A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-β Signaling and Activation of cAMP/PKA Signaling

 , , , and
, , , and
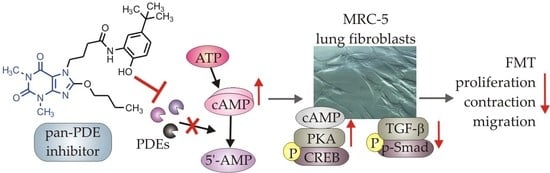
Abstract
:
1. Introduction
2. Results
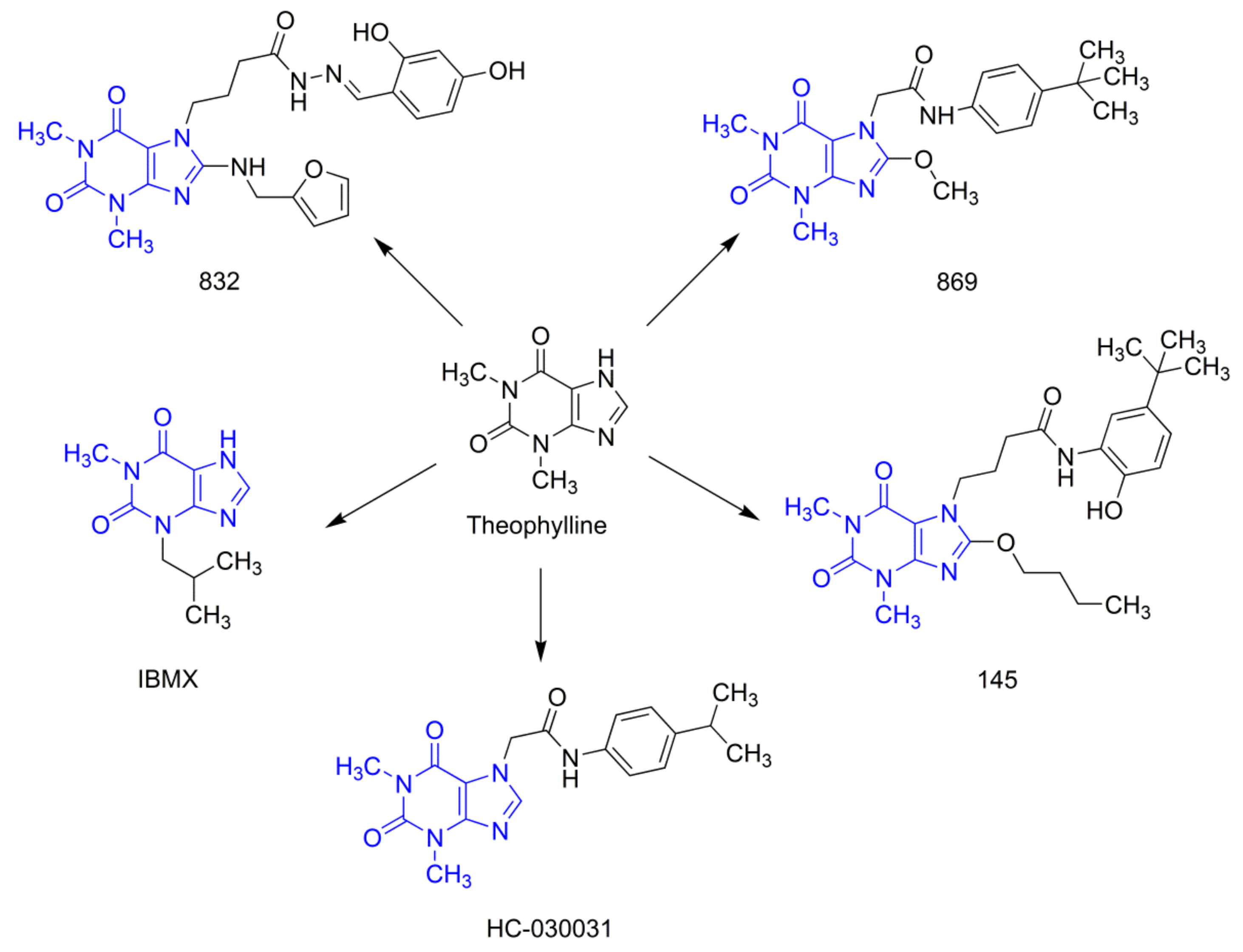
2.1. Compounds 832 and 145 but not 869 are Prominent, Pan-PDE Inhibitors
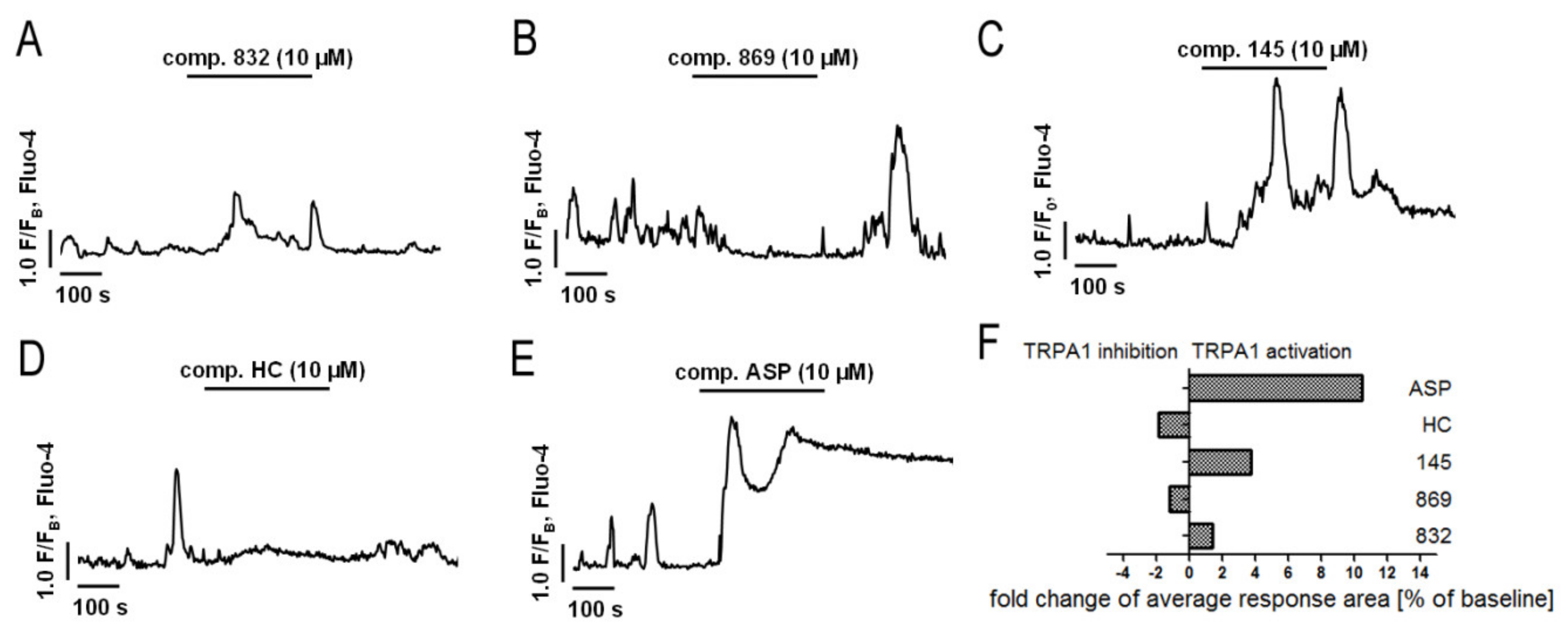
2.2. Compound 869 is a Representative Antagonist of the TRPA1 Channel
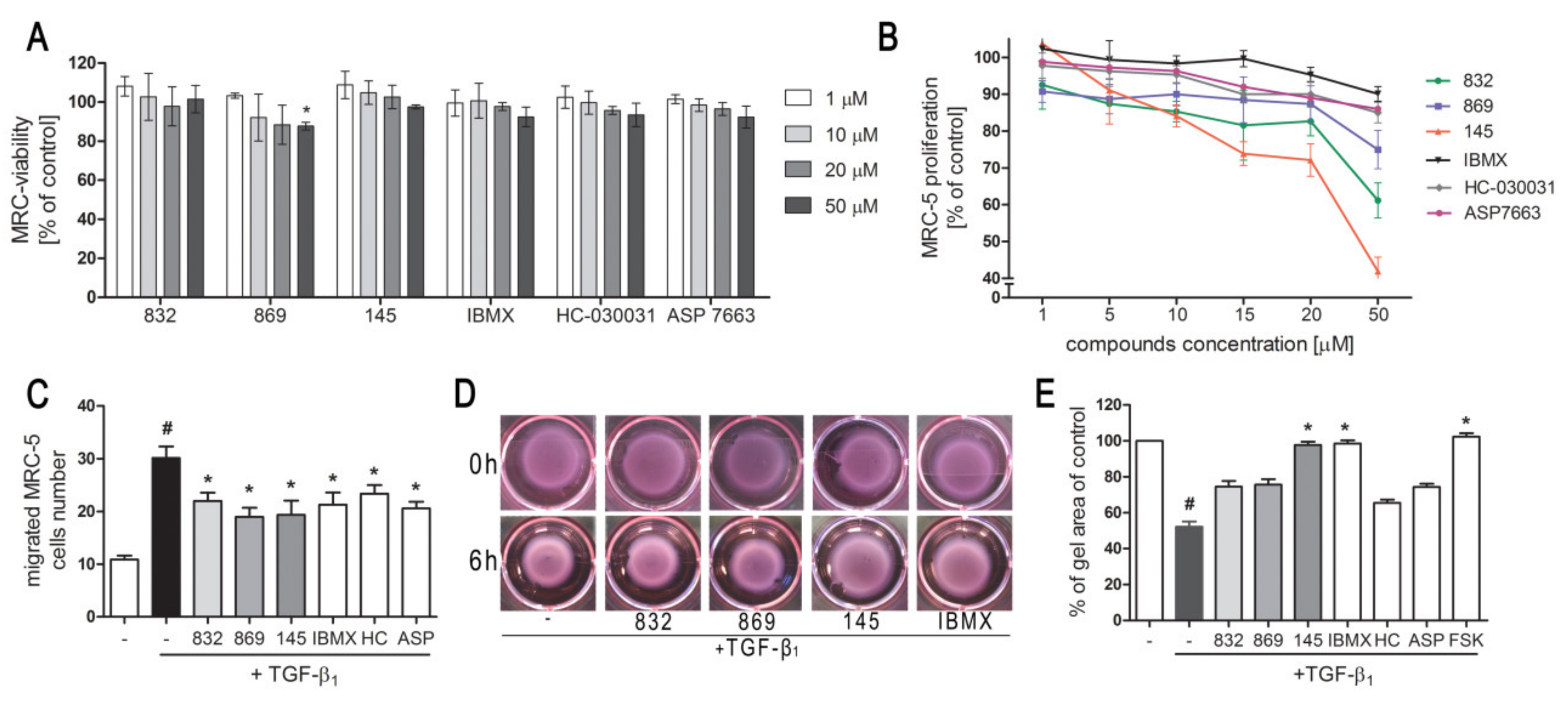
2.3. 7,8-Disubstituted Purine-2,6-dione Derivatives Selectively Affect Activated Lung Fibroblasts’ Proliferation and Migration
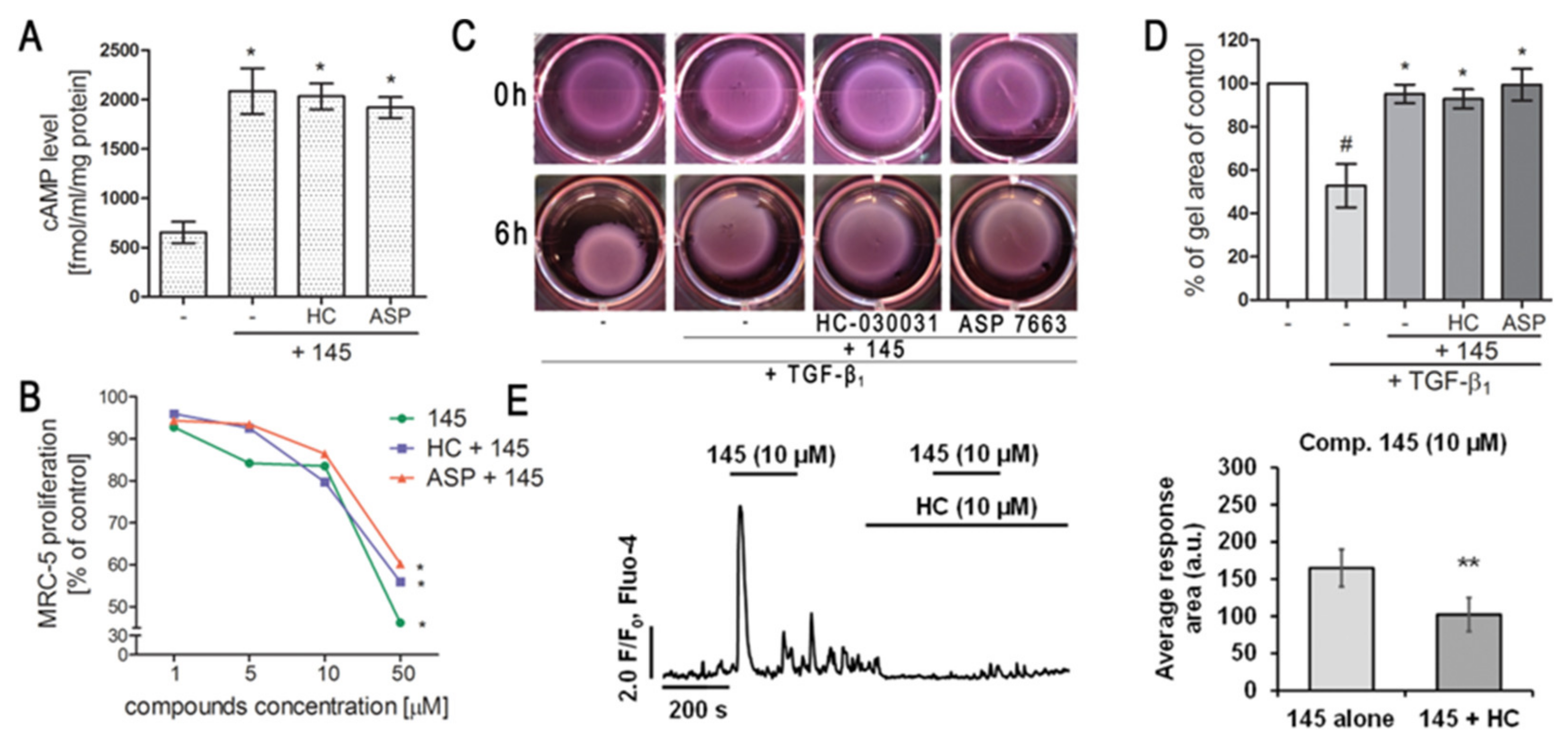
2.4. Compound 145 Significantly Limits TGF-β1-Induced Lung Fibroblast to Myofibroblast Transition
2.5. Both Inhibition and Activation of TRPA1 Channels Affect Ca2+ Signals in MRC-5
2.6. Modulation of TRPA1 Ion Channel Does Not Affect Compound 145 Anti-Fibrotic Properties
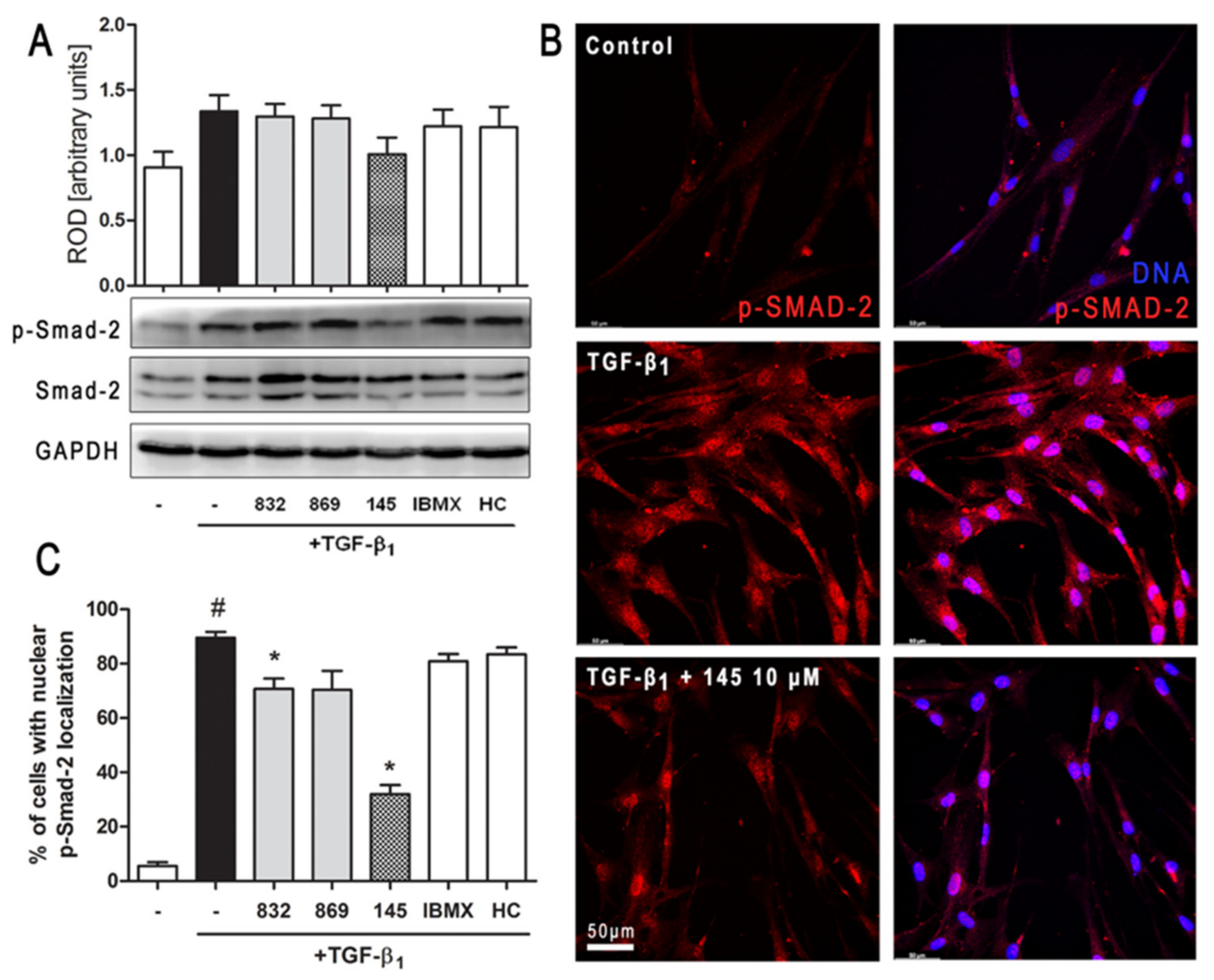
2.7. Compound 145 Attenuates Smad-2 Phosphorylation in TGF-β1-Induced Lung Fibroblasts
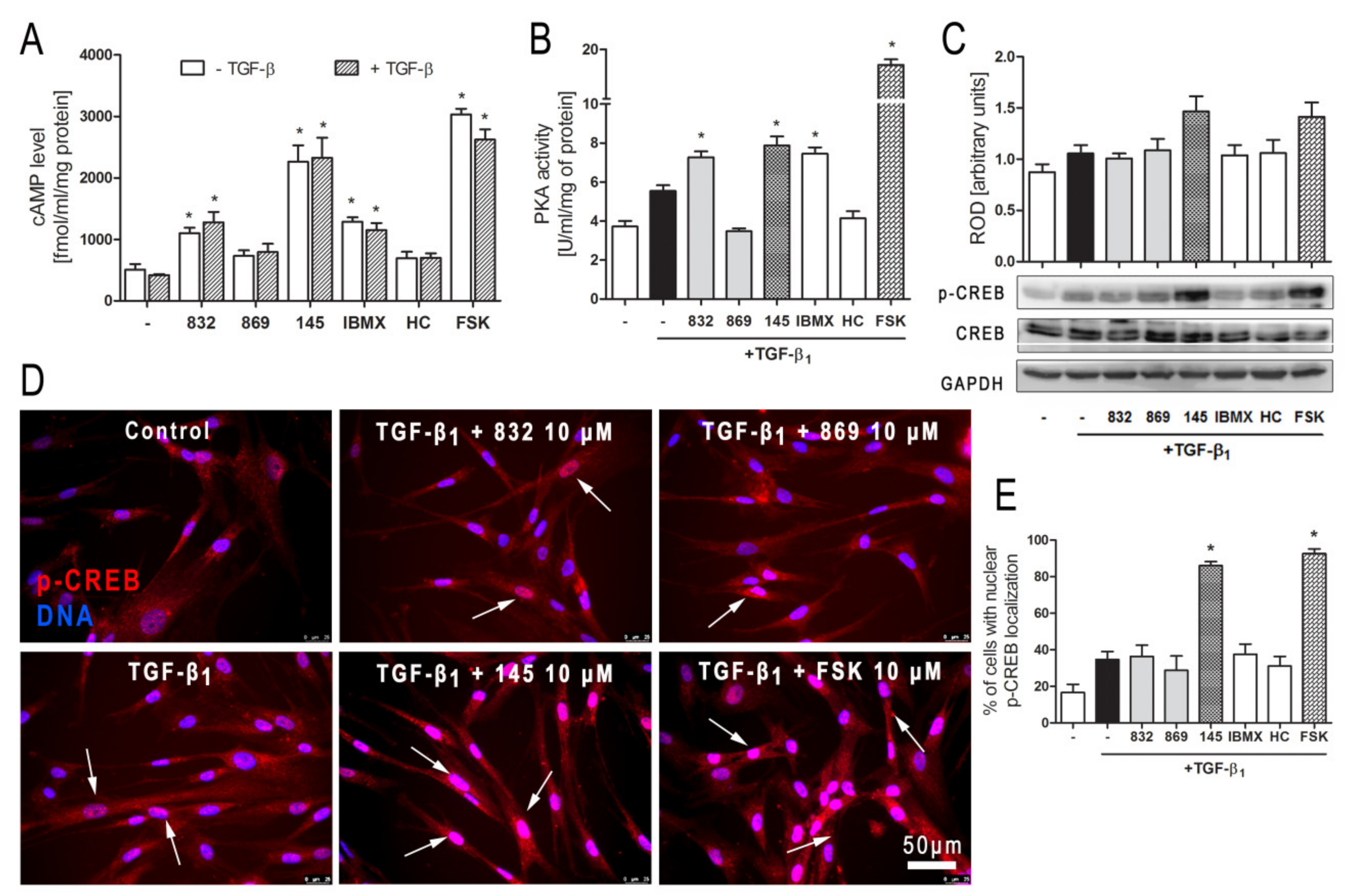
2.8. Compound 145 Activates cAMP/PKA/CREB Dependent Signaling in TGF-β1-Induced Lung Fibroblasts
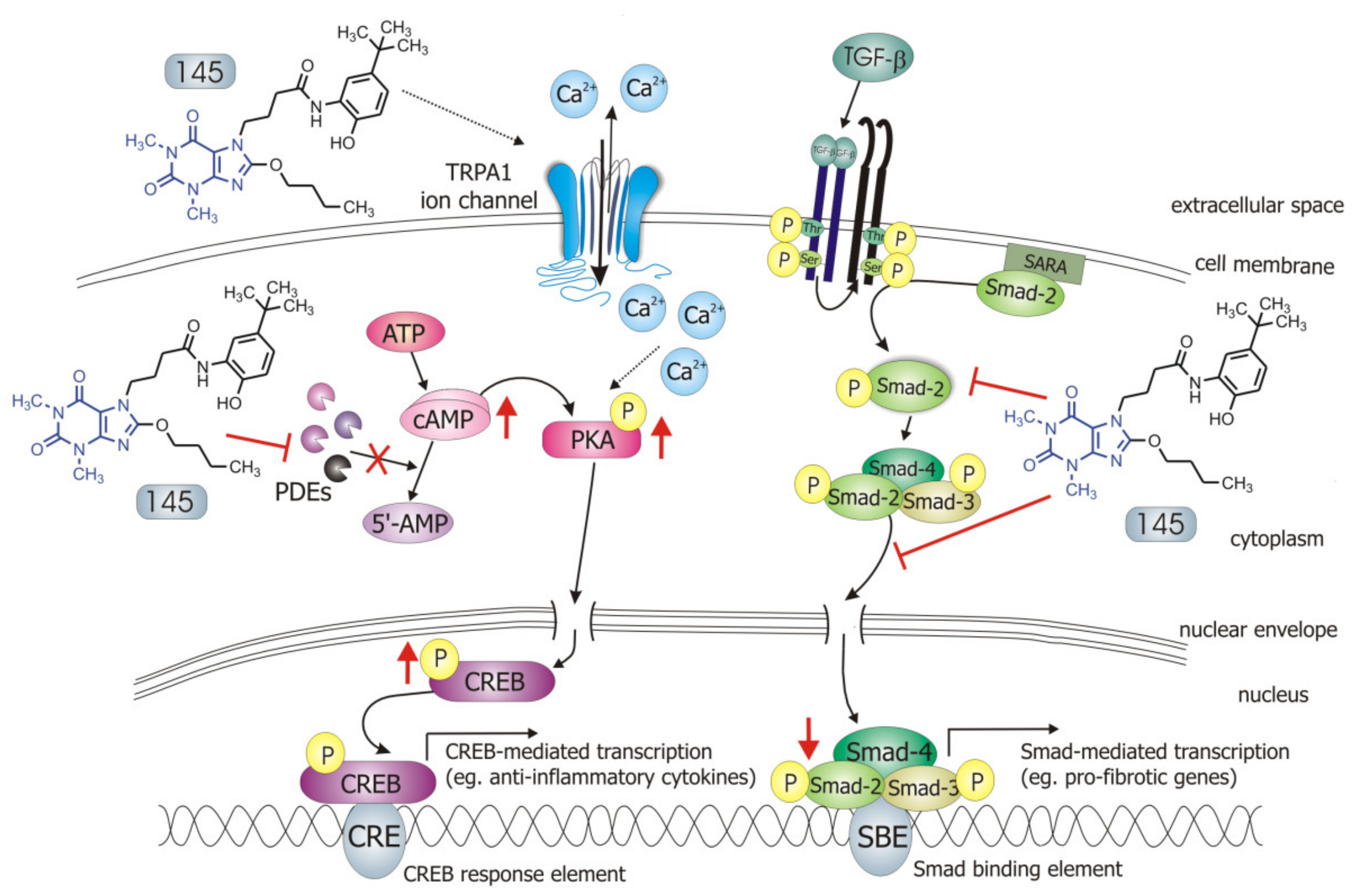
3. Discussion
4. Materials and Methods
4.1. Tested Compounds
4.2. PDE Subtype Selectivity
4.3. TRPA1 Assay
4.4. In Vitro Lung Fibroblast Culture
4.5. Viability and Proliferation Assays
4.6. Transwell Migration Assay
4.7. Cell Contraction Assay
4.8. Intracellular Calcium Measurements
4.9. cAMP ELISA
4.10. In-Cell ELISA
4.11. Immunofluorescence Labelling
4.12. Quantitative PCR
4.13. Western Blotting
4.14. Protein Kinase A Activity
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, C.P. Phosphodiesterase Inhibitors for the Treatment of Asthma and Chronic Obstructive Pulmonary Disease. Int. Arch. Allergy Immunol. 2014, 165, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Cattani-Cavalieri, I.; Musheshe, N.; Nikolaev, V.O.; Schmidt, M. Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol. Ther. 2019, 197, 225–242. [Google Scholar] [CrossRef]
- Billington, C.K.; Ojo, O.O.; Penn, R.B.; Ito, S.; Pharmacol, P.; Author, T. cAMP Regulation of Airway Smooth Muscle Function. Pulm. Pharmacol. Ther. 2013, 26, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Ntontsi, P.; Detta, A.; Bakakos, P.; Loukides, S.; Hillas, G. Experimental and investigational phosphodiesterase inhibitors in development for asthma. Expert Opin. Investig. Drugs 2019, 28, 261–266. [Google Scholar] [CrossRef]
- Glynos, C.; Dupont, L.L.; Vassilakopoulos, T.; Papapetropoulos, A.; Brouckaert, P.; Giannis, A.; Joos, G.F.; Bracke, K.R.; Brusselle, G.G. The Role of Soluble Guanylyl Cyclase in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2013, 188, 789–799. [Google Scholar] [CrossRef]
- Paul, T.; Salazar-Degracia, A.; Peinado, V.I.; Tura-Ceide, O.; Blanco, I.; Barreiro, E.; Barberà, J.A. Soluble guanylate cyclase stimulation reduces oxidative stress in experimental Chronic Obstructive Pulmonary Disease. PLoS ONE 2018, 13, e0190628. [Google Scholar] [CrossRef] [Green Version]
- Zuo, H.; Faiz, A.; van den Berge, M.; Mudiyanselage, S.N.H.R.; Borghuis, T.; Timens, W.; Nikolaev, V.O.; Burgess, J.K.; Schmidt, M. Cigarette smoke exposure alters phosphodiesterases in human structural lung cells. Am. J. Physiol. Cell. Mol. Physiol. 2019, 318, L59–L64. [Google Scholar] [CrossRef]
- Barnes, P.J. Theophylline. Am. J. Respir. Crit. Care Med. 2013, 188, 901–906. [Google Scholar] [CrossRef]
- Oñatibia-Astibia, A.; Martínez-Pinilla, E.; Franco, R. The potential of methylxanthine-based therapies in pediatric respiratory tract diseases. Respir. Med. 2016, 112, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.E. Inhaled Phosphodiesterase 4 (PDE4) Inhibitors for Inflammatory Respiratory Diseases. Front. Pharmacol. 2020, 11, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Beeh, K.M.; Colgan, B.; Kornmann, O.; Leaker, B.; Watz, H.; Lucci, G.; Geraci, S.; Emirova, A.; Govoni, M.; et al. Effect of the inhaled PDE4 inhibitor CHF6001 on biomarkers of inflammation in COPD. Respir. Res. 2019, 20, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenburger, A.; Maarsingh, H.; Schmidt, M. Multiple facets of cAMP signalling and physiological impact: cAMP compartmentalization in the lung. Pharmaceuticals 2012, 5, 1291–1331. [Google Scholar] [CrossRef] [Green Version]
- Grace, M.S.; Baxter, M.; Dubuis, E.; Birrell, M.A.; Belvisi, M.G. Transient receptor potential (TRP) channels in the airway: Role in airway disease. Br. J. Pharmacol. 2014, 171, 2593–2607. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, S. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel and Neurogenic Inflammation in Pathogenesis of Asthma. Med. Sci. Monit. 2016, 22, 2917–2923. [Google Scholar] [CrossRef] [Green Version]
- Belvisi, M.G.; Birrell, M.A. The emerging role of transient receptor potential channels in chronic lung disease. Eur. Respir. J. 2017, 50, 1601357. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, A. Modulators of Transient Receptor Potential (TRP) Channels as Therapeutic Options in Lung Disease. Pharmaceuticals 2019, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Wójcik-Pszczoła, K.; Chłoń-Rzepa, G.; Jankowska, A.; Ellen, E.; Świerczek, A.; Pociecha, K.; Koczurkiewicz, P.; Piska, K.; Gawędzka, A.; Wyska, E.; et al. Novel phosphodiesterases inhibitors from the group of purine-2,6-dione derivatives as potent modulators of airway smooth muscle cell remodelling. Eur. J. Pharmacol. 2019, 865, 172779. [Google Scholar] [CrossRef]
- Chłoń-Rzepa, G.; Ślusarczyk, M.; Jankowska, A.; Gawalska, A.; Bucki, A.; Kołaczkowski, M.; Świerczek, A.; Pociecha, K.; Wyska, E.; Zygmunt, M.; et al. Novel amide derivatives of 1,3-dimethyl-2,6-dioxopurin-7-yl-alkylcarboxylic acids as multifunctional TRPA1 antagonists and PDE4/7 inhibitors: A new approach for the treatment of pain. Eur. J. Med. Chem. 2018, 158, 517–533. [Google Scholar] [CrossRef]
- Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Świerczek, A.; Pociecha, K.; Wyska, E.; Bucki, A.; Gawalska, A.; Kołaczkowski, M.; Pawłowski, M. Novel butanehydrazide derivatives of purine-2,6-dione as dual PDE4/7 inhibitors with potential anti-inflammatory activity: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 146, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Świerczek, A.; Wyska, E.; Baś, S.; Woyciechowska, M.; Mlynarski, J. PK/PD studies on non-selective PDE inhibitors in rats using cAMP as a marker of pharmacological response. Naunyn. Schmiedebergs. Arch. Pharmacol. 2017, 390, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicalini, I.; De Filippis, B.; Gambacorta, N.; Di Michele, A.; Valentinuzzi, S.; Ammazzalorso, A.; Della Valle, A.; Amoroso, R.; Nicolotti, O.; Del Boccio, P.; et al. Development of a Rapid Mass Spectrometric Determination of AMP and Cyclic AMP for PDE3 Activity Study: Application and Computational Analysis for Evaluating the Effect of a Novel 2-oxo-1,2-dihydropyridine-3-carbonitrile Derivative as PDE-3 Inhibitor. Molecules 2020, 25, 1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.-I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [Green Version]
- Michalik, M.; Wojcik-Pszczola, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pekala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018, 75, 3943–3961. [Google Scholar] [CrossRef] [Green Version]
- Lambers, C.; Boehm, P.M.; Karabacak, Y.; Samaha, E.; Benazzo, A.; Jaksch, P.; Roth, M. Combined Activation of Guanylate Cyclase and Cyclic AMP in Lung Fibroblasts as a Novel Therapeutic Concept for Lung Fibrosis. Biomed Res. Int. 2019, 2019, 1345402. [Google Scholar] [CrossRef]
- Dunkern, T.R.; Feurstein, D.; Rossi, G.A.; Sabatini, F.; Hatzelmann, A. Inhibition of TGF-β induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur. J. Pharmacol. 2007, 572, 12–22. [Google Scholar] [CrossRef]
- Sabatini, F.; Petecchia, L.; Boero, S.; Silvestri, M.; Klar, J.; Tenor, H.; Beume, R.; Hatzelmann, A.; Rossi, G.A. A phosphodiesterase 4 inhibitor, roflumilast N-oxide, inhibits human lung fibroblast functions in vitro. Pulm. Pharmacol. Ther. 2010, 23, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Yoshida, M.; Hoshino, S.; Inoue, K.; Kida, H.; Yanagita, M.; Takimoto, T.; Hirata, H.; Kijima, T.; Kumagai, T.; et al. Anti-fibrotic effects of theophylline on lung fibroblasts. Biochem. Biophys. Res. Commun. 2006, 341, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Hińcza, K.; Wnuk, D.; Kądziołka, D.; Koczurkiewicz, P.; Sanak, M.; Madeja, Z.; Pękala, E.; Michalik, M. Pentoxifylline and its active metabolite lisofylline attenuate transforming growth factor β1-induced asthmatic bronchial fibroblast-to-myofibroblast transition. Acta Biochim. Pol. 2016, 63, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Selige, J.; Hatzelmann, A.; Dunkern, T. The differential impact of PDE4 subtypes in human lung fibroblasts on cytokine-induced proliferation and myofibroblast conversion. J. Cell. Physiol. 2011, 226, 1970–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togo, S.; Liu, X.; Wang, X.; Sugiura, H.; Kamio, K.; Kawasaki, S.; Kobayashi, T.; Ertl, R.F.; Ahn, Y.; Holz, O.; et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGF-β1-stimulated fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L959–L969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Kim, T.J.; Peng, D.H.; Duan, D.; Gibbons, D.L.; Yamauchi, M.; Jackson, J.R.; Le Saux, C.J.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J. Clin. Investig. 2017, 127, 3675–3688. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [Green Version]
- Fehrholz, M.; Speer, C.P.; Kunzmann, S. Caffeine and rolipram affect Smad signalling and TGF-β1 stimulated CTGF and transgelin expression in lung epithelial cells. PLoS ONE 2014, 9, e97357. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Huang, K.; Lv, Z.; Gao, S.; Su, C.; Li, X.; Zhou, H. Comparison of in-vitro anti-fibrotic effects of pirfenidone and nintedanib. Eur. Respir. J. 2019, 54, PA1282. [Google Scholar]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, G.; Wu, J.-J.; Wang, L.; Uhler, M.; Simeone, D. Protein Kinase A Modulates Transforming Growth Factor-β Signaling through a Direct Interaction with Smad4 Protein. J. Biol. Chem. 2013, 288, 8737–8749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-β1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. 2015, 36, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, S.Q.; Hassid, A.; Ostrom, R.S. cAMP Inhibits Transforming Growth Factor-β-Stimulated Collagen Synthesis via Inhibition of Extracellular Signal-Regulated Kinase 1/2 and Smad Signaling in Cardiac Fibroblasts. Mol. Pharmacol. 2006, 70, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xu, H.; Geng, Y.; Xu, D.; Zhang, L.; Yang, Y.; Wei, Z.; Zhang, B.; Li, S.; Gao, X.; et al. Dibutyryl-cAMP attenuates pulmonary fibrosis by blocking myofibroblast differentiation via PKA/CREB/CBP signaling in rats with silicosis. Respir. Res. 2017, 18, 38. [Google Scholar] [CrossRef] [Green Version]
- Schiller, M.; Dennler, S.; Anderegg, U.; Kokot, A.; Simon, J.C.; Luger, T.A.; Mauviel, A.; Böhm, M. Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J. Biol. Chem. 2010, 285, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Cardinaux, J.R.; Notis, J.C.; Zhang, Q.; Vo, N.; Craig, J.C.; Fass, D.M.; Brennan, R.G.; Goodman, R.H. Recruitment of CREB binding protein is sufficient for CREB-mediated gene activation. Mol. Cell. Biol. 2000, 20, 1546–1552. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Lam, S.S.; Srinath, H.; Schiffer, C.A.; Royer, W.E.; Lin, K. Competition between Ski and CREB-binding Protein for Binding to Smad Proteins in Transforming Growth Factor-β Signaling. J. Biol. Chem. 2007, 282, 11365–11376. [Google Scholar] [CrossRef] [Green Version]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ni, L.; Chang, D.; Lu, H.; Jiang, Y.; Kim, B.-S.; Wang, A.; Liu, X.; Zhong, B.; Yang, X.; et al. Cyclic AMP-Responsive Element-Binding Protein (CREB) is Critical in Autoimmunity by Promoting Th17 but Inhibiting Treg Cell Differentiation. EBioMedicine 2017, 25, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yan, Z.; Yang, S.; Cai, J.; Robinson, H.; Ke, H. Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry 2008, 47, 12760–12768. [Google Scholar] [CrossRef] [Green Version]
- Vang, A.G.; Basole, C.; Dong, H.; Nguyen, R.K.; Housley, W.; Guernsey, L.; Adami, A.J.; Thrall, R.S.; Clark, R.B.; Epstein, P.M.; et al. Differential Expression and Function of PDE8 and PDE4 in Effector T cells: Implications for PDE8 as a Drug Target in Inflammation. Front. Pharmacol. 2016, 7, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, A.; Steinritz, D.; Gudermann, T. Transient receptor potential (TRP) channels as molecular targets in lung toxicology and associated diseases. Cell Calcium 2017, 67, 123–137. [Google Scholar] [CrossRef]
- Mukhopadhyay, I.; Kulkarni, A.; Khairatkar-Joshi, N. Blocking TRPA1 in Respiratory Disorders: Does It Hold a Promise? Pharmaceuticals 2016, 9, 70. [Google Scholar] [CrossRef]
- Nagatomo, K.; Kubo, Y. Caffeine activates mouse TRPA1 channels but suppresses human TRPA1 channels. Proc. Natl. Acad. Sci. USA 2008, 105, 17373–17378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurahara, L.H.; Hiraishi, K.; Hu, Y.; Koga, K.; Onitsuka, M.; Doi, M.; Aoyagi, K.; Takedatsu, H.; Kojima, D.; Fujihara, Y.; et al. Activation of Myofibroblast TRPA1 by Steroids and Pirfenidone Ameliorates Fibrosis in Experimental Crohn’s Disease. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 299–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, L.J.; Mukherjee, S.; Ask, K. Calcium Homeostasis and Ionic Mechanisms in Pulmonary Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015, 53, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.J.; Broome, R.E.; Kent, T.C.; Charlton, S.J.; Rosethorne, E.M. The inhibition of human lung fibroblast proliferation and differentiation by Gs-coupled receptors is not predicted by the magnitude of cAMP response. Respir. Res. 2018, 19, 56. [Google Scholar] [CrossRef]
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 151–199. [Google Scholar]
- Frredholm, B.B. On the Mechanism of Action of Theophylline and Caffeine. Acta Med. Scand. 1985, 217, 149–153. [Google Scholar] [CrossRef]
- Monteiro, J.P.; Alves, M.G.; Oliveira, P.F.; Silva, B.M. Structure-Bioactivity Relationships of Methylxanthines: Trying to Make Sense of All the Promises and the Drawbacks. Molecules 2016, 21, 974. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PDE-IC50 [µM] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1A | 1B | 1C | 3A | 3B | 4A | 4B | 4D | 5A | 7A | 8A | |
| 832 | 7.24 | 0.04 | 89.18 | 0.125 | 0.42 | 4.75 | 4.4 3 | n.a. | n.a. | 10.5 3 | n.a. |
| 869 | n.a. | n.a. | 172.18 | n.a. | 156.70 | 58.05 | n.a. | n.a. | n.a. | n.a. | n.a. |
| 145 | n.a. | 2.69 1 | 48.52 | 0.18 1 | 2.05 | 1.59 | 5.43 2 | 13.85 1 | 103.88 1 | 5.07 2 | 73.18 |
| IBMX | 0.11 | 35.70 1 | 12.52 | 0.71 1 | 0.31 | 16.39 | 46.60 4 | 81.94 | 45.09 1 | 77.70 4 | n.a. |
| Compound | TRPA1 Inhibition (% of control) | TRPA1 Activation (% of Control) | ||
|---|---|---|---|---|
| 10 µM | 50 µM | 10 µM | 50 µM | |
| 832 | 12.4 | 8.8 | 3.8 | 2.4 |
| 869 | 74.9 | 98.7 | 0.0 | 2.2 |
| 145 | 12.3 | 4.8 | 0.2 | 4.6 |
| HC-030031 | 91.8 | 98.8 | - | - |
| ASP 7663 | - | - | 75.8 | 76.1 |
| Gene | Forward Sequence (5′→3′) | Reverse Sequence (5′→3′) |
|---|---|---|
| ACTA2 | CCGGGAGAAAATGACTCAAA | GAAGGAATAGCCACGCTCAG |
| MYH11 | CAGATGCTGGACCTTGAAGA | TCCATGACCAGGATCTCATC |
| SM22 | CGCGAAGTGCAGTCCAAAATCG | GGGCTGGTTCTTCTTCAATGGGC |
| COL1A1 | AGCCAGCAGATCGAGAACAT | TCTTGTCCTTGGGGTTCTTG |
| TNC | CCAGCGACCATCAACGCAGC | GGGGCTTGTTCAGTGGATGCCT |
| FN1 | TCGAGGAGGAAATTCCAATG | ACACACGTCCACCTCATCAT |
| VCAN | GGGAACCTGGTGAAGAAACA | CTTCCACAGTGGGTGGTCTT |
| 18S rRNA | CGCCGCTAGAGGTGAAATTC | TTGGCAAATGCTTTCGCTC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wójcik-Pszczoła, K.; Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Ferdek, P.E.; Kusiak, A.A.; Świerczek, A.; Pociecha, K.; Koczurkiewicz-Adamczyk, P.; Wyska, E.; et al. A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-β Signaling and Activation of cAMP/PKA Signaling. Int. J. Mol. Sci. 2020, 21, 4008. https://doi.org/10.3390/ijms21114008
Wójcik-Pszczoła K, Chłoń-Rzepa G, Jankowska A, Ślusarczyk M, Ferdek PE, Kusiak AA, Świerczek A, Pociecha K, Koczurkiewicz-Adamczyk P, Wyska E, et al. A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-β Signaling and Activation of cAMP/PKA Signaling. International Journal of Molecular Sciences. 2020; 21(11):4008. https://doi.org/10.3390/ijms21114008
Chicago/Turabian StyleWójcik-Pszczoła, Katarzyna, Grażyna Chłoń-Rzepa, Agnieszka Jankowska, Marietta Ślusarczyk, Paweł E Ferdek, Agnieszka A Kusiak, Artur Świerczek, Krzysztof Pociecha, Paulina Koczurkiewicz-Adamczyk, Elżbieta Wyska, and et al. 2020. "A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-β Signaling and Activation of cAMP/PKA Signaling" International Journal of Molecular Sciences 21, no. 11: 4008. https://doi.org/10.3390/ijms21114008