Apelin Controls Angiogenesis-Dependent Glioblastoma Growth
by
 ,
,
Anne Frisch
1,†, 
Stefanie Kälin
1,‡,
Raymond Monk
1,§,
Josefine Radke
1,2,
Frank L. Heppner
1 and
Roland E. Kälin
1,*,‖ 1
Department of Neuropathology, Charité–Universitätsmedizin, 10117 Berlin, Germany
2
German Cancer Consortium (DKTK), Partner Site Charité Berlin AND Berlin Institute of Health (BIH), 10178 Berlin, Germany
*
Author to whom correspondence should be addressed.
†
Present Address: Department of Radiology, Charité–Universitätsmedizin, 10117 Berlin, Germany.
‡
Present Address: BSL BIOSERVICE Scientific Laboratories Munich GmbH, 82152 Planegg, Germany.
§
Present Address: Institute of Experimental Endocrinology, Charité–Universitätsmedizin, 13353 Berlin, Germany.
‖
Present Address: Neurosurgical Research, Department of Neurosurgery, University Hospital and Walter-Brendel-Centre of Experimental Medicine, Faculty of Medicine, LMU Munich, 81377 Munich, Germany.
Int. J. Mol. Sci. 2020, 21(11), 4179; https://doi.org/10.3390/ijms21114179
Submission received: 28 May 2020
/
Accepted: 9 June 2020
/
Published: 11 June 2020
(This article belongs to the Special Issue Advances of Molecular Biology and Translational Aspects in CNS Tumors)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Glioblastoma (GBM) present with an abundant and aberrant tumor neo-vasculature. While rapid growth of solid tumors depends on the initiation of tumor angiogenesis, GBM also progress by infiltrative growth and vascular co-option. The angiogenic factor apelin (APLN) and its receptor (APLNR) are upregulated in GBM patient samples as compared to normal brain tissue. Here, we studied the role of apelin/APLNR signaling in GBM angiogenesis and growth. By functional analysis of apelin in orthotopic GBM mouse models, we found that apelin/APLNR signaling is required for in vivo tumor angiogenesis. Knockdown of tumor cell-derived APLN massively reduced the tumor vasculature. Additional loss of the apelin signal in endothelial tip cells using the APLN-knockout (KO) mouse led to a further reduction of GBM angiogenesis. Direct infusion of the bioactive peptide apelin-13 rescued the vascular loss-of-function phenotype specifically. In addition, APLN depletion massively reduced angiogenesis-dependent tumor growth. Consequently, survival of GBM-bearing mice was significantly increased when APLN expression was missing in the brain tumor microenvironment. Thus, we suggest that targeting vascular apelin may serve as an alternative strategy for anti-angiogenesis in GBM.
1. Introduction
Glioblastoma (GBM) is the most frequent and most aggressive primary brain tumor [1], and, -despite intensive efforts including recent advances of anti-GBM treatment modalities, there are still only limited therapeutic options, offering patients only an average survival time of one year after diagnosis, at best [2,3]. Hallmarks of GBM biology include strong invasiveness of tumor cells and the abundance of an aberrant vasculature [4]. GBM cells infiltrate normal brain tissue and, as a result, surgical resection is always incomplete [3,5,6]. Regardless of aggressive surgery and radio and chemotherapy, GBM remain uniformly fatal in patients. Hence, alternative therapeutic approaches for treatment against GBM are urgently needed.
The finding that highly vascularized tumors are rapidly growing while tumors with low vessel density are rather dormant led to the idea that tumor angiogenesis was required for tumor progression [7]. Vascular endothelial growth factor—A (VEGFA), which was first identified as a vascular permeability factor—turned out to be a potent endothelial mitogen [8]. The treatment of xenografted mice with a specific monoclonal VEGFA antibody demonstrated that VEGFA was a strong in vivo pro-angiogenic factor and that its blockade suppressed tumor growth [9]. This led to the first successful anti-angiogenic therapy with a humanized monoclonal antibody that blocks VEGFA (bevacizumab) in colorectal cancer and resulted in FDA approval [10]. However, recent clinical studies performed in patients with GBM using bevacizumab (AVAglio, RTOG-0825) did not show an improved overall survival [11,12]. Possible explanations for treatment resistance to bevacizumab are the ability of GBM cells for vascular co-option to obtain access to blood supply [13] or the upregulation of alternative angiogenic factors [14]. In the search for such additional factors involved in tumor angiogenesis, Masiero and colleagues [15] analyzed the expression profile of more than 1000 well-vascularized primary human cancers (head and neck squamous cell carcinomas, breast cancers, and clear cell renal cell carcinomas) for genes correlating with that of several well-recognized angiogenesis signature genes. The angiogenesis core signature they identified contained a set of 40 genes, including the apelin receptor (APLNR; also known as APJ). The human APLNR gene was coincidently cloned with primers designed to obtain vasopressin receptors in 1993 and found to encode a 7 transmembrane receptor related to the angiotensin II receptor type (AGTRL1) [16]. By screening various tissue extracts for ligands of orphan G-protein coupled receptors (GPCR) in an extracellular acidification assay, Tatemoto et al. (1998) [17] identified the apelin isoforms apelin-36, apelin-17, apelin-13, and the pyro-glutamylated (Pyr)apelin-13 as bioactive peptides encoded by the apelin gene (APLN). It is now established that the different apelin isoforms are the cognate ligands for APLNR with different binding properties [18].
In our previous work, we described a proangiogenic role of apelin acting as chemoattractant during vertebrate development [19]. That apelin signaling is instrumental for cell attraction was first shown for APLNR-transfected cell lines [20,21] and then verified for immortalized and primary vascular endothelial cells [19,22,23], vascular smooth muscle cells [24,25], lymphatic endothelial cells [26], and one tumor cell line (lung adenocarcinoma cell line A549) [27]. The functional importance of apelin in angiogenesis and its epistatic relationship with VEGFA signaling was established in a series of loss- and gain-of-function experiments in Xenopus tadpoles [19,22] and further confirmed in other organisms, like zebrafish and mouse [28]. In early vessel development, apelin appears to act in a paracrine fashion to first induce intersomitic blood vessel outgrowth shaping the primitive vascular plexus [19,29,30]. Next, during vessel sprouting, APLN-expression gets confined to endothelial tip cells, while the receptor is also found in the stalk cells controlling vessel-guidance and -maturation [19,28,31]. APLN can thus be used as a tip cell marker for ongoing angiogenesis and has a role in sprouting angiogenesis in pathology [32]. While APLNR mutant mice are born at sub-mendelian ratios, APLN knockout (KO) mice are viable and fertile [33,34,35]. The lethality observed in APLNR-KO mice is due to growth retardations and cardiac malformations during embryogenesis [36].
By in situ hybridization on GBM patient specimens, we found the first indication of a role for apelin signaling in tumor development. While APLN RNA was undetectable and only low level of APLNR RNA was found in normal brain vessels, we detected a dramatic upregulation of both within GBM-associated microvascular proliferations, particularly in areas of vessel sprouting and branching as assessed by in situ hybridization [19]. Co-expression of ligand and receptor in the tumor vasculature suggested an autocrine mode of signaling, similar to the one observed during embryonic development [19]. In addition, we also found abundant APLN expression in the pseudo-palisading areas of GBM. In these hypoxic regions, APLN is co-expressed with VEGFA [19,37], suggesting a cooperative function of apelin and VEGFA in paracrine signaling from tumor to endothelial cells during GBM angiogenesis.
All these data hint towards a vascular function for apelin/APLNR signaling during GBM growth. To study the role of apelin in establishing the structure and function of the GBM vascular beds and its implications to tumor growth, we here used established GBM mouse models. Intracerebral implantation of GBM cell lines with different APLN expression levels showed that tumor-derived apelin is required for the formation of the GBM neo-vasculature. Moreover, loss-of-APLN in the tumor microenvironment of APLN-KO mice showed that APLN expression controls patterning of the glioma vasculature. We also show, for the first time, that this sprouting angiogenesis effect is specific to APLN gene function as the vascular patterning phenotype was rescued by adding back the bioactive apelin-13 peptide. Furthermore, we found that the loss-of-APLN expression in the tumor microenvironment reduced angiogenesis-dependent tumor growth and increased the survival of GBM-bearing mice. Together, our data demonstrates that the endothelial APLN signal can serve as an alternative target to reduce GBM growth by specifically blocking sprouting angiogenesis.
2. Results
2.1. APLN Is Expressed at Variable Levels in GBM Cells and Upregulated in the Pathologic Neo-Vasculature
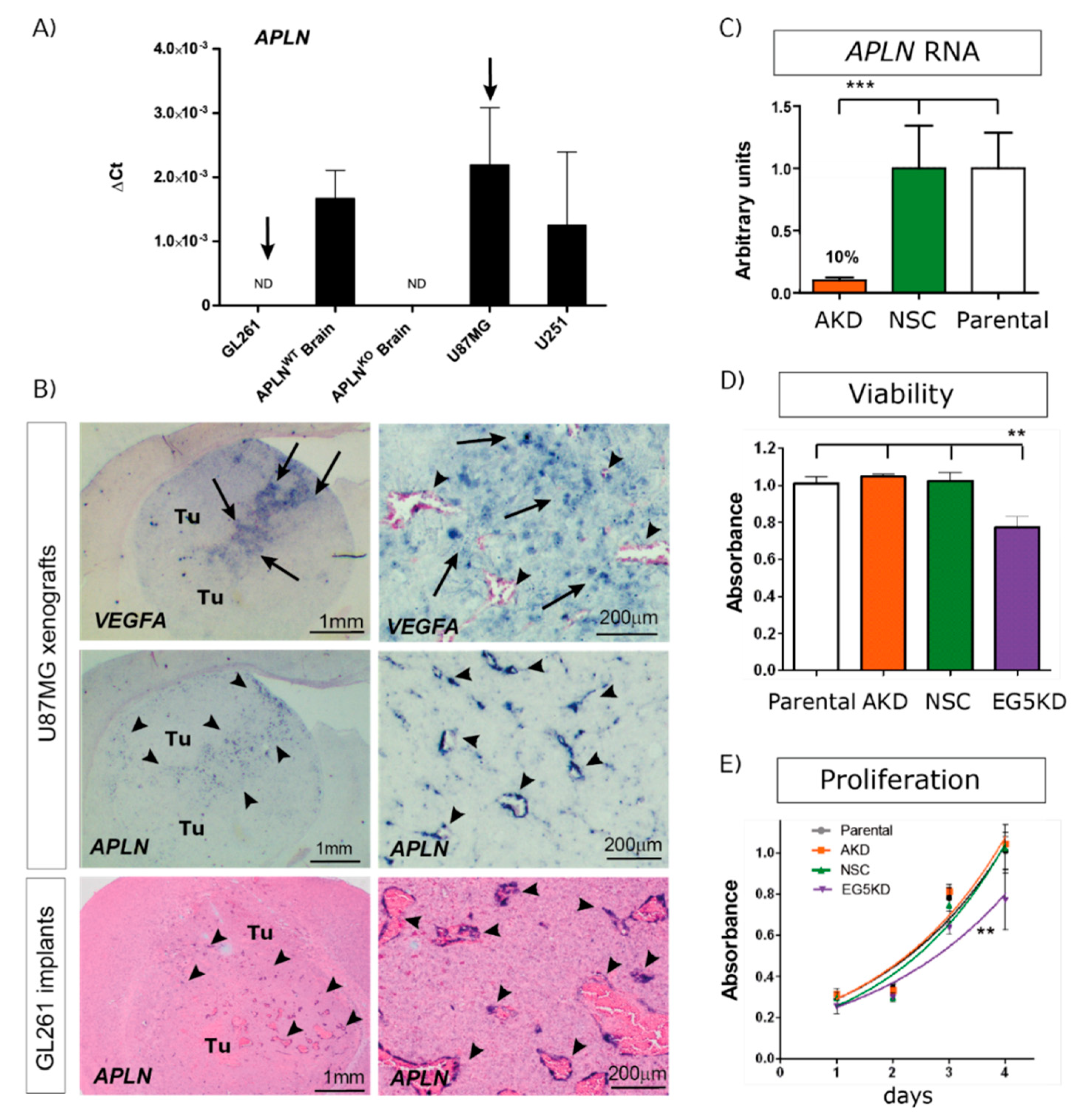
To study if tumor cell-derived APLN controls angiogenesis-dependent GBM growth, we first tested widely used GBM cell lines for APLN expression, revealing differing levels of expression. Using APLN expression levels in mouse wildtype (WT) brains as a comparison, we characterized the human cell-line U87MG as having high levels of APLN expression and the human U251 (formerly known as U373MG) cell line expression as being lower, whilst in the murine GL261 glioma cell-line APLN expression was not detectable (n.d.; Figure 1A).
In the next step, we orthotopically implanted human, as well as murine, GBM cells into immunodeficient or immunocompetent mice, respectively, to assess APLN expression in in situ pathology. In a previous study, we had found that APLN was not only expressed in GBM cells but was also upregulated in the vascular proliferates of the disease [19]. Thus, we also tested our experimental GBM models for vascular APLN expression and found that APLN expression was upregulated in newly forming tumor vessels (Figure 1B; arrowheads) of the xenografts. Interestingly, the vascular APLN RNA signal was highest in the tumor regions where human VEGFA RNA was also detected. VEGFA expression is a marker for hypoxic tumor areas in pseudopalisading necrosis in GBM. In addition, in murine GL261 glioma, APLN expression became localized to newly forming tumor vessels but remained absent from GL261 tumor cells (Figure 1B). To also address the role of GBM cell-derived APLN-levels, we selected the high APLN-expressing U87MG cell line, in addition, and lentivirally transduced it with shRNA against APLN RNA to create tumor cells with stable reduction of APLN RNA expression by 90% (U87AKD) compared to the non-silencing control (NSC) shRNA-transduced cells (U87NSC) or to untransduced parental U87MG cells (Figure 1C). Viability, as well as in vitro proliferation, of the manipulated cells was tested and turned out to be unchanged compared to their parental controls (Figure 1D,E). In contrast, cells transduced with the shRNA against EG5 kinesin (encoded by the Kinesin Family Member 11 gene KIF11 involved in nuclear division and cell growth [38]) caused reduced viability and proliferation rates (Figure 1D,E).
2.2. APLN Expression Is Required for the Formation of the GBM Neo-Vasculature
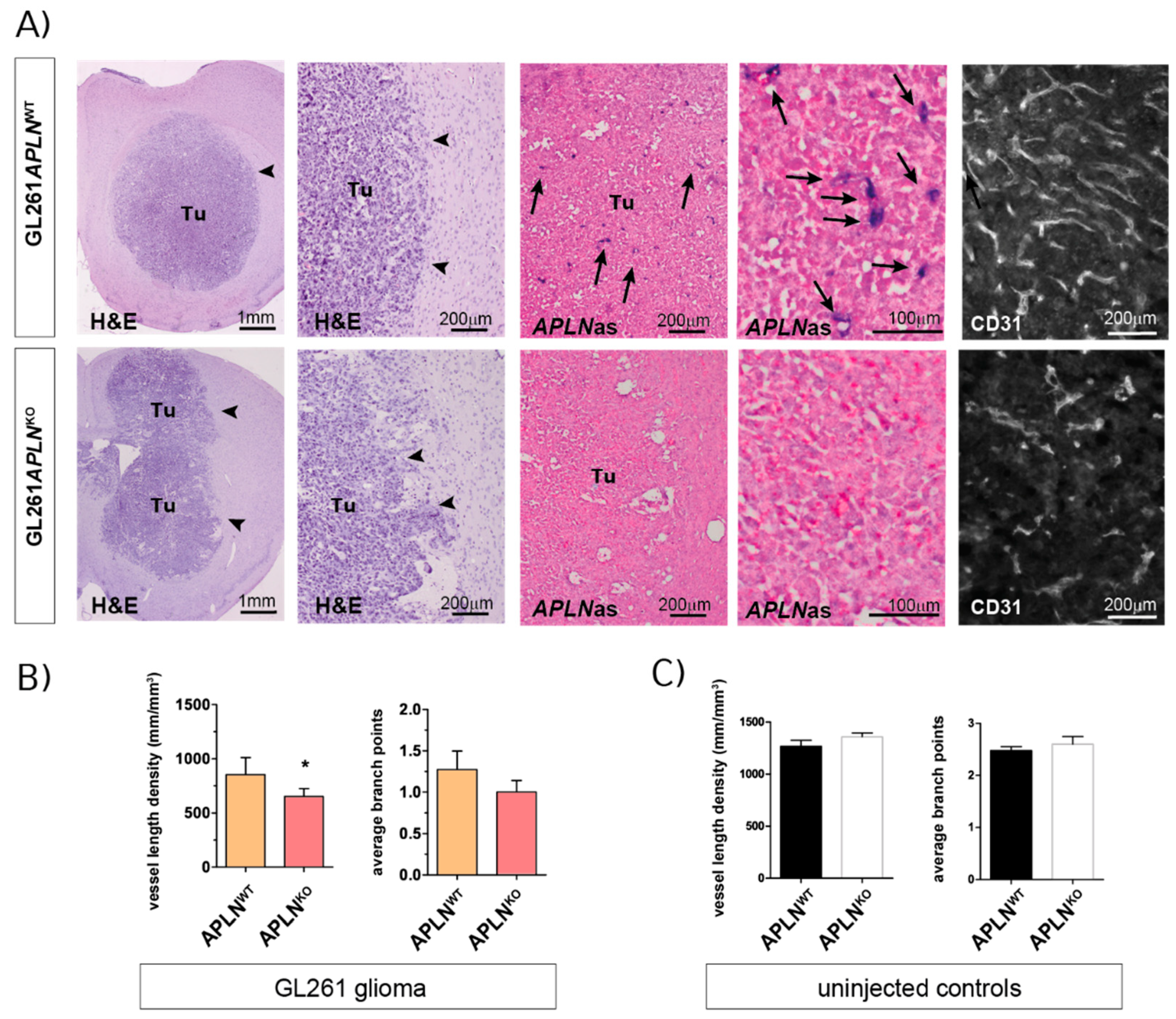
In agreement with a range of previous reports [39,40], we observed that orthotopic implantation of GL261 cells generated compact glioma mass 21 days post-implantation (dpi) with an abundant but aberrant tumor vasculature (Figure 2A, upper panel). To investigate the impact of APLN expression derived from the tumor microenvironment, namely APLN expressed on endothelial tip cells [19,28,31,32] during sprouting angiogenesis (Figure 1B), we implanted GL261 cells into APLNKO mice. We found that the tumor vasculature was significantly attenuated compared to APLNWT control mice (Figure 2A,B). Specifically, we found by stereological assessment of the tumor tissues that the vessel length density (VLD), as a read-out for the extent of tumor vascularization independent of tumor-size [41,42,43], was 856 mm/mm3 in controls, while this number significantly decreased to 653 mm/mm3 in APLNKO glioma (Figure 2B). Note that in situ hybridization for mouse APLN RNA shows that APLN is absent in tumor cells, as well as in the GBM microenvironment, of APLNKO mice, while APLN expression in APLNWT mice is specifically upregulated in endothelia of the tumor neo-vasculature (Figure 2A; arrows). Interestingly, the VLD determined in GL261 implants is smaller than the values observed for a healthy tumor-free brain. In the caudate putamen, which is the brain region targeted by the tumor cell implantation, we measured a VLD of 1269 mm/mm3 for APLNWT mice and 1359 mm/mm3 for APLNKO mice, without the difference being significant (Figure 2C). Similar values for VLD were also previously reported for the tumor-free hippocampus (1100 mm/mm3) of adult mice [44]. This finding of reduced vessel density in GBM as compared to healthy brain can be explained by the neoplastic growth and the higher cell density inside tumors. While the VLD of tumor-free brain is unchanged in APLNKO mice, the VLD of the glioma is reduced in APLNKO mice compared to APLNWT controls. In addition, loss-of host-derived APLN also resulted in reduced vascular complexity, serving as an indicator of the extent of angiogenic sprouting [45]. We found that the average number of vessel branch-points (ABP), which measured 1.25 in APLNWT glioma, was reduced to 1 in tumor vessels of APLNKO mice. In addition, the ABP values in the tumor were thus lower than the values observed in tumor-free striatum (Figure 2C; ABP = 2.5 in APLNWT and 2.6 in APLNKO). This means that the vascular complexity in APLNKO GBM is low to nearly linear, and this indicates that no more angiogenic sprouting took place during tumor growth.
Together, these results show that autocrine signaling of endothelial apelin to APLNR-expressing vessels is required for sprouting angiogenesis to take place and shape the complex tumor neo-vasculature.
2.3. Paracrine and Autocrine APLN Signaling Controls GBM Angiogenesis
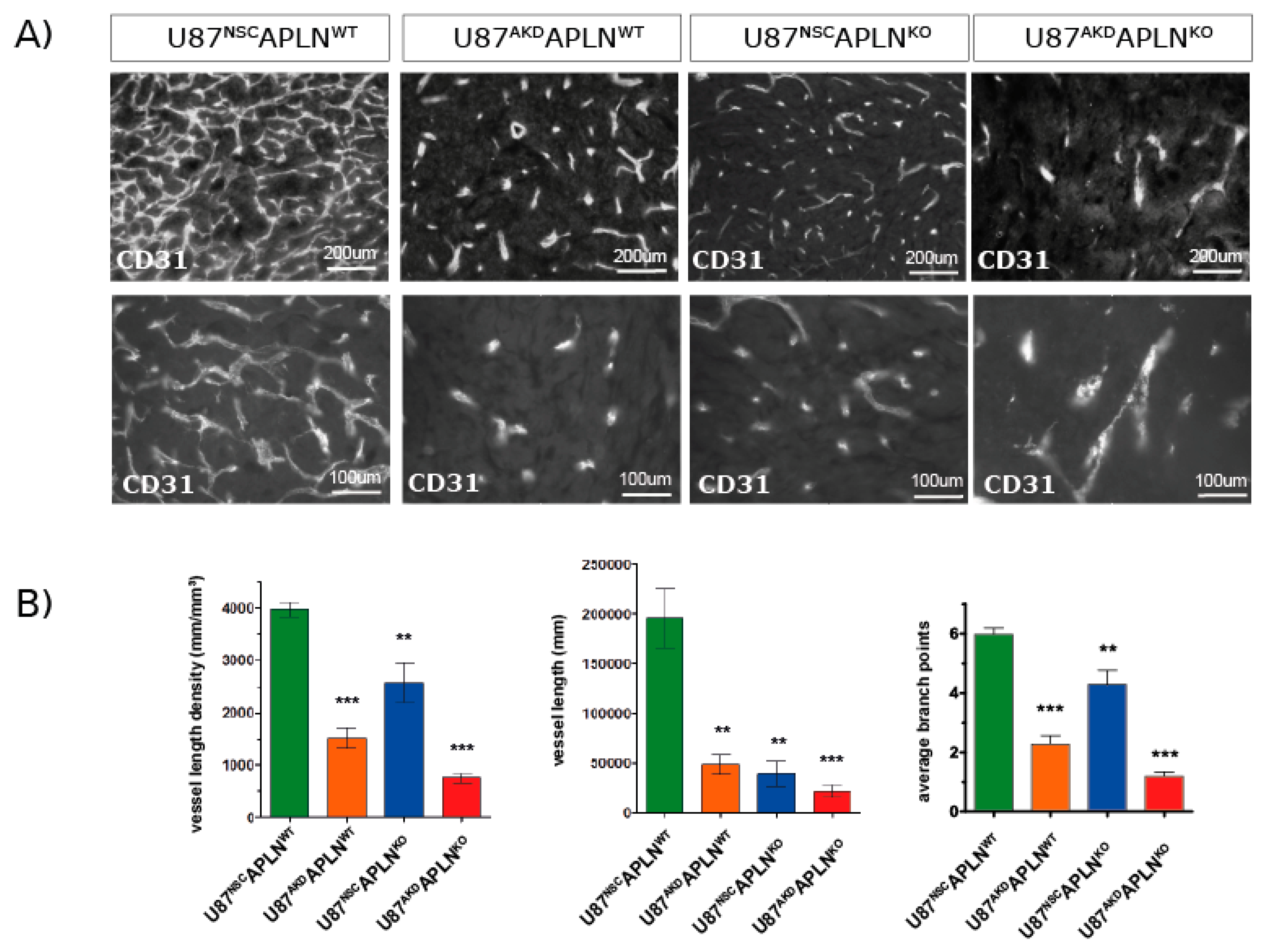
Next, we wanted to study the contribution of GBM cell-derived APLN to tumor angiogenesis and thus we compared vascular patterns generated by U87MG cells with a stable knock-down for APLN (U87AKD) to non-silencing control cells (U87NSC; Figure 1C–E). The modulation of human GBM-derived APLN is pathologically meaningful in mouse models as the amino acid sequence of the bioactive peptide apelin-13 of human or mouse origin is 100% identical [19]. Four weeks after tumor-induction with U87NSC cells in immune-deficient WT mice (U87NSCAPLNWT), we observed an extremely dense tumor vasculature (Figure 3A) with a VLD of 3937 mm/mm3 (Figure 3B), while the VLD of U87AKD-induced GBM in WT mice (U87AKDAPLNWT) was 1519 mm/mm3 (62% reduced as compared to U87NSCAPLNWT). Injection of U87NSC into APLNKO mice (U87NSCAPLNKO) resulted in a VLD of 2601 mm/mm3, whereas combined ablation of APLN expression in U87MG cells and in the host (implantation of U87AKD cells into immune-deficient APLNKO mice; U87AKDAPLNKO) largely blocked tumor angiogenesis and resulted in a VLD of 766 mm/mm3, which is lower than in tumor-free brain-tissue (Figure 2C and Figure 3B). While vessel branching reflects the picture observed by VLD assessment, the total vessel length (that is the VLD multiplied by the individual tumor volume) was highly reduced in all three xenografts with reduction of APLN expression (Figure 3B). At the same time, the change in APLN-levels did not affect the extent of pericyte cell coverage as assessed by CD31/Desmin double-immunofluorescence (Figure S1A). BrdU in vivo labeling of the tumor vasculature further demonstrated that endothelial cells are still able to proliferate upon APLN level reduction (Figure S1B).
These data showed, on one hand, that the high expression of APLN in the U87MG (and U87NSC control) cells induces a very dense and complex tumor vasculature as compared to U87AKD cells depleted in APLN-expression, as well as the APLN-non expressing GL261 cells. On the other hand, the loss-of-APLN in both GBM cell models in the tumor microenvironment decreased vascular sprouting to a level even lower than the vessel density observed throughout the healthy brain.
Together, these experiments indicate that paracrine (from the tumor) and autocrine (from the host microenvironment) APLN release has profound effects on the extent of GBM angiogenesis and thus the vascular pattern generated.
2.4. Apelin-13 Specifically Controls Vessel Density in the GBM Neo-Vasculature
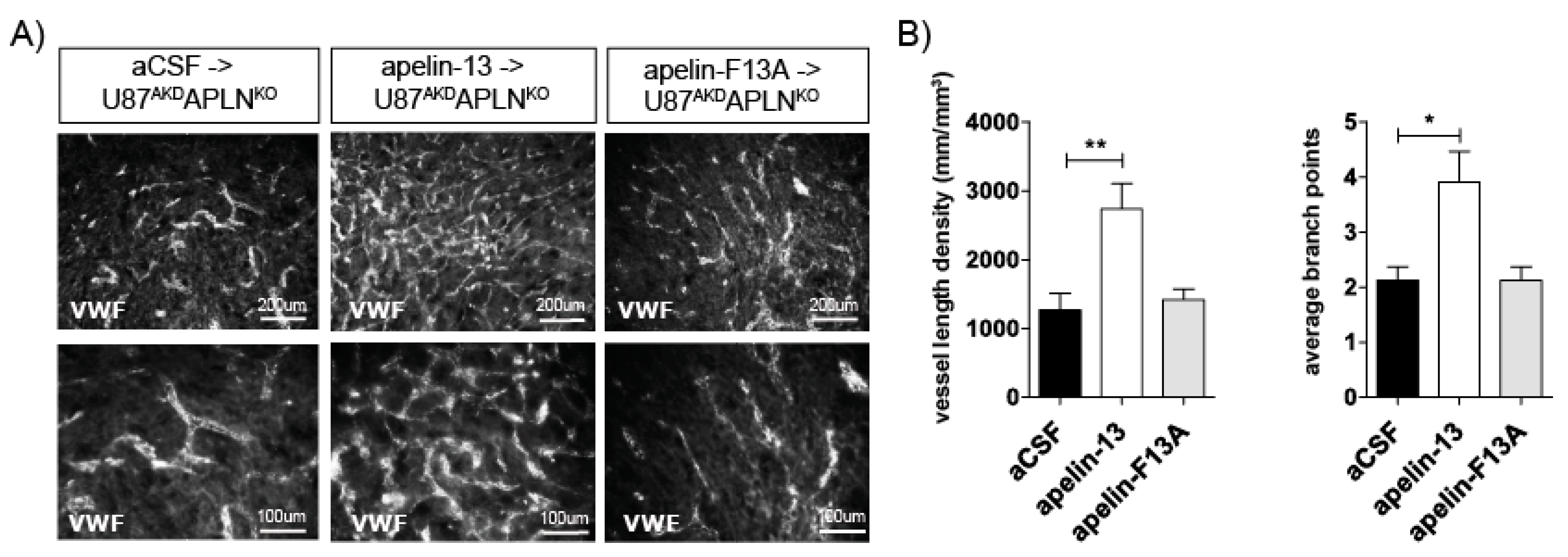
In the following experiment, we addressed the question if APLN-deficiency in our mouse model (i.e., in the APLNKO mice) would lead to, e.g., developmentally regulated alterations in the signal transduction pathways controlling angiogenesis or if APLN-responsiveness by APLNR is maintained in the absence of the ligand. Therefore, we infused the bioactive apelin-13 peptide into the U87AKDAPLNKO xenograft model and were indeed able to obtain a partial rescue of the tumor neo-vasculature increasing VLD from 766 mm/mm3 (infusion of artificial cerebrospinal fluid; aCSF control) to 2573 mm/mm3 (Figure 4). In contrast, the C-terminally mutated peptide apelin-F13A (which had been used as an antagonist for physiological functions on APLNR before [46]) was not able to rescue tumor angiogenesis. Instead, when apelin-F13A was infused into U87NSCAPLNWT GBMs expressing endogenous APLN levels, it significantly reduced VLD working as a competitive antagonist on APLNR (Figure S1C). Together, this part of our study supports the notion that APLNKO mice are devoid of endogenous APLN but can respond to exogenous or tumor-derived APLN. This is a prerequisite to explain the gradual anti-angiogenic responses to APLN-ablation in our GBM models. Here, we observed that APLN -deficiency in the GBM-cells or in the host generates a tumor mass with a reduced vascularization, as compared to tumor with unmodulated APLN levels. Ablation of APLN in both the GBM cells and in the tumor-bearing animals resulted in a striking cooperative anti-angiogenic effect. The partial rescue of the vascular GBM phenotype by addition of apelin-13 peptide to APLN-depleted xenografts shows that these effects are specifically mediated by apelin-signaling.
2.5. Loss of APLN Reduces Angiogenesis-Dependent Tumor Growth
To compare the effect of APLN on tumor growth, we performed a classical test for in vivo anti-angiogenic pharmacological function [9,47]. By generating subcutaneous xenografts, we found that APLN knockdown in tumor cells, as well as the loss-of-APLN, in the tumor microenvironment significantly reduced VLD by 36% (U87AKDAPLNWT) and 48% (in U87NSCAPLNKO), respectively, as compared to U87NSCAPLNWT controls (Figure 5). The depletion of both, tumor and host APLN in U87AKDAPLNKO xenografts further reduced VLD by 61% as compared to U87NSCAPLNWT control. Interestingly, the tumor volume was reduced by 50% when tumor-cell derived APLN was depleted compared to APLN-expressing control cells. Strikingly, if APLN was missing in the tumor microenvironment, the tumor volume decreased to less than 15% compared to WT controls. We also assessed if the tumor cells were growing equally well in all mice. We found that tumor take was similar for APLN-knockdown (87% for U87AKDAPLNWT) compared to U87NSC control cells (90% for U87NSCAPLNWT) when implanted in WT mice and only slightly reduced when APLN was missing in the tumor microenvironment in APLNKO but still present in the tumor cells (72% for U87NSCAPLNKO xenografts). However, when APLN expression was fully depleted as in U87AKDAPLNKO xenografts, only 50% of tumor cell implantation led to successful tumor growth.
This experiment demonstrated that apelin is important for tumor angiogenesis and that compact subcutaneous tumors depend on the apelin-induced neo-vasculature for their growth.
2.6. Loss-of-APLN Expression in the Tumor Microenvironment Increases Survival of Glioma-Bearing Mice
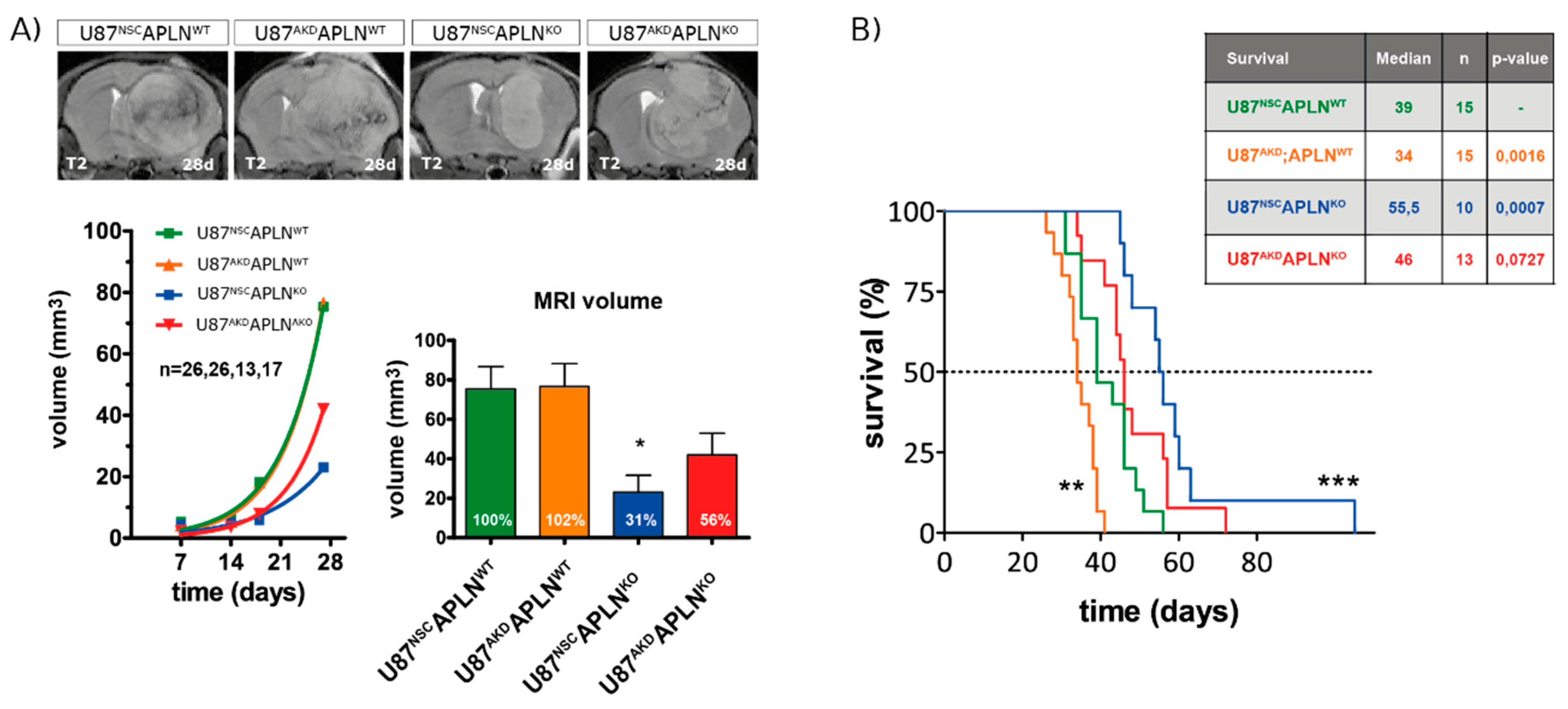
Finally, to assess the impact of apelin-controlled tumor angiogenesis on survival, we implanted the human GBM cells into APLNWT or APLNKO mice orthotopically. To follow in vivo tumor growth over time, we performed longitudinal magnetic resonance imaging (MRI) and found that all four groups of xenografts expanded exponentially however at a lower rate in APLNKO mice (Figure 6A). At 28 dpi, the U87NSCAPLNKO glioma showed a significantly smaller tumor volume of 31% compared to U87NSCAPLNWT tumors (defined as 100%). These results are pathologically relevant as we could confirm them in the survival experiment in which the APLNKO mice bearing U87NSCAPLNKO xenografts survived longest (Figure 6B). Survival of this group increased by 42.3% compared to the U87NSCAPLNWT control group. Surprisingly, APLNWT mice receiving the apelin-deficient xenografts (U87AKDAPLNWT) survived by 14.7%, significantly shorter than controls (U87NSCAPLNWT). Interestingly, when APLN levels were decreased stepwise in the tumor microenvironment and the tumor cells, the cell behavior of the GBM cells changed towards increased invasiveness (Figure S2).
In summary, we found that the absence of APLN expression from the tumor microenvironment showed a beneficial effect on the survival of GBM-bearing mice.
3. Discussion
In this study, we show, for the first time, that apelin peptide specifically and directly controls in vivo GBM angiogenesis. Moreover, we demonstrate that compact GBM growth depends on apelin-directed angiogenesis. Interestingly, the absence of apelin upregulation in the tumor vasculature of APLNKO mice led to smaller tumor volume and a significant extension of the survival time for GBM-bearing mice. The results of our study fit with previous in vivo observations made, where ectopic apelin overexpression enhanced angiogenesis and tumor cell proliferation in subcutaneous tumor models [26,48,49]. Moreover, recent results obtained in a mouse model of breast cancer showed that loss-of-APLN markedly reduced tumor angiogenesis leading to impaired tumor growth and, consequently, improved survival of these animals [50,51].
In line with these findings, we found in our GBM mouse model that the absence of APLN expression from the tumor vasculature showed a beneficial effect on the survival of GBM-bearing mice; however, the reduction of APLN expression from the tumor cell had an ambiguous effect on in vivo GBM volume. Specifically, we found that tumor-derived, as well as ectopically infused, apelin-13 could rescue partially the vascular phenotype. However, the tumor volume did not increase in these models (Figure S2D). Thus, vascular APLN expression seems to be essential for the formation of a mature tumor neo-vasculature that supports GBM growth. One explanation for this finding might be that the apelin-peptide is present in many different isoforms that could confer additional biological effects [17,52].
Another possibility could be that certain effects might be mediated by the second APLNR ligand APELA that was recently identified to be expressed in GBM [53]. APELA expression was associated with poor patient survival and correlated with glioma grade. APELA had been identified as an early APLNR ligand during embryogenesis being essential for early cardiovascular development and controlling cellular movements through APLNR [54,55]. In GBM, APELA expression was detected in stem cell niches and may thus drive tumorigenesis by supporting GSC growth [53]. A role in GBM angiogenesis has not been described yet.
Previously, ectopic expression of APLN in subcutaneous tumors was described to stabilize tumor vessels and support vascular maturation [56]. Moreover, overexpression of APLN reduced the leakiness of vessels in models of ischemia [35]. Our finding that the in vivo volume assessed by MRI was smallest when APLN was expressed by the tumor cells, but not by the vessels, might be explained by improved tightness of the neo-vasculature and, consequently, improved function and reduced formation of brain edema. In contrast, the loss-of-APLN expression in the tumor cells and the tumor vessels may lead to less functional vessels that finally induce the tumor cells to change their growth behavior. Indications for such a change in tumor cell behavior we recently found in a study of highly invasive glioblastoma stem-like cells [57]. Using these infiltrative GBM mouse models, we found increased in vivo, as well as in vitro, invasion, when the tumor cells lost autocrine apelin signaling [57]. Interestingly, U87AKD cells with low apelin expression also showed increased invasion in vitro as compared to U87NSC cells [57]. Thus, we re-inspected the U87MG xenografts (Figure S2A–C) and found that single invasive cells were detectable detaching from the compact GBM mass when APLN expression was knocked down in tumor cells (U87AKD). Hence, the notoriously low invasiveness that is generally observed with the U87MG cell line (Figure 1B) seemed to be increased upon loss-of tumor-derived apelin/APLNR signaling. We also observed in our second immunocompetent GBM model, the APLN-non-expressing GL261 cells, that GBMs growing in APLNKO mice exhibited a drastic increase in glioma cell-invasion (Figure 2A; see arrowheads in Gl261 APLNKO samples) as compared to controls. While tumor volume slightly decreased, the invasive score significantly increased from 1.2 to 2.8 (Figure S2E). Moreover, by confocal microscopy, we observed that the GBM cells in the invasive U87AKDAPLNKO GBMs adhere to remaining tumor vessels instead of being equally distributed amongst the highly dense vascular network in the U87AKDAPLNKO control GBMs. And, finally, we found in a qPCR screen for angiogenesis and invasion-related genes that loss-of-apelin directed angiogenesis led to a change in their expression Figure S2F). Marker gene expression levels for angiogenesis like KDR, VEGFA, FGF2, HIF1a, and APLN itself were decreased. Instead Timp1, the inhibitor of the matrix metalloproteinase 2 MMP2, was downregulated in U87AKDAPLNKO GBMs compared to the wildtype situation, and MMP2 itself, a proteinase degrading the extracellular matrix to support tumor cell invasion, was upregulated. Taken together, it seems that APLN-deficient GBM cells can change their cellular behavior to adhere to the remaining tumor vessels supporting tumor growth by increased invasiveness into the tumor-free brain. Such a shift towards a more invasive behavior of the tumor might be a reason for GBM recurrence after VEGFA-centered anti-angiogenic therapy, in addition to utilizing alternative pro-angiogenic factors [14], as previously observed in glioma models after blockage of angiogenesis by inhibition of VEGFA signaling [58,59,60]. We also showed that anti-VEGF therapy resulted in decreased APLN expression in the glioblastoma-stem-like model, resulting in increased tumor cell invasiveness [57]. To avoid this pro-invasive side effect, we used an alternative approach to target apelin/APLNR signaling by infusion of APLNR antagonists [57]. We, and others, found that the use of such competitive antagonistic peptides, like apelin-F13A or MM54, indeed led to reduced vascularization (Figure S1C), attenuated tumor progression, and increased survival in GBM mouse models, as well as models of breast cancer [51,57,61]. In the breast cancer model, tumor cell invasion was also changed and led to reduced metastatic growth [51]. What hampers the clinical application of such a peptidic agent is their usually low penetration into the target tissue.
Moreover, clinical trials of GBM therapies have highlighted the need to identify predictive markers for clinical outcomes of new therapeutic strategies [2,62]. APLN expression levels inversely correlate with overall survival in breast, colorectal, and lung cancer by directly correlating with increased angiogenesis in these tumors [48,51,63,64]. Furthermore, APLNR is a marker of increased angiogenesis in many cancer types [15]. However, in GBM, we found that the ratio between APLN and APLNR expression is an important indicator of therapy success, as low APLN and high APLNR expression correlated with increased GBM cell invasiveness [57]. Interestingly, targeting vascular APLNR by conditional ablation of APLNR-positive tumor vessels led to a significant reduction of tumor growth [65].
Thus, an overall depletion of apelin in GBM might not be the therapy of choice. Instead, together with our here-presented results, we propose that a more targeted-strategy to specifically deplete vascular apelin (e.g., using an apelin-specific antibody delivered intravenously) can open a new treatment avenue. This strategy would specifically block sprouting angiogenesis by not targeting the established brain vasculature and may lead to additional normalization of the tumor vasculature [50] also in GBM. In addition to the anti-angiogenic effect such an approach would circumvent the increased GBM cell invasion and carry the potential to reduce tumor hypoxia and edema formation, as well [66].
In summary, we found that apelin expression correlated with enhanced vascularization in GBM. Reduction of apelin expression in tumor cells attenuated tumor angiogenesis and ectopic infusion of apelin-13 peptide specifically reversed this vascular GBM phenotype. Moreover, loss-of-APLN in the mouse brain further reduced GBM vessel density below normal levels that are observed in the striatum of a healthy mouse brain, underlining the switch to an avascular tumor growth. By this blockade of APLN-dependent tumor angiogenesis, tumor growth was reduced and survival of GBM-bearing mice was, in turn, increased. Thus, we believe that targeting vascular apelin/APLNR signaling in the GBM microenvironment specifically offers a promising new opportunity to overcome resistance to anti-angiogenic therapy, as observed by VEGFA blockade, in GBM.
4. Materials and Methods
4.1. Cell Culture
U87MG and U373 cells were obtained from the American Type Culture Collection (ATCC), GL261 cells were obtained from the National Cancer Institute, NCI-Frederick (Tumor Cell Repository) and all cells were cultured under adherent conditions in DMEM containing 1× MEM non-essential amino acids, 5% penicillin-streptomycin and 10% FBS (all Thermo Fisher Scientific, Waltham, MA, USA). All cell lines were maintained at 37 °C in a humidified atmosphere of 95% O2 and 5% CO2.
4.2. Cell Transduction
For gene silencing of U87MG cells, lentiviral shRNAmir constructs for APLN (AKD; cat. RHS4430: V3LHS_401190), EG5 positive control (EG5KD; cat. RHS4480), or non-silencing control (NSC; cat. RHS4346) were produced in HEK293T cells using the TransLenti Viral GIPZ Packaging System (cat. TPLP4614, all, Dharmacon GE Life Sciences, Lafayette, CO, USA) according to the manufacturer´s instructions. Virus particle-containing supernatant was harvested two days after transfection, filtered with a 0.22 μm filter to avoid cellular contamination and stored at −80 °C. U87MG cells were detached with Trypsin/EDTA (Merck Millipore, Burlington, MA, USA). Eight by ten [4] cells were incubated with 500 μL of virus particles with an multiplicity of infection of 0.6–0.7 for six hours in a 24-well plate; then, 1 mL of medium was added, and cells were left overnight at 37 °C. The day after, cells were centrifuged and resuspended in fresh medium. After cell recovery, selection with puromycin (Sigma Aldrich, St.Louis, MO, USA) was performed for up to three weeks. Concentration of antibiotics had been previously determined by a kill curve. More than 99% efficiency of transduction/selection was confirmed by FACS and immunofluorescence.
4.3. Animals
All experiments were performed in compliance with the National Guidelines for Animal Protection, Germany, with approval of the local animal care committee of the “Landesamt für Gesundheit und Soziales” (LaGeSO) in Berlin, and every experiment was conducted after the guidelines of the UK Co-ordinating Committee on Cancer Research [67]. APLNKO mice were obtained from J.P. (Vienna, Austria) [34] and crossed to Rag2KO mice (B6.129S6-Rag2tm1Fwa) [68] kindly provided by Prof. G. Willimsky/Charité–Univeritätsmedizin Berlin previously purchased from Taconic (Rensselaer, NY, USA) (all on C57Bl/6J background). All mice were kept in a 12 h light/dark cycle with ad libitum access to food and water. Mice were sacrificed at defined presymptomatic time points or at humane end-point for the survival experiment.
4.4. Tumor Implantation
Mice were anesthetized with 7 μL/g of body weight of a mixture of Xylazine (Rompun 2%; Bayer Health Care, Leverkusen, Germany) and Ketamin (Ketavet; Zoetis, Berlin, Germany) in 0.9% NaCl, immobilized on a stereotactic frame (David Kopf Instruments, Tujunga, CA, USA) in flat-skull position and kept warm. A midline incision was made with a scalpel. One by ten [5] cells/μL in medium without supplements were implanted by stereotactic injection 1 mm anterior and 1.5 mm right to the bregma with a 22-gauge Hamilton syringe (Hamilton, Bonaduz, Switzerland) after drilling a whole into the skull with a 23G needle. At a depth of 4 mm, cells were slowly injected within 2 min and, after a settling period of another minute, the needle was removed in 1 mm step/minute. The incision was sutured and patched with Opsite spray dressing (Smith&Nephew, London, UK).
4.5. Intracerebral Drug Application
One day before implantation, micro-osmotic pumps were filled with 30 μg pyr-apelin-13 or apelin-F13A for 14 days (micro-osmotic pumps cat. 1002; Alzet; Charles River) in artificial cerebrospinal fluid (aCSF; as described by Alzet) or aCSF alone to be primed overnight in aCSF at 37 °C. Pump implantation was performed under anesthesia using the brain infusion kit 3 (Alzet) inserting the needle into the previously drilled hole after initial orthotopic tumor establishment as observed by magnetic resonance imaging. At the end of pump performance, animals were sacrificed.
4.6. Magnetic Resonance Imaging
MRI was performed using a 7 Tesla rodent scanner (Pharmascan 70⁄16AS, Bruker BioSpin Bruker, Billerica, MA, USA) with a 16 cm horizontal bore magnet and a 9 cm (inner diameter) shielded gradient with a H-resonance-frequency of 300 MHz and a maximum gradient strength of 300 mT/m. For imaging, a 1H-RF quadrature-volume resonator with an inner diameter of 20 mm was used. Data acquisition and image processing were carried out with the Bruker software Paravision 5.1. During the examinations, mice were placed on a heated circulating water blanket to ensure constant body temperature of 37 °C. Anesthesia was induced with 3% and maintained with 1.5–2.0% isoflurane (Forene, Abbot) delivered in 0.5 L/min of 100% O2 via a facemask under constant ventilation monitoring (Small Animal Monitoring & Gating System, SA Instruments, Stony Brook, NY, USA). For imaging the mouse brain, T2-weighted 2D turbo spin-echo sequence was used (imaging parameters for T2 TR/TE = 4200/36 ms, RARE factor 8.4 averages). Twenty axial slices with a slice thickness of 0.5 mm, a field of view of 2.60 × 2.60 cm and a matrix of 256 × 256 were positioned over the brain. Calculation of lesion volume was carried out with the program Analyze 5.0 (AnalyzeDirect, Inc., Mayo Clinic, MI, USA). The hyperintense tumor areas were assigned with a region of interest tool. This enables a threshold-based segmentation by connecting all pixels within a specified threshold range about the selected seed pixel and results in a 3D object map of the whole tumor region. Further, the total volume of the whole object map was automatically calculated.
4.7. HE Tumor Volume Analysis
Tumor volumes were obtained from Hematoxylin & Eosin (H&E)-stained tumor sections measuring the area of every 9th section and calculated by the Cavalieri Method [69].
4.8. Quantitative PCR
RNA extraction was performed using Trizol (Thermo Fisher Scientific) according to the manufacturer´s instructions. One microgram total RNA was reverse-transcribed into cDNA using QuantiTect Reverse Transcription Kit (Qiagen) and the cDNA was analyzed by quantitative PCR using TaqMan Gene Expression Assays for APLN (Hs00936329_m1; Mm00443562_m1; Rn00581093_m1), EG5 (Hs00189698_m1), and GAPDH (Hs99999905_m1; Mm99999915_g1; Rn01775763_g1) with TaqMan Gene Expression Master Mix (Cat. 4369016) in a StepOnePlus Instrument (all Thermo Fisher Scientific). Samples were amplified with the standard running method provided by StepOne Software v2.2.2, increasing the cycle numbers to 45. In every run, gene-of-interest expression levels were normalized to the house-keeping gene GAPDH.
4.9. Viability and Proliferation Assays
Six thousand cells/well were plated in 96-well plates in DMEM-F12 medium on day 0. Cell viability was measured after 24, 48, 72, and 96 h using a MTT assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay, Promega, Fitchburg, WI, USA) for cell metabolic activity according to manufacturer´s instruction, incubating the cells one hour with the “Stop Mix” solution. Absorbance was measured with Versa Max microplate reader and SoftMax Pro software (Molecular Devices) with a reference wavelength of 630 nm. Background absorbance from wells containing no cells was subtracted from all measurements, and six replicate samples were used in each experiment. Three experiments per cell type were performed.
4.10. In Situ Hybridization
Solutions were prepared with RNAse free water and sterilized. Sections on slides were deparaffinized by serial passages into Roti-Histol and graded alcohol (100–25%). Tissue was permeabilized with 10 min incubation in 10 μg/mL of Proteinase K (PeqLab, VWR). Slides were fixed for 10 min in 4% paraformaldehyde (PFA) and blocked for 10 min with Acetic Anhydride (0.25%; Sigma Aldrich) in Triethanolamine (1.5%; Sigma Aldrich). Sections were dried for 2 h at room temperature, then incubated overnight at 65 °C in a humidified chamber with DIG-labeled (DIG RNA labeling; Roche Diagnostics) antisense or sense probes at a final concentration of 7 μg/mL, and diluted in a hybridization solution containing ssDNA (100 μg/mL, Salmon sperm DNA; Ambion, Thermo Fisher Scientific) to mask unspecific binding and co-precipitant RNA (100 μg/mL, Yeast RNA; Ambion). RNA probes were generated from human VEGFA and mouse APLN cDNA as previously described [19]. Probe-containing hybridization solution was boiled at 95 °C for 10 min before application. On day two, unspecific signal was removed with graded stringency washes with saline sodium citrate from 20× to 0.1× and incubated with alkaline phosphatase conjugated anti-DIG antibody (Roche Diagnostics) overnight at 4 °C. On day three, slides were washed in PBT (0.1% Tween in 1×PBS) and incubated with BCIP/NBT substrate (Vector Laboratories, Burlingham, CA, USA) at 37 °C for up to four days. For counterstaining with Eosin, slides underwent serial passages in graded alcohol (70–100%) till Roti-Histol and were then mounted with Entellan (Merck Millipore). Pictures were taken under an Axioskop2 microscope with Axiocam 105 Color and Axiovision SE64 Rel. 4.9 software (Carl Zeiss, Jena, Germany).
4.11. Immunofluorescence and Vessel Density Quantification
Mice were transcardially perfused under Narcoren (Merial) anesthesia with 1x PBS followed by 4% phosphate buffered PFA. Brains were post-fixed for two days in 4% PFA and then left in 30% sucrose for at least 24 h at 4 °C. Freezing was performed embedding the tissue in Cryomatrix (Thermo Fisher Scientific) and brains were preserved at −20 °C. Tissue was sectioned horizontally in 40 μm-thick slices on a microtome. Floating sections were blocked for 1 h at room temperature in 1x PBS containing 5% normal donkey serum (NDS; cat. 017-000-121; Jackson Immuno-Research,) and 0.3% Triton-X (cat. 93418; Fluka) and incubated overnight at 4 °C with primary antibodies rat anti-CD31 (1:50, cat. 550274; BD Biosciences) or rabbit anti-VWF (1:400, cat. A0082; Dako, Agilent Technologies, Santa Clara, CA, USA). The next day, sections were incubated for 3 h at room temperature with the secondary antibodies biotin donkey anti-rabbit, anti-rat (1:250, cat. 711-065-152; 712-065-150), and/or 2 h at room temperature with Streptavidin-AF488 or -AF594 (1:500, cat. 016-540-084; 016-580-084, Jackson Immuno-Research). Alternatively, sections were directly incubated for 2 h at room temperature with the secondary antibodies donkey anti-rabbit AF488 or AF594 (1:500, cat. A-21206, A-21207 Thermo Fisher Scientific). All antibodies were diluted in blocking solution. After staining, tissue was mounted in Fluorescent Mounting Medium (Dako) and pictures taken at Axiovert25 microscope with Axiocam MRm and Axiovision Rel 4.8 software (Zeiss) or with confocal laser scanning microscopy at the Leica TCS SP5 Confocal with LAS AF software (Leica Microsystems). Stereological analysis of vessel density was performed in green fluorescent protein (GFP)-positive tumor area of the CD31- or VWF-positive red fluorescent vessels on every 9th section using the space ball method of the StereoInvestigator Software 10.21.1 (MicroBrightField Bioscience, Williston, VT, USA) connected to an Olympus-BX53-microscope (Olympus Europe, Hamburg, Germany, USA) and a motorized object table MicroBrightField Bioscience.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/11/4179/s1. Figure S1. Modulation of APLN-signaling changes vessel density but not pericyte coverage and endothelial proliferation. (A) U87MG was implanted into immunodeficient mice and grown to big xenografts within 28 dpi. Double-fluorescent immunostaining for CD31+ endothelial cells and Desmin+ pericytes was performed. Double-fluorescent staining confirms decreased vessel density upon reduction of APLN expression in tumour cells (U87AKDAPLNWT), the microenvironment (U87NSCAPLNKO) or both (U87AKDAPLNKO) compared to control (U87NSCAPLNWT). The number of pericyte-covered vessel objects was assessed by the “Count and measure” function in the Cellsense software (Olympus Life Science) on 20× micrographs of different tumour areas of three different tumour sections per animal (six animals per group) and no significant difference of the experimental group compared to the control group was observed (graph not shown). (B) U87MG were grown for 28 dpi and mice intravenously injected with the proliferation marker BrdU three hours before sacrifice. Tumours were stained by double-immuofluorescence for BrdU and Isolectin B4 (IB4) and confocal images were taken. In all four groups BrdU+ endothelial cells (IB4+) could be observed. (C) U87NSCAPLNWT GBM cells were treated with aCSF or the antagonistic peptide apelin-F13A by intracerebral infusion for 14 days and tumours assessed 28 dpi for vessel density by stereomorphometry (n = 6.5). Vessel length density significantly decreased compared to controls when apelin-F13A was used. Data are reported as mean +/-SEM; statistical significance (students t-test) is indicated ** p < 0.01. Figure S2. Decrease in APLN-expression correlates with increased GBM cell invasion. (A) U87MG cells generally form a very compact tumour 28 dpi (see U87NSCAPLNWT control in the left panel) but single invading cells (arrows) are especially detectable in U87AKDAPLNKO xenografts (right panel) by the GFP reporter. By H&E staining a gradual loosening of the tumor border (arrowhead) from left to right panel can be observed (arrows). (B) To assess the extent of GBM cell-invasion, we analysed every 8th tumour section per mouse assigning it an invasive score from 0 to 3 (where 0 is no histological sign of cell invasion from the tumour mass, 1 describes a larger, connected group of invading GBM cells, 2 indicates smaller scattered groups of invading GBM cells and 3 highlights single scattered highly invasive GBM cells). While the U87NSCAPLNWT control tumours grow fully compact, the invasive score gradually increases when APLN expression is knocked down in the tumor cells (U87AKDAPLNWT), the microenvironment (U87NSCAPLNKO) or both (U87AKDAPLNKO). In particular the U87NSCAPLNKO GBMs exhibit a strong and inter-individually heavily heterogeneous invasive behavior. Here, one set of tumours had moderate levels of invasion (invasive scores below 1, which are comparable to the invasive pattern in the U87AKDAPLNWT or U87NSCAPLNKO group), while other gliomas generated by the U87NSCAPLNKO cells were strongly invasive (invasive scores above 2). (C) Double immunofluorescent staining for GFP-positive tumor (green) and CD31-positive endothelial cells (red) of U87 tumours 28 dpi were performed for U87NSCAPLNWT or U87AKDAPLNKO xenografts and merged confocal micrographs (left panels) or a Volocity representation of the confocal z-stacks (right panels) are shown. Note that In the APLN-deficient U87AKDAPLNKO xenografts the GBM cells largely associate with the vasculature which may support directed invasion towards the tumour-free brain. (D) Infusion of Apelin-13 peptide intracerebrally decreased T2-weighted MRI tumour volume of U87AKDAPLNKO xenografts (n = 5) compared to control aCSF treated mice (n = 6). (E) Orthotopic GL261 tumor cell implants in APLNWT or APLNKO mice 21 days post implantation show a reduced tumour volume assessed by the Cavalieri Estimator method (StereoInvestigator) and increased tumor cell invasiveness (arrowheads) as quantified on H&E sections (n = 9.6 mice). Data are reported as mean +/-SEM; statistical significance (students t-test) is indicated *** p < 0.001. (F) Gene expression screen for change in 84 angiogenesis and invasion related genes. RNA was isolated from microdissected orthotpic xenografts of all four U87 GBM groups (n = 4 per group) 28 dpi with different APLN expression levels and a qPCR screen using an “RT2 Profile PCR Array Mouse Angiogenesis” (Qiagen PAMM 024Z) was performed. Marker genes involved in angiogenesis were downregulated when APLN expression was decreaed in the tumour cells or the microenvironment. Instead the marker for cell invasion MMP2 was found to be upregulated, while its inhibitor TIMP1 was downregulated. Data are reported as mean +/-SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated * p < 0.05, ** p < 0.01.
Author Contributions
Conceptualization, F.L.H. and R.E.K.; methodology, A.F., S.K., R.M., and R.E.K.; formal analysis, A.F., S.K., R.M., J.R. and R.E.K.; resources, R.E.K. and F.L.H.; data curation, R.E.K. and S.K.; writing R.E.K.; review and editing, all authors; visualization, R.E.K; supervision, F.L.H. and R.E.K.; funding acquisition, F.L.H. and R.E.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funding of the “Berliner Krebsgesellschaft”, the “Wilhelm Sander-Stiftung”, the “Anni-Hofmann Stiftung” and the “Familie Mehdorn Stiftung”.
Acknowledgments
We are thankful to Li Fohs-Li, Giorgia Mastrella and Mengzhuo Hou for technical assistance with in situ hybridizations, immunohistochemisty, and genotyping. We would especially like to thank Susanne Müller for great support with MRI imaging and analysis and Josef Penninger for providing the APLN-KO strain.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABP | Average branch points |
| aCSF | Artificial cerebrospinal fluid |
| AKD | Apelin knock-down |
| APLNKO | Apelin knock-out |
| APLNWT | Apelin wildtype |
| APLN | Apelin |
| APLNR | Apelin Receptor |
| dpi | days post implantation |
| GBM | Glioblastoma |
| GFP | Green fluorescent protein |
| NSC | Non-silencing control |
| VEGFA | Vascular endothelial growth factor-A |
| VLD | Vessel length density |
| VWF | von Willebrand factor |
| WT | Wild-type |
References
- Kleihues, P.; Cavenee, W.K. Pathology and Genetics of Tumours of the Nervous System; IARC Press: Lyon, France, 2000. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W.; et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; van den Bent, M.J.; Hegi, M.E. Optimal role of temozolomide in the treatment of malignant gliomas. Curr. Neurol. Neurosci. Rep. 2005, 5, 198–206. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Masiero, M.; Simoes, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Habata, Y.; Fukusumi, S.; Hosoya, M.; Fujii, R.; Hinuma, S.; Nishizawa, N.; Kitada, C.; Onda, H.; Nishimura, O.; et al. Molecular properties of apelin: Tissue distribution and receptor binding. Biochim. Biophys. Acta 2001, 1538, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Kälin, R.E.; Kretz, M.P.; Meyer, A.M.; Kispert, A.; Heppner, F.L.; Brändli, A.W. Paracrine and autocrine mechanisms of apelin signaling govern embryonic and tumor angiogenesis. Dev. Biol. 2007, 305, 599–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Ishida, J.; Yamamoto, R.; Fujiwara, K.; Asada, S.; Kasuya, Y.; Mochizuki, N.; Fukamizu, A. G protein-coupled APJ receptor signaling induces focal adhesion formation and cell motility. Int. J. Mol. Med. 2005, 16, 787–792. [Google Scholar]
- Hosoya, M.; Kawamata, Y.; Fukusumi, S.; Fujii, R.; Habata, Y.; Hinuma, S.; Kitada, C.; Honda, S.; Kurokawa, T.; Onda, H.; et al. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J. Biol. Chem. 2000, 275, 21061–21067. [Google Scholar] [CrossRef] [Green Version]
- Cox, C.M.; D’Agostino, S.L.; Miller, M.K.; Heimark, R.L.; Krieg, P.A. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev. Biol. 2006, 296, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Kasai, A.; Shintani, N.; Oda, M.; Kakuda, M.; Hashimoto, H.; Matsuda, T.; Hinuma, S.; Baba, A. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem. Biophys. Res. Commun. 2004, 325, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Samura, M.; Morikage, N.; Suehiro, K.; Tanaka, Y.; Nakamura, T.; Nishimoto, A.; Ueno, K.; Hosoyama, T.; Hamano, K. Combinatorial Treatment with Apelin-13 Enhances the Therapeutic Efficacy of a Preconditioned Cell-Based Therapy for Peripheral Ischemia. Sci. Rep. 2016, 6, 19379. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wen, J.; Zhou, Y.; Li, L.; Cui, X.; Wang, J.; Pan, L.; Ye, Z.; Liu, P.; Wu, L. Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci. Rep. 2015, 5, 8170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berta, J.; Hoda, M.A.; Laszlo, V.; Rozsas, A.; Garay, T.; Torok, S.; Grusch, M.; Berger, W.; Paku, S.; Renyi-Vamos, F.; et al. Apelin promotes lymphangiogenesis and lymph node metastasis. Oncotarget 2014, 5, 4426–4437. [Google Scholar] [CrossRef] [Green Version]
- Lv, D.; Li, L.; Lu, Q.; Li, Y.; Xie, F.; Li, H.; Cao, J.; Liu, M.; Wu, D.; He, L.; et al. PAK1-cofilin phosphorylation mediates human lung adenocarcinoma cells migration induced by apelin-13. Clin. Exp. Pharmacol. Physiol. 2016, 43, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Del Toro, R.; Prahst, C.; Mathivet, T.; Siegfried, G.; Kaminker, J.S.; Larrivee, B.; Breant, C.; Duarte, A.; Takakura, N.; Fukamizu, A.; et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood 2010, 116, 4025–4033. [Google Scholar] [CrossRef] [Green Version]
- Kasai, A.; Shintani, N.; Kato, H.; Matsuda, S.; Gomi, F.; Haba, R.; Hashimoto, H.; Kakuda, M.; Tano, Y.; Baba, A. Retardation of retinal vascular development in apelin-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1717–1722. [Google Scholar] [CrossRef]
- Kidoya, H.; Ueno, M.; Yamada, Y.; Mochizuki, N.; Nakata, M.; Yano, T.; Fujii, R.; Takakura, N. Spatial and temporal role of the apelin/APJ system in the caliber size regulation of blood vessels during angiogenesis. EMBO J. 2008, 27, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Strasser, G.A.; Kaminker, J.S.; Tessier-Lavigne, M. Microarray analysis of retinal endothelial tip cells identifies CXCR4 as a mediator of tip cell morphology and branching. Blood 2010, 115, 5102–5110. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Hu, T.; He, L.; Huang, X.; Tian, X.; Zhang, H.; He, L.; Pu, W.; Zhang, L.; Sun, H.; et al. Genetic targeting of sprouting angiogenesis using Apln-CreER. Nat. Commun. 2015, 6, 6020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, A.Y.; Chun, H.J.; Glassford, A.J.; Kundu, R.K.; Kutschka, I.; Ardigo, D.; Hendry, S.L.; Wagner, R.A.; Chen, M.M.; Ali, Z.A.; et al. F genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H88–H98. [Google Scholar] [CrossRef] [Green Version]
- Kuba, K.; Zhang, L.; Imai, Y.; Arab, S.; Chen, M.; Maekawa, Y.; Leschnik, M.; Leibbrandt, A.; Markovic, M.; Schwaighofer, J.; et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ. Res. 2007, 101, e32–e42. [Google Scholar] [CrossRef] [Green Version]
- Kidoya, H.; Naito, H.; Takakura, N. Apelin induces enlarged and nonleaky blood vessels for functional recovery from ischemia. Blood 2010, 115, 3166–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charo, D.N.; Ho, M.; Fajardo, G.; Kawana, M.; Kundu, R.K.; Sheikh, A.Y.; Finsterbach, T.P.; Leeper, N.J.; Ernst, K.V.; Chen, M.M.; et al. Endogenous regulation of cardiovascular function by apelin-APJ. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1904–H1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef]
- Weil, D.; Garcon, L.; Harper, M.; Dumenil, D.; Dautry, F.; Kress, M. Targeting the kinesin Eg5 to monitor siRNA transfection in mammalian cells. Biotechniques 2002, 33, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Machein, M.R.; Renninger, S.; de Lima-Hahn, E.; Plate, K.H. Minor contribution of bone marrow-derived endothelial progenitors to the vascularization of murine gliomas. Brain Pathol. 2003, 13, 582–597. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Dockery, P.; Fraher, J. The quantification of vascular beds: A stereological approach. Exp. Mol. Pathol. 2007, 82, 110–120. [Google Scholar] [CrossRef]
- Lokkegaard, A.; Nyengaard, J.R.; West, M.J. Stereological estimates of number and length of capillaries in subdivisions of the human hippocampal region. Hippocampus 2001, 11, 726–740. [Google Scholar] [CrossRef]
- Mouton, P.R.; Gokhale, A.M.; Ward, N.L.; West, M.J. Stereological length estimation using spherical probes. J. Microsc. 2002, 206, 54–64. [Google Scholar] [CrossRef]
- Franciosi, S.; Gama Sosa, M.A.; English, D.F.; Oler, E.; Oung, T.; Janssen, W.G.; De Gasperi, R.; Schmeidler, J.; Dickstein, D.L.; Schmitz, C.; et al. Novel cerebrovascular pathology in mice fed a high cholesterol diet. Mol. Neurodegener. 2009, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Walchli, T.; Mateos, J.M.; Weinman, O.; Babic, D.; Regli, L.; Hoerstrup, S.P.; Gerhardt, H.; Schwab, M.E.; Vogel, J. Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat. Protoc. 2015, 10, 53–74. [Google Scholar] [CrossRef]
- Lee, D.K.; Saldivia, V.R.; Nguyen, T.; Cheng, R.; George, S.R.; O’Dowd, B.F. Modification of the terminal residue of apelin-13 antagonizes its hypotensive action. Endocrinology 2005, 146, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Berta, J.; Kenessey, I.; Dobos, J.; Tovari, J.; Klepetko, W.; Jan Ankersmit, H.; Hegedus, B.; Renyi-Vamos, F.; Varga, J.; Lorincz, Z.; et al. Apelin expression in human non-small cell lung cancer: Role in angiogenesis and prognosis. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2010, 5, 1120–1129. [Google Scholar] [CrossRef]
- Sorli, S.C.; Le Gonidec, S.; Knibiehler, B.; Audigier, Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene 2007, 26, 7692–7699. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L. What is normal? Apelin and VEGFA, drivers of tumor vessel abnormality. EMBO Mol. Med. 2019, 11, e10892. [Google Scholar] [CrossRef]
- Uribesalgo, I.; Hoffmann, D.; Zhang, Y.; Kavirayani, A.; Lazovic, J.; Berta, J.; Novatchkova, M.; Pai, T.P.; Wimmer, R.A.; Laszlo, V.; et al. Apelin inhibition prevents resistance and metastasis associated with anti-angiogenic therapy. EMBO Mol. Med. 2019, 11, e9266. [Google Scholar] [CrossRef] [PubMed]
- Kälin, S.; Kälin, R.E. Adipocitokines, Energy Balance and Cancer—Energy Balance and Cancer; Reizes, O., Berger, N.A., Eds.; Springer International Publishing Switzerland: Cham, Switzerland, 2017; pp. 137–166. [Google Scholar]
- Ganguly, D.; Cai, C.; Sims, M.M.; He Yang, C.; Thomas, M.; Cheng, J.; Saad, A.G.; Pfeffer, L.M. APELA Expression in Glioma, and Its Association with Patient Survival and Tumor Grade. Pharmaceuticals 2019, 12, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, S.C.; Ho, L.; Tian, J.; Reversade, B. Elabela: A hormone essential for heart development signals via the apelin receptor. Dev. Cell 2013, 27, 672–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, A.; Norris, M.L.; Valen, E.; Chew, G.L.; Gagnon, J.A.; Zimmerman, S.; Mitchell, A.; Ma, J.; Dubrulle, J.; Reyon, D.; et al. Toddler: An embryonic signal that promotes cell movement via apelin receptors. Science 2014, 343, 1248636. [Google Scholar] [CrossRef] [PubMed]
- Kidoya, H.; Kunii, N.; Naito, H.; Muramatsu, F.; Okamoto, Y.; Nakayama, T.; Takakura, N. The apelin/APJ system induces maturation of the tumor vasculature and improves the efficiency of immune therapy. Oncogene 2011, 31, 3254–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrella, G.; Hou, M.; Li, M.; Stoecklein, V.M.; Zdouc, N.; Volmar, M.N.M.; Miletic, H.; Reinhard, S.; Herold-Mende, C.C.; Kleber, S.; et al. Targeting APLN/APLNR Improves Antiangiogenic Efficiency and Blunts Proinvasive Side Effects of VEGFA/VEGFR2 Blockade in Glioblastoma. Cancer Res. 2019, 79, 2298–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, P.; Ulbricht, U.; Bohlen, P.; Brockmann, M.A.; Fillbrandt, R.; Stavrou, D.; Westphal, M.; Lamszus, K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001, 61, 6624–6628. [Google Scholar]
- Lamszus, K.; Kunkel, P.; Westphal, M. Invasion as limitation to anti-angiogenic glioma therapy. Acta Neurochir. Suppl. 2003, 88, 169–177. [Google Scholar]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Harford-Wright, E.; Andre-Gregoire, G.; Jacobs, K.A.; Treps, L.; Le Gonidec, S.; Leclair, H.M.; Gonzalez-Diest, S.; Roux, Q.; Guillonneau, F.; Loussouarn, D.; et al. Pharmacological targeting of apelin impairs glioblastoma growth. Brain 2017, 140, 2939–2954. [Google Scholar] [CrossRef] [Green Version]
- Haynes, H.R.; Camelo-Piragua, S.; Kurian, K.M. Prognostic and predictive biomarkers in adult and pediatric gliomas: Toward personalized treatment. Front. Oncol. 2014, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Heo, K.; Kim, Y.H.; Sung, H.J.; Li, H.Y.; Yoo, C.W.; Kim, J.Y.; Park, J.Y.; Lee, U.L.; Nam, B.H.; Kim, E.O.; et al. Hypoxia-induced up-regulation of apelin is associated with a poor prognosis in oral squamous cell carcinoma patients. Oral Oncol. 2012, 48, 500–506. [Google Scholar] [CrossRef]
- Zuurbier, L.; Rahman, A.; Cordes, M.; Scheick, J.; Wong, T.J.; Rustenburg, F.; Joseph, J.C.; Dynoodt, P.; Casey, R.; Drillenburg, P.; et al. Apelin: A putative novel predictive biomarker for bevacizumab response in colorectal cancer. Oncotarget 2017, 8, 42949–42961. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Tian, X.; He, L.; Li, Y.; Pu, W.; Liu, Q.; Tang, J.; Wu, J.; Cheng, X.; Liu, Y.; et al. Apj (+) Vessels Drive Tumor Growth and Represent a Tractable Therapeutic Target. Cell Rep. 2018, 25, 1241–1254. [Google Scholar] [CrossRef] [Green Version]
- Amoozgar, Z.; Jain, R.K.; Duda, D.G. Role of Apelin in Glioblastoma Vascularization and Invasion after Anti-VEGF Therapy: What Is the Impact on the Immune System? Cancer Res. 2019, 79, 2104–2106. [Google Scholar] [CrossRef] [PubMed]
- United Kingdom Co-Ordinating Committee on Cancer. Guidelines for the welfare of animals in experimental neoplasia. Cancer Metastasis Rev. 1989, 8, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Rathbun, G.; Lam, K.P.; Oltz, E.M.; Stewart, V.; Mendelsohn, M.; Charron, J.; Datta, M.; Young, F.; Stall, A.M.; et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- Mayhew, T.M.; Mwamengele, G.L.; Dantzer, V. Stereological and allometric studies on mammalian cerebral cortex with implications for medical brain imaging. J. Anat. 1996, 189, 177–184. [Google Scholar] [PubMed]
Figure 1.
Angiogenic factor apelin (APLN) expression in glioblastoma (GBM) cells and the tumor neo-vasculature. (A) Expression analysis of APLN RNA was performed by qPCR on the human cells U87MG and U251, the murine GL261 cells and on APLNWT or APLNKO mouse brain tissue. APLN RNA expression was not detectable (n.d.) in GL261 cells but high in U87MG cells. (B) In situ hybridization against mouse APLN showed no expression in implanted mouse GL261 tumor cells but an upregulation of APLN in the tumor vessels of GL261 or U87MG gliomas (arrowheads). The right panel is the magnifications within the tumor that is depicted in the overview panel on the left. Note that vascular APLN RNA expression is highest in hypoxic regions marked by human vascular endothelial growth factor (VEGFA) RNA expression (arrows) in the U87MG xenografts. (C) Lentiviral transduction of U87MG parental cells caused shRNAmir-mediated stable APLN knock-down (AKD) by 90% (as analyzed by qPCR) compared to the non-silencing shRNA control (NSC) transduced cells. (D) Cell viability, as well as in vitro proliferation (E), was unchanged in U87AKD cells compared to U87NSC control or untransduced U87MG cells. In contrast, U87 cells transduced with the shRNA against the kinesin EG5 significantly reduced viability and proliferation of U87E5KD cells compared to U87NSC control. Data are obtained from more than 3 independent experiments each and reported as mean +/-SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated ** p < 0.005, *** p < 0.0005. WT = wildtype; KO = knockout.
Figure 1.
Angiogenic factor apelin (APLN) expression in glioblastoma (GBM) cells and the tumor neo-vasculature. (A) Expression analysis of APLN RNA was performed by qPCR on the human cells U87MG and U251, the murine GL261 cells and on APLNWT or APLNKO mouse brain tissue. APLN RNA expression was not detectable (n.d.) in GL261 cells but high in U87MG cells. (B) In situ hybridization against mouse APLN showed no expression in implanted mouse GL261 tumor cells but an upregulation of APLN in the tumor vessels of GL261 or U87MG gliomas (arrowheads). The right panel is the magnifications within the tumor that is depicted in the overview panel on the left. Note that vascular APLN RNA expression is highest in hypoxic regions marked by human vascular endothelial growth factor (VEGFA) RNA expression (arrows) in the U87MG xenografts. (C) Lentiviral transduction of U87MG parental cells caused shRNAmir-mediated stable APLN knock-down (AKD) by 90% (as analyzed by qPCR) compared to the non-silencing shRNA control (NSC) transduced cells. (D) Cell viability, as well as in vitro proliferation (E), was unchanged in U87AKD cells compared to U87NSC control or untransduced U87MG cells. In contrast, U87 cells transduced with the shRNA against the kinesin EG5 significantly reduced viability and proliferation of U87E5KD cells compared to U87NSC control. Data are obtained from more than 3 independent experiments each and reported as mean +/-SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated ** p < 0.005, *** p < 0.0005. WT = wildtype; KO = knockout.

Figure 2.
Endothelial APLN expression controls formation of a complex GBM vasculature. Orthotopic GL261 implants in APLNWT or APLNKO mice 21 days post-implantation (dpi). (A) The panels on the left indicate the tumor (tumor boarder highlighted by arrowheads) in the right brain hemisphere in overview and close up view on Hematoxylin & Eosin (H&E) sections. The in situ hybridization panels in the middle show the loss-of vascular APLN expression (arrows) in the tumor in overview and close up view when comparing APLNWT to APLNKO mice. The CD31-immunostaining shown in the panel on the right illustrates the reduced vessel density in APLNKO mice. (B) Vessel length density (VLD) was assessed on CD31 immunofluorescent brain slides and demonstrated a significant decrease in APLNKO mice. In addition, vascular complexity measured by the average branch points (ABP) was reduced in APLNKO as compared to APLNWT. (C) In the healthy brain, VLD and ABP do not differ in APLNKO compared to APLNWT mice. Data of n = 9 APLNWT vs. 6 APLNKO mice are reported as mean +/-SEM; statistical significance (students test) is indicated * p < 0.05.
Figure 2.
Endothelial APLN expression controls formation of a complex GBM vasculature. Orthotopic GL261 implants in APLNWT or APLNKO mice 21 days post-implantation (dpi). (A) The panels on the left indicate the tumor (tumor boarder highlighted by arrowheads) in the right brain hemisphere in overview and close up view on Hematoxylin & Eosin (H&E) sections. The in situ hybridization panels in the middle show the loss-of vascular APLN expression (arrows) in the tumor in overview and close up view when comparing APLNWT to APLNKO mice. The CD31-immunostaining shown in the panel on the right illustrates the reduced vessel density in APLNKO mice. (B) Vessel length density (VLD) was assessed on CD31 immunofluorescent brain slides and demonstrated a significant decrease in APLNKO mice. In addition, vascular complexity measured by the average branch points (ABP) was reduced in APLNKO as compared to APLNWT. (C) In the healthy brain, VLD and ABP do not differ in APLNKO compared to APLNWT mice. Data of n = 9 APLNWT vs. 6 APLNKO mice are reported as mean +/-SEM; statistical significance (students test) is indicated * p < 0.05.

Figure 3.
GBM-and endothelial cell-derived APLN are both controlling sprouting angiogenesis. (A) U87MG was implanted into immunodeficient mice and grown to big xenografts within 28 dpi. Fluorescent immunostaining for CD31 was performed and is depicted in an overview and a close up view for every xenograft. (B) The microvasculature in the green fluorescent protein (GFP)-positive tumor area was analyzed by stereomorphology. In comparison to U87NSCAPLNWT controls (n = 8), VLD in U87AKDAPLNWT xenografts (n = 13) was reduced by 62% and vessel length by 75%. Additional loss-of-APLN expression in the tumor neo-vasculature of the APLNKO mouse (U87AKDAPLNKO, n = 10) lead to further reduction of the tumor vasculature. VLD was also reduced when only the tumor cells express APLN (U87NSCAPLNKO, n = 9) but was the highest of all manipulated xenografts. While vascular complexity measured by vascular branch points (ABP values are 6, 2.3; 4.3; and 1.2, respectively) reflects the results seen by VLD, the total vessel length was highly reduced in all three xenografts with reduction of APLN expression. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni´s post hoc test) is indicated ** p < 0.005, *** p < 0.0005.
Figure 3.
GBM-and endothelial cell-derived APLN are both controlling sprouting angiogenesis. (A) U87MG was implanted into immunodeficient mice and grown to big xenografts within 28 dpi. Fluorescent immunostaining for CD31 was performed and is depicted in an overview and a close up view for every xenograft. (B) The microvasculature in the green fluorescent protein (GFP)-positive tumor area was analyzed by stereomorphology. In comparison to U87NSCAPLNWT controls (n = 8), VLD in U87AKDAPLNWT xenografts (n = 13) was reduced by 62% and vessel length by 75%. Additional loss-of-APLN expression in the tumor neo-vasculature of the APLNKO mouse (U87AKDAPLNKO, n = 10) lead to further reduction of the tumor vasculature. VLD was also reduced when only the tumor cells express APLN (U87NSCAPLNKO, n = 9) but was the highest of all manipulated xenografts. While vascular complexity measured by vascular branch points (ABP values are 6, 2.3; 4.3; and 1.2, respectively) reflects the results seen by VLD, the total vessel length was highly reduced in all three xenografts with reduction of APLN expression. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni´s post hoc test) is indicated ** p < 0.005, *** p < 0.0005.

Figure 4.
Apelin peptide rescues the vascular loss-of-function phenotype. (A) Intracerebral infusion of 30 µg of apelin-13 peptide (n = 9) increased glioma angiogenesis in U87AKDAPLNKO xenografts compared to infusion of artificial cerebrospinal fluid (aCSF, n = 8) or apelin-F13A (n = 6) antagonist as shown by von Willebrand factor (VWF) staining. (B) Quantification on a Stereoinvestigator resulted in a VLD of 1269 mm/mm3 in U87AKDAPLNKO xenografts infused with aCSF only; 2573 mm/mm3 with the APLN receptor (APLNR) agonist apelin-13 peptide or 1419 mm/mm3 with the APLNR antagonist apelin-F13A. ABP obtained were 2.2; 3.9 and 2.2, respectively. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni´s post hoc test) is indicated. * p < 0.05, ** p < 0.005.
Figure 4.
Apelin peptide rescues the vascular loss-of-function phenotype. (A) Intracerebral infusion of 30 µg of apelin-13 peptide (n = 9) increased glioma angiogenesis in U87AKDAPLNKO xenografts compared to infusion of artificial cerebrospinal fluid (aCSF, n = 8) or apelin-F13A (n = 6) antagonist as shown by von Willebrand factor (VWF) staining. (B) Quantification on a Stereoinvestigator resulted in a VLD of 1269 mm/mm3 in U87AKDAPLNKO xenografts infused with aCSF only; 2573 mm/mm3 with the APLN receptor (APLNR) agonist apelin-13 peptide or 1419 mm/mm3 with the APLNR antagonist apelin-F13A. ABP obtained were 2.2; 3.9 and 2.2, respectively. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni´s post hoc test) is indicated. * p < 0.05, ** p < 0.005.

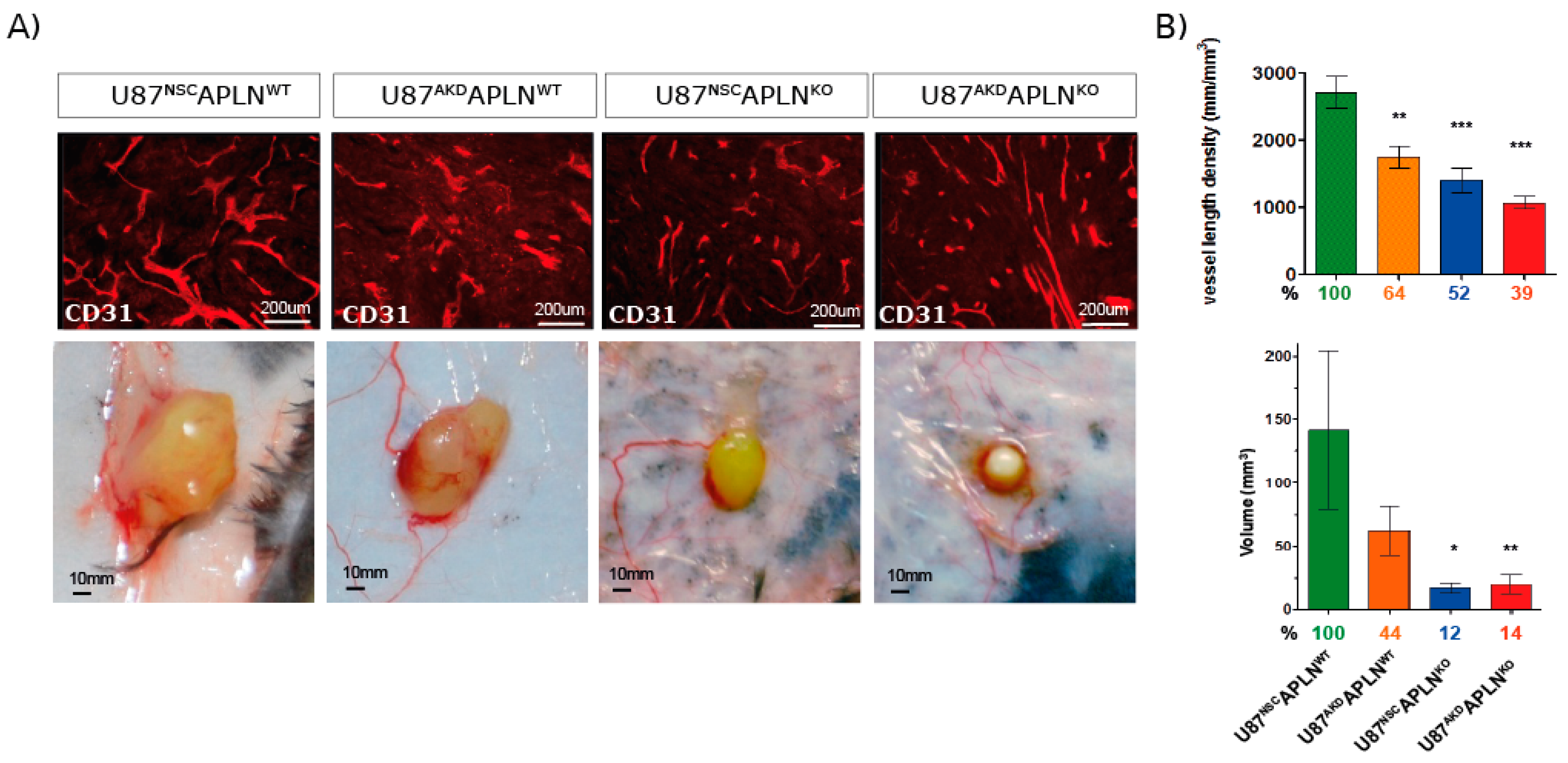
Figure 5.
Apelin is required for compact tumor growth. Mice were inoculated subcutaneously with U87 cells and grown for 28 dpi. (A) Tumor volumes were measured, and vessel density was quantified on CD31-immunostained sections. Representative pictures of subcutaneous tumors are shown. (B) VLD and tumor volume was significantly attenuated upon reduction of APLN expression as compared to U87NSCAPLNWT controls (percentages compared to the control group are indicated). Tumor volumes were reduced to 50% in APLN knockdown U87AKDAPLNWT xenografts. Further reduction to less than 15% was observed in APLNKO mice. Number of cell implantations for U87NSCAPLNWT, U87AKDAPLNWT, U87NSCAPLNKO, U87AKDAPLNKO were n = 38, 31, 18, 24, while tumor take was 87%, 90% 72%, 50%, respectively. For VLD analysis n = 7, 6, 7, 6 of the respective xenografts were analyzed. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated * p < 0.05, ** p < 0.005, *** p < 0.0005.
Figure 5.
Apelin is required for compact tumor growth. Mice were inoculated subcutaneously with U87 cells and grown for 28 dpi. (A) Tumor volumes were measured, and vessel density was quantified on CD31-immunostained sections. Representative pictures of subcutaneous tumors are shown. (B) VLD and tumor volume was significantly attenuated upon reduction of APLN expression as compared to U87NSCAPLNWT controls (percentages compared to the control group are indicated). Tumor volumes were reduced to 50% in APLN knockdown U87AKDAPLNWT xenografts. Further reduction to less than 15% was observed in APLNKO mice. Number of cell implantations for U87NSCAPLNWT, U87AKDAPLNWT, U87NSCAPLNKO, U87AKDAPLNKO were n = 38, 31, 18, 24, while tumor take was 87%, 90% 72%, 50%, respectively. For VLD analysis n = 7, 6, 7, 6 of the respective xenografts were analyzed. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated * p < 0.05, ** p < 0.005, *** p < 0.0005.

Figure 6.
Loss-of-APLN in the tumor microenvironment increases survival of GBM mice. Human U87 cells modulated for APLN levels were orthotopically implanted into APLNWT or APLNKO mice. (A) T2-weighted magnetic resonance imaging (MRI) was performed weekly and MRI volumes were measured producing an in vivo growth curve for every xenograft. After 28 days, U87NSC or U87AKD cells in APLNKO mice showed reduced tumor volume by 69% and 44%, respectively, compared to U87NSCAPLNWT controls. Number of mice per group are indicated. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated * p < 0.05. (B) A mouse survival experiment with orthotopically implanted tumor cells was performed and mice sacrificed at humane endpoints. Median survival differed significantly in U87AKDAPLNWT (shorter survival) and U87NSCAPLNKO (longer survival) mice compared to U87NSCAPLNWT control mice. U87AKD xenografts in APLNKO mice showed a trend to longer survival. Number of mice per group are indicated. Survival data are shown as Kaplan-Meier Curves and significant differences between the experimental groups and the U87NSCAPLNWT control group assessed by long-rank (Mantel-Cox) test is given in the table and is indicated in the graph by * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 6.
Loss-of-APLN in the tumor microenvironment increases survival of GBM mice. Human U87 cells modulated for APLN levels were orthotopically implanted into APLNWT or APLNKO mice. (A) T2-weighted magnetic resonance imaging (MRI) was performed weekly and MRI volumes were measured producing an in vivo growth curve for every xenograft. After 28 days, U87NSC or U87AKD cells in APLNKO mice showed reduced tumor volume by 69% and 44%, respectively, compared to U87NSCAPLNWT controls. Number of mice per group are indicated. Data are reported as mean +/−SEM; statistical significance (one-way ANOVA plus Bonferroni’s post hoc test) is indicated * p < 0.05. (B) A mouse survival experiment with orthotopically implanted tumor cells was performed and mice sacrificed at humane endpoints. Median survival differed significantly in U87AKDAPLNWT (shorter survival) and U87NSCAPLNKO (longer survival) mice compared to U87NSCAPLNWT control mice. U87AKD xenografts in APLNKO mice showed a trend to longer survival. Number of mice per group are indicated. Survival data are shown as Kaplan-Meier Curves and significant differences between the experimental groups and the U87NSCAPLNWT control group assessed by long-rank (Mantel-Cox) test is given in the table and is indicated in the graph by * p < 0.05, ** p < 0.01, *** p < 0.001.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Frisch, A.; Kälin, S.; Monk, R.; Radke, J.; Heppner, F.L.; Kälin, R.E. Apelin Controls Angiogenesis-Dependent Glioblastoma Growth. Int. J. Mol. Sci. 2020, 21, 4179. https://doi.org/10.3390/ijms21114179
AMA Style
Frisch A, Kälin S, Monk R, Radke J, Heppner FL, Kälin RE. Apelin Controls Angiogenesis-Dependent Glioblastoma Growth. International Journal of Molecular Sciences. 2020; 21(11):4179. https://doi.org/10.3390/ijms21114179
Chicago/Turabian StyleFrisch, Anne, Stefanie Kälin, Raymond Monk, Josefine Radke, Frank L. Heppner, and Roland E. Kälin. 2020. "Apelin Controls Angiogenesis-Dependent Glioblastoma Growth" International Journal of Molecular Sciences 21, no. 11: 4179. https://doi.org/10.3390/ijms21114179
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.