patients.
5. Alternative Roles of GSK-3 in AD and Seizures
As previously discussed, the occurrence of seizures may accelerate the progress of AD. Some evidence showed that GSK-3 might also be involved in this process. It is clear that GSK-3 plays an important role in the pathogenesis of AD through the phosphorylation of tau [56,91] and that inhibiting GSK-3 decelerates the progression of AD phenotypes in animal models [86]. Therefore, it is possible that seizures aggravate AD phenotypes by increasing GSK-3 activity and hyper-phosphorylated tau. The activation of GSK-3 and phosphorylation of tau have been shown in chemical and electrical induced epilepsy models [54,92,93]. However, the results of GSK-3 activation are not always consistent in post-seizure models. One study showed that GSK-3 was not activated 24 h after kainate-induced seizures [94], which might be explained by the observation that the activation of GSK-3 might last for less than 8 h [95]. Furthermore, some studies showed that there might be some regulatory factors working against the activation of GSK-3 after seizures. It is reported that GSK-3 could not be activated 24 h after seizures, while the activation of GSK-3 lasted longer than 24 h in Dopamine D2 receptor (D2R) knockout mice [94,96]. Interestingly, the loss of D2R was also reported in AD patients [97,98]. Lines of evidence suggest that D2R might suppress the activation of GSK-3 while decrease of D2R releases the suppression and causes the activation of GSK-3. However, the relevance of these results to AD still needs to be considered cautiously, as they were not conducted in AD animal models. More importantly, these studies do not specify whether it is seizures per se that directly activate GSK-3, as it might be the exogenous stimuli such as chemical or electrical stimulations that increase the activity of GSK-3. Therefore, it is too early to draw the conclusion that seizures exacerbate AD through GSK-3.
Based on the current evidences, it may be hypothesized that GSK-3 contributes to the “vicious cycle” between seizure and AD. After being activated in AD, GSK-3 might induce the occurrence of epileptic seizures, which could further increase the activity of GSK-3 and establish a positive feedback mechanism. Therefore, regulation of GSK-3 activity might be a potential intervention for both AD and epileptic seizures.
There are also multiple studies that link GSK-3 to seizures in non-AD models. One study showed that GSK-3β is an essential regulator of the cystine/glutamate antiporter xc-, a system that imports cystine and exports glutamate [97]. The disturbance of GSK-3β activity might lead to the spillover of glutamate, which may trigger spontaneous seizures [99]. Inhibition of GSK-3 showed anticonvulsant effect in rodents and zebrafish models of temporal lobe epilepsy [100]. However, some studies suggest that simply decreasing the activity of GSK-3 might not be the appropriate solution. Engel and colleagues showed that both increase and decrease of GSK-3β activity acutely aggravated the development of seizures in acquired seizure model [95]. Furthermore, a recent research showed that modified GSK-3β with increased activity decreased the progression of kainate-induced epileptogenesis [101], which was contradictory to many other results. Therefore, the specific role of GSK-3 in epileptic seizures and AD-associated epileptic seizures has to be further clarified.
6. Future Research Directions
There is increasing evidence supporting the cooccurrence of AD and seizures. To improve the understanding of both fields and to develop novel therapeutic approaches targeting AD-associated seizures, there are many questions to be addressed. Firstly, GSK-3 activity in AD models has not been well studied. Although some studies have found that GSK-3 could be activated in transgenic human APP mouse models [79,84], the results need to be interpreted cautiously. Some studies inferred GSK-3 activity by measuring protein levels of either activated GSK-3 (Y216/Y279) or inactivated GSK-3 (S9/S21), which might not necessarily correlate with the enzymatic activity of the protein. Therefore, it is important to accurately measure GSK-3 activity through a quantitative kinase activity assay.
Importantly, whether and at what time point GSK-3 is activated in AD models requires further study. To date, no study has reported the chronological analysis on the activity of GSK-3 in animal models carrying AD-relevant mutations. It is possible that GSK-3 activity is increased at an early age due to the gene mutation or the overexpression of genes, such as APP and AICD. The former is more likely to be relevant to human condition while the latter is limited to animal models, so carefully designed studies need to exclude simple overexpression of APP as a driving factor. In addition, a concern raised by Saito et al. suggested the possibility that some phenotypes in APP-overexpressing mice might come from membrane protein overexpression [102]. Furthermore, it is also likely that GSK-3 activity fluctuates over time in animal models. Notably, some studies showed that unprovoked seizures and epileptiform events could occur in AD transgenic models [14,31]. As epileptic seizures might also disturb the activity of GSK-3, it is possible that the GSK-3 activity also changes with the occurrence of seizures. Therefore, the variables that can change GSK-3 activity in AD have to be considered cautiously. It might be AD-associated seizures instead of AD mutations that induce the activation of GSK-3. Investigating GSK-3 activity at different timepoints with the monitoring of seizures might help us unravel the cause and effect relationship between AD phenotypes and GSK-3 activation.
Second, the roles of GSK-3 in AD-associated seizure need to be clarified. Although GSK-3 has been recognised as one of the contributors of AD pathogenesis, whether GSK-3 causes or accelerates the development of epilepsy in AD has not been well studied. One study reported that genetically modified GSK-3 with increased activity decelerates kainate-induced epileptogenesis [101], which suggests that the role of GSK-3 in epileptogenesis can also be different in AD and non-AD animal models. Furthermore, presuming that GSK-3 contributes to epileptogenesis does not necessarily mean that reducing GSK-3 activity can impede this process. Therefore, inhibiting GSK-3 activity, as most studies proposed, might not be the optimal intervention for AD-associated seizure.
Last but not least, the pathway that involves GSK-3 in AD-associated epileptic seizures requires further investigation. GSK-3 is involved in multiple cell signalling pathways [54], which suggests that GSK-3 might not be implicated directly in the development of seizure and epilepsy. It might be another downstream molecule, such as Fyn and tau, that directly contributes to the onset of seizures, but more evidence is needed to establish the pathway and to verify the therapeutic potential of regulating the targets involved. Therefore, it would be valuable to pinpoint the position of GSK-3 in the pathway that leads to AD-associated seizure.
To summarise, emerging evidence suggests that GSK-3 may play a crucial but complex role in the pathogenesis of epileptic seizures in AD. Unravelling this relationship could potentially open up new therapeutic strategies.
Author Contributions
Conceptualization, R.L.; methodology, R.L., N.C.J., P.K.; validation, R.L., N.C.J., P.K.; formal analysis, R.L..; investigation, R.L., N.C.J., P.K.; resources, N.C.J., P.K.; data curation, R.L., N.C.J., P.K.; writing—original draft preparation, R.L.; writing—review and editing, N.C.J., P.K.; supervision, N.C.J., P.K.; project administration, N.C.J., P.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 2007, 3, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Hesdorffer, D.C.; Hauser, W.A.; Annegers, J.F.; Kokmen, E.; Rocca, W.A. Dementia and adult-onset unprovoked seizures. Neurology 1996, 46, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.A.; Morris, M.L.; Heston, L.L.; Anderson, V.E. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986, 36, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, M.F.; Morris, J.C.; Ashkin, K.; Coben, L.A. Advanced Alzheimer’s disease is a risk factor for late-onset seizures. Arch Neurol. 1990, 47, 847–850. [Google Scholar] [CrossRef]
- Bakker, A.; Krauss, G.L.; Albert, M.S.; Speck, C.L.; Jones, L.R.; Stark, C.E.; Yassa, M.A.; Bassett, S.S.; Shelton, A.L.; Gallagher, M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 2012, 74, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Rabinowicz, A.L.; Starkstein, S.E.; Leiguarda, R.C.; Coleman, A.E. Transient epileptic amnesia in dementia: A treatable unrecognized cause of episodic amnestic wandering. Alzheimer Dis. Assoc. Disord. 2000, 14, 231–233. [Google Scholar] [CrossRef]
- Scharfman, H.E. Alzheimer’s disease and epilepsy: Insight from animal models. Future Neurol 2012, 7, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.A. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009, 47, 289–299. [Google Scholar]
- Sperfeld, A.D.; Collatz, M.B.; Baier, H.; Palmbach, M.; Storch, A.; Schwarz, J.; Tatsch, K.; Reske, S.; Joosse, M.; Heutink, P.; et al. FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 1999, 46, 708–715. [Google Scholar] [CrossRef]
- Garcia-Cabrero, A.M.; Guerrero-Lopez, R.; Giraldez, B.G.; Llorens-Martin, M.; Avila, J.; Serratosa, J.M.; Sanchez, M.P. Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis. 2013, 58, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.; Verret, L.; Juan, C.; Remaud, J.; Halley, H.; Rampon, C.; Dahan, L. Early onset of hypersynchronous network activity and expression of a marker of chronic seizures in the Tg2576 mouse model of Alzheimer’s disease. PLoS ONE 2015, 10, e0119910. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fulop, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkanen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef]
- Dunning, C.J.; McGauran, G.; Willen, K.; Gouras, G.K.; O’Connell, D.J.; Linse, S. Direct High Affinity Interaction between Abeta42 and GSK3alpha Stimulates Hyperphosphorylation of Tau. A New Molecular Link in Alzheimer’s Disease? ACS Chem. Neurosci. 2016, 7, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Muyllaert, D.; Kremer, A.; Jaworski, T.; Borghgraef, P.; Devijver, H.; Croes, S.; Dewachter, I.; Van Leuven, F. Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav. 2008, 7 (Suppl. S1), 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.C.; Dove, G.; Cascino, G.D.; Petersen, R.C. Recurrent seizures in patients with dementia: Frequency, seizure types, and treatment outcome. Epilepsy Behav. 2009, 14, 118–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, W.A.; Annegers, J.F.; Kurland, L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993, 34, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Cabrejo, L.; Guyant-Marechal, L.; Laquerriere, A.; Vercelletto, M.; De la Fourniere, F.; Thomas-Anterion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype associated with APP duplication in five families. Brain 2006, 129, 2966–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.E.; Bird, T.D. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- de Jonghe-Rouleau, A.P.; Pot, A.M.; de Jonghe, J.F. Self-injurious behaviour in nursing home residents with dementia. Int. J. Geriatr. Psychiatry 2005, 20, 651–657. [Google Scholar] [CrossRef]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Ziyatdinova, S.; Ronnback, A.; Gurevicius, K.; Miszczuk, D.; Graff, C.; Winblad, B.; Pitkanen, A.; Tanila, H. Increased Epileptiform EEG Activity and Decreased Seizure Threshold in Arctic APP Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Corbett, B.F.; Leiser, S.C.; Ling, H.P.; Nagy, R.; Breysse, N.; Zhang, X.; Hazra, A.; Brown, J.T.; Randall, A.D.; Wood, A.; et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer’s disease. J. Neurosci. 2013, 33, 7020–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Born, H.A.; Kim, J.Y.; Savjani, R.R.; Das, P.; Dabaghian, Y.A.; Guo, Q.; Yoo, J.W.; Schuler, D.R.; Cirrito, J.R.; Zheng, H.; et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer’s disease. J. Neurosci. 2014, 34, 3826–3840. [Google Scholar] [CrossRef] [PubMed]
- Vogt, D.L.; Thomas, D.; Galvan, V.; Bredesen, D.E.; Lamb, B.T.; Pimplikar, S.W. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol. Aging 2011, 32, 1725–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvan, V.; Gorostiza, O.F.; Banwait, S.; Ataie, M.; Logvinova, A.V.; Sitaraman, S.; Carlson, E.; Sagi, S.A.; Chevallier, N.; Jin, K.; et al. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl. Acad. Sci. USA 2006, 103, 7130–7135. [Google Scholar] [CrossRef] [Green Version]
- Saganich, M.J.; Schroeder, B.E.; Galvan, V.; Bredesen, D.E.; Koo, E.H.; Heinemann, S.F. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J. Neurosci. 2006, 26, 13428–13436. [Google Scholar] [CrossRef]
- Volicer, L.; Smith, S.; Volicer, B.J. Effect of seizures on progression of dementia of the Alzheimer type. Dementia 1995, 6, 258–263. [Google Scholar] [CrossRef]
- Lott, I.T.; Doran, E.; Nguyen, V.Q.; Tournay, A.; Movsesyan, N.; Gillen, D.L. Down syndrome and dementia: Seizures and cognitive decline. J. Alzheimers Dis. 2012, 29, 177–185. [Google Scholar] [CrossRef]
- Nygaard, H.B.; Wagner, A.F.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; Strittmatter, K.A.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P.; et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Holmes, G.L.; Lenck-Santini, P.P. Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 2006, 8, 504–515. [Google Scholar] [CrossRef]
- Kleen, J.K.; Scott, R.C.; Holmes, G.L.; Lenck-Santini, P.P. Hippocampal interictal spikes disrupt cognition in rats. Ann. Neurol. 2010, 67, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, H.B.; Kaufman, A.C.; Sekine-Konno, T.; Huh, L.L.; Going, H.; Feldman, S.J.; Kostylev, M.A.; Strittmatter, S.M. Brivaracetam, but not ethosuximide, reverses memory impairments in an Alzheimer’s disease mouse model. Alzheimers Res. Ther. 2015, 7, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.Y.; Zheng, C.Y.; Zou, M.M.; Zhu, J.W.; Zhang, Y.; Wang, J.; Liu, C.F.; Li, Q.F.; Xiao, Z.C.; Li, S.; et al. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol. Aging 2014, 35, 2713–2725. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Q.; Wang, B.R.; Tian, Y.Y.; Xu, J.; Gao, L.; Zhao, S.L.; Jiang, T.; Xie, H.G.; Zhang, Y.D. Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci. Ther. 2013, 19, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, K.; Shiratsuchi, A.; Sato, S.; Omori, A.; Arioka, M.; Kobayashi, S.; Uchida, T.; Imahori, K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993, 325, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Tian, F.F.; Chen, J.M.; Guo, T.H.; Ma, Y.F.; Fang, J.; Dang, J.; Song, M.Y. GSK-3beta may be involved in hippocampal mossy fiber sprouting in the pentylenetetrazole-kindling model. Mol. Med. Rep. 2013, 8, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Ryder, J.; Su, Y.; Liu, F.; Li, B.; Zhou, Y.; Ni, B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem. Biophys. Res. Commun. 2003, 312, 922–929. [Google Scholar] [CrossRef]
- Avila, J.; Wandosell, F.; Hernandez, F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 2010, 10, 703–710. [Google Scholar] [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9 (Suppl. S3), 309–317. [Google Scholar] [CrossRef]
- Ishiguro, K.; Takamatsu, M.; Tomizawa, K.; Omori, A.; Takahashi, M.; Arioka, M.; Uchida, T.; Imahori, K. Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J. Biol. Chem. 1992, 267, 10897–10901. [Google Scholar]
- Wagner, U.; Utton, M.; Gallo, J.M.; Miller, C.C. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J. Cell Sci. 1996, 109, 1537–1543. [Google Scholar]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [Green Version]
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell Mol. Neurobiol. 2018, 38, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Mucke, L. Tau Phosphorylation-Much More than a Biomarker. Neuron 2016, 92, 265–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hall, A.M.; Kelinske, M.; Roberson, E.D. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol. Aging 2014, 35, 2617–2624. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Shen, Y.; Shultz, S.R.; Nguyen, A.; Hovens, C.; Adlard, P.A.; Bush, A.I.; Chan, J.; Kwan, P.; O’Brien, T.J.; et al. Accelerated kindling epileptogenesis in Tg4510 tau transgenic mice, but not in tau knockout mice. Epilepsia 2017, 58, e136–e141. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Zheng, P.; Wright, D.K.; Dezsi, G.; Braine, E.; Nguyen, T.; Corcoran, N.M.; Johnston, L.A.; Hovens, C.M.; Mayo, J.N.; et al. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 2016, 139, 1919–1938. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.C.; Nguyen, T.; Corcoran, N.M.; Velakoulis, D.; Chen, T.; Grundy, R.; O’Brien, T.J.; Hovens, C.M. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol. Dis. 2012, 45, 897–901. [Google Scholar] [CrossRef]
- Bennecib, M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000, 485, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Menezes, M.E.; Pannell, L.K.; Mulekar, M.S.; Honkanen, R.E.; Shevde, L.A.; Samant, R.S. DNAJB6 chaperones PP2A mediated dephosphorylation of GSK3beta to downregulate beta-catenin transcription target, osteopontin. Oncogene 2012, 31, 4472–4483. [Google Scholar] [CrossRef] [Green Version]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, A.P.; Ali Hussaini, S.M.; Krishna, N.H.; Babu, B.N.; Kamal, A. Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem. 2018, 144, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: A patent update (2014–2015). Expert Opin. Ther. Pat. 2017, 27, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Lesort, M.; Jope, R.S.; Johnson, G.V. Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 1999, 72, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Shen, F.; Tian, K.; Wang, M.; Xi, Y.; Li, J.; Huang, Z. Triptolide induces oxidative damage in NRK-52E cells through facilitating Nrf2 degradation by ubiquitination via the GSK-3beta/Fyn pathway. Toxicol In Vitro 2019, 58, 187–194. [Google Scholar] [CrossRef]
- Kojima, N.; Ishibashi, H.; Obata, K.; Kandel, E.R. Higher seizure susceptibility and enhanced tyrosine phosphorylation of N-methyl-D-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem. 1998, 5, 429–445. [Google Scholar]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef]
- Miyamoto, T.; Stein, L.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 41. [Google Scholar] [CrossRef]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Torrance, M.; Adame, A.; Mante, M.; Bar-on, P.; Rose, J.B.; Crews, L.; Masliah, E. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 2007, 27, 1981–1991. [Google Scholar] [CrossRef]
- Ishizawa, T.; Sahara, N.; Ishiguro, K.; Kersh, J.; McGowan, E.; Lewis, J.; Hutton, M.; Dickson, D.W.; Yen, S.H. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am. J. Pathol. 2003, 163, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, E.M.; Lee, J.P.; Park, C.H.; Kim, S.; Seo, J.H.; Chang, K.A.; Yu, E.; Jeong, S.J.; Chong, Y.H.; et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 2003, 17, 1951–1953. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.A.; Kim, H.S.; Ha, T.Y.; Ha, J.W.; Shin, K.Y.; Jeong, Y.H.; Lee, J.P.; Park, C.H.; Kim, S.; Baik, T.K.; et al. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol. Cell Biol. 2006, 26, 4327–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.A.; Pimplikar, S.W. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. J. Cell Biol. 2005, 171, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Pimplikar, S.W. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiol. Aging 2011, 32, 2320 e1-9. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [Green Version]
- von Rotz, R.C.; Kohli, B.M.; Bosset, J.; Meier, M.; Suzuki, T.; Nitsch, R.M.; Konietzko, U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 2004, 117, 4435–4448. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, D.M.; Miller, C.C. The FE65 proteins and Alzheimer’s disease. J. Neurosci. Res. 2008, 86, 744–754. [Google Scholar] [CrossRef]
- Jaworski, T.; Dewachter, I.; Lechat, B.; Gees, M.; Kremer, A.; Demedts, D.; Borghgraef, P.; Devijver, H.; Kugler, S.; Patel, S.; et al. GSK-3alpha/beta kinases and amyloid production in vivo. Nature 2011, 480, E4–E5. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Takahashi, M.; Hayashi, S.; Kakita, A.; Wakabayashi, K.; Fukuda, M.; Kameyama, S.; Tanaka, R.; Takahashi, H.; Nawa, H. Patients with temporal lobe epilepsy show an increase in brain-derived neurotrophic factor protein and its correlation with neuropeptide Y. Brain Res 1999, 818, 579–582. [Google Scholar] [CrossRef]
- Gangarossa, G.; Sakkaki, S.; Lory, P.; Valjent, E. Mouse hippocampal phosphorylation footprint induced by generalized seizures: Focus on ERK, mTORC1 and Akt/GSK-3 pathways. Neuroscience 2015, 311, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Biel, N.; Canudas, A.M.; Camins, A.; Pallas, M. Kainate induces AKT, ERK and cdk5/GSK3beta pathway deregulation, phosphorylates tau protein in mouse hippocampus. Neurochem. Int. 2007, 50, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.P.; Santorufo, G.; Brilli, E.; Borrelli, E.; Bozzi, Y. Kainic acid-induced seizures activate GSK-3beta in the hippocampus of D2R-/- mice. Neuroreport 2010, 21, 846–850. [Google Scholar] [CrossRef]
- Engel, T.; Gomez-Sintes, R.; Alves, M.; Jimenez-Mateos, E.M.; Fernandez-Nogales, M.; Sanz-Rodriguez, A.; Morgan, J.; Beamer, E.; Rodriguez-Matellan, A.; Dunleavy, M.; et al. Bi-directional genetic modulation of GSK-3beta exacerbates hippocampal neuropathology in experimental status epilepticus. Cell Death Dis. 2018, 9, 969. [Google Scholar] [CrossRef]
- Joyce, J.N.; Myers, A.J.; Gurevich, E. Dopamine D2 receptor bands in normal human temporal cortex are absent in Alzheimer’s disease. Brain Res. 1998, 784, 7–17. [Google Scholar] [CrossRef]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.A.; Halatsch, M.E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2alpha and activating transcription factor 4—A pathway active in glioblastomas and epilepsy. Antioxid Redox Signal 2014, 20, 2907–2922. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.N.; Kaeger, C.; Ryoo, H.; Goldsmith, S. Dopamine D2 receptors in the hippocampus and amygdala in Alzheimer’s disease. Neurosci. Lett. 1993, 154, 171–174. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Aourz, N.; Serruys, A.K.; Chabwine, J.N.; Balegamire, P.B.; Afrikanova, T.; Edrada-Ebel, R.; Grey, A.I.; Kamuhabwa, A.R.; Walrave, L.; Esguerra, C.V.; et al. Identification of GSK-3 as a Potential Therapeutic Entry Point for Epilepsy. ACS Chem. Neurosci. 2019, 10, 1992–2003. [Google Scholar] [CrossRef]
- Urbanska, M.; Kazmierska-Grebowska, P.; Kowalczyk, T.; Caban, B.; Nader, K.; Pijet, B.; Kalita, K.; Gozdz, A.; Devijver, H.; Lechat, B.; et al. GSK3beta activity alleviates epileptogenesis and limits GluA1 phosphorylation. EBioMedicine 2019, 39, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Matsuba, Y.; Yamazaki, N.; Hashimoto, S.; Saido, T.C. Calpain Activation in Alzheimer’s Model Mice Is an Artifact of APP and Presenilin Overexpression. J. Neurosci. 2016, 36, 9933–9936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
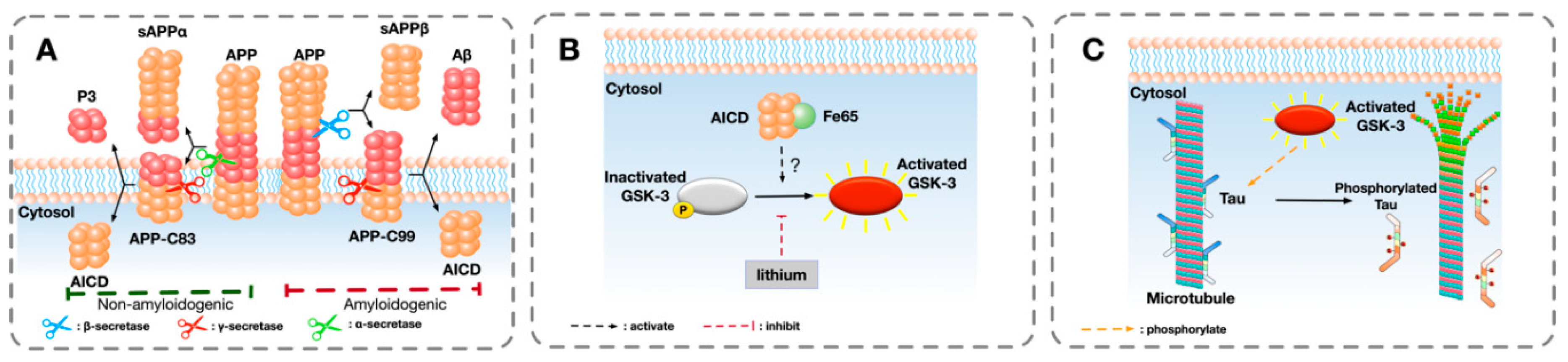
Figure 1.
A schematic overview of a hypothesised pathway that GSK-3 is involved in the phosphorylation of tau: (A) The sequential cleavage process of amyloid precursor protein (APP) in the amyloidogenic and non-amyloidogenic pathway in Alzheimer’s disease (AD). In the amyloidogenic pathway, the extracellular domain of APP is firstly cleaved by β-secretase, liberating a soluble extracellular N-terminus fragment sAPPβ and a transmembrane fragment AP-C99. The remaining APP-C99 is further cleaved by γ-secretase, generating β-amyloid (Aβ) and releasing APP intracellular domain (AICD) into cytosol. In the non-amyloidogenic pathway, APP is cleaved by α-secretase and γ-secretase, releasing sAPPα, P3, and AICD. (B) One possible pathway of the activation of GSK-3. After binding with Fe65, AICD directly or indirectly activates GSK-3 by removing the phosphorylation from serine site (Ser9 of GSK-3β and Ser21 of GSK-3α). The activation of GSK-3 can be inhibited by GSK-3 inhibitor such as lithium. (C) GSK-3 phosphorylates microtubule-associated protein tau (MAPT). Tau is involved in the stabilisation of microtubules and axonal transport. After being abnormally phosphorylated by activated GSK-3, hyper-phosphorylated tau detaches from microtubules and causes the dissociation of microtubules.
Figure 1.
A schematic overview of a hypothesised pathway that GSK-3 is involved in the phosphorylation of tau: (A) The sequential cleavage process of amyloid precursor protein (APP) in the amyloidogenic and non-amyloidogenic pathway in Alzheimer’s disease (AD). In the amyloidogenic pathway, the extracellular domain of APP is firstly cleaved by β-secretase, liberating a soluble extracellular N-terminus fragment sAPPβ and a transmembrane fragment AP-C99. The remaining APP-C99 is further cleaved by γ-secretase, generating β-amyloid (Aβ) and releasing APP intracellular domain (AICD) into cytosol. In the non-amyloidogenic pathway, APP is cleaved by α-secretase and γ-secretase, releasing sAPPα, P3, and AICD. (B) One possible pathway of the activation of GSK-3. After binding with Fe65, AICD directly or indirectly activates GSK-3 by removing the phosphorylation from serine site (Ser9 of GSK-3β and Ser21 of GSK-3α). The activation of GSK-3 can be inhibited by GSK-3 inhibitor such as lithium. (C) GSK-3 phosphorylates microtubule-associated protein tau (MAPT). Tau is involved in the stabilisation of microtubules and axonal transport. After being abnormally phosphorylated by activated GSK-3, hyper-phosphorylated tau detaches from microtubules and causes the dissociation of microtubules.

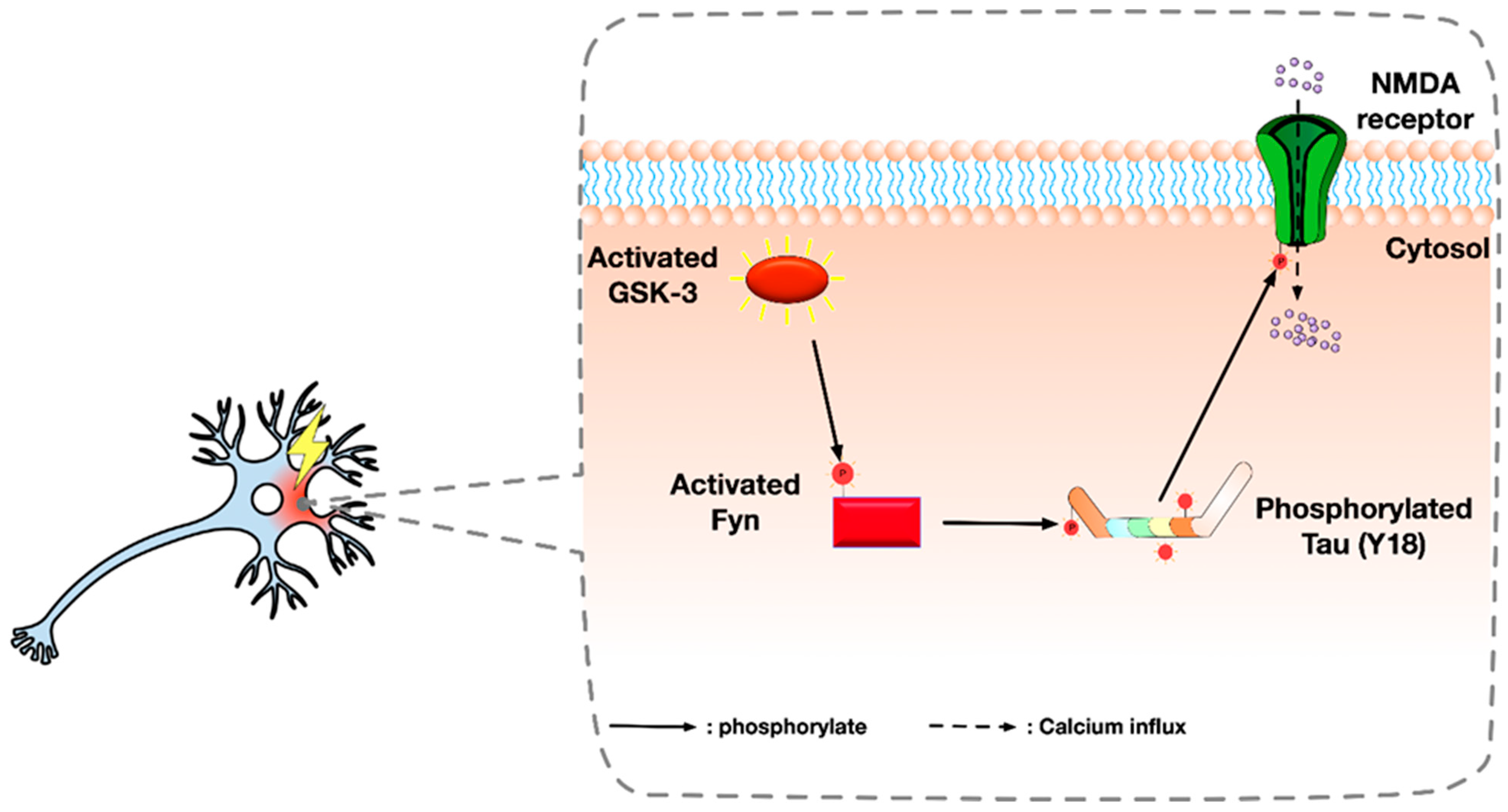
Figure 2.
Illustration summarising an alternative pathway of Glycogen synthase kinase-3 (GSK-3): It is possible that GSK-3 activates Fyn by phosphorylation. Activated Fyn then binds to tau and mediates the phosphorylation of tau at tyrosine 18 (Y18). Y18 phosphorylated tau induces the phosphorylation of the NR2 subunit of NMDA receptor at tyrosine 1472 (Y1472) near the C-terminus, which causes the abnormal activation of NMDA receptor and excessive Ca2+ influx and excitotoxicity.
Figure 2.
Illustration summarising an alternative pathway of Glycogen synthase kinase-3 (GSK-3): It is possible that GSK-3 activates Fyn by phosphorylation. Activated Fyn then binds to tau and mediates the phosphorylation of tau at tyrosine 18 (Y18). Y18 phosphorylated tau induces the phosphorylation of the NR2 subunit of NMDA receptor at tyrosine 1472 (Y1472) near the C-terminus, which causes the abnormal activation of NMDA receptor and excessive Ca2+ influx and excitotoxicity.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lin, R.; Jones, N.C.; Kwan, P. Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures. Int. J. Mol. Sci. 2020, 21, 3676. https://doi.org/10.3390/ijms21103676
AMA Style
Lin R, Jones NC, Kwan P. Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures. International Journal of Molecular Sciences. 2020; 21(10):3676. https://doi.org/10.3390/ijms21103676
Chicago/Turabian StyleLin, Runxuan, Nigel Charles Jones, and Patrick Kwan. 2020. "Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures" International Journal of Molecular Sciences 21, no. 10: 3676. https://doi.org/10.3390/ijms21103676
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.