Capsaicin-Sensitive Sensory Nerves and the TRPV1 Ion Channel in Cardiac Physiology and Pathologies

 ,
,
Abstract
:1. Introduction and Background
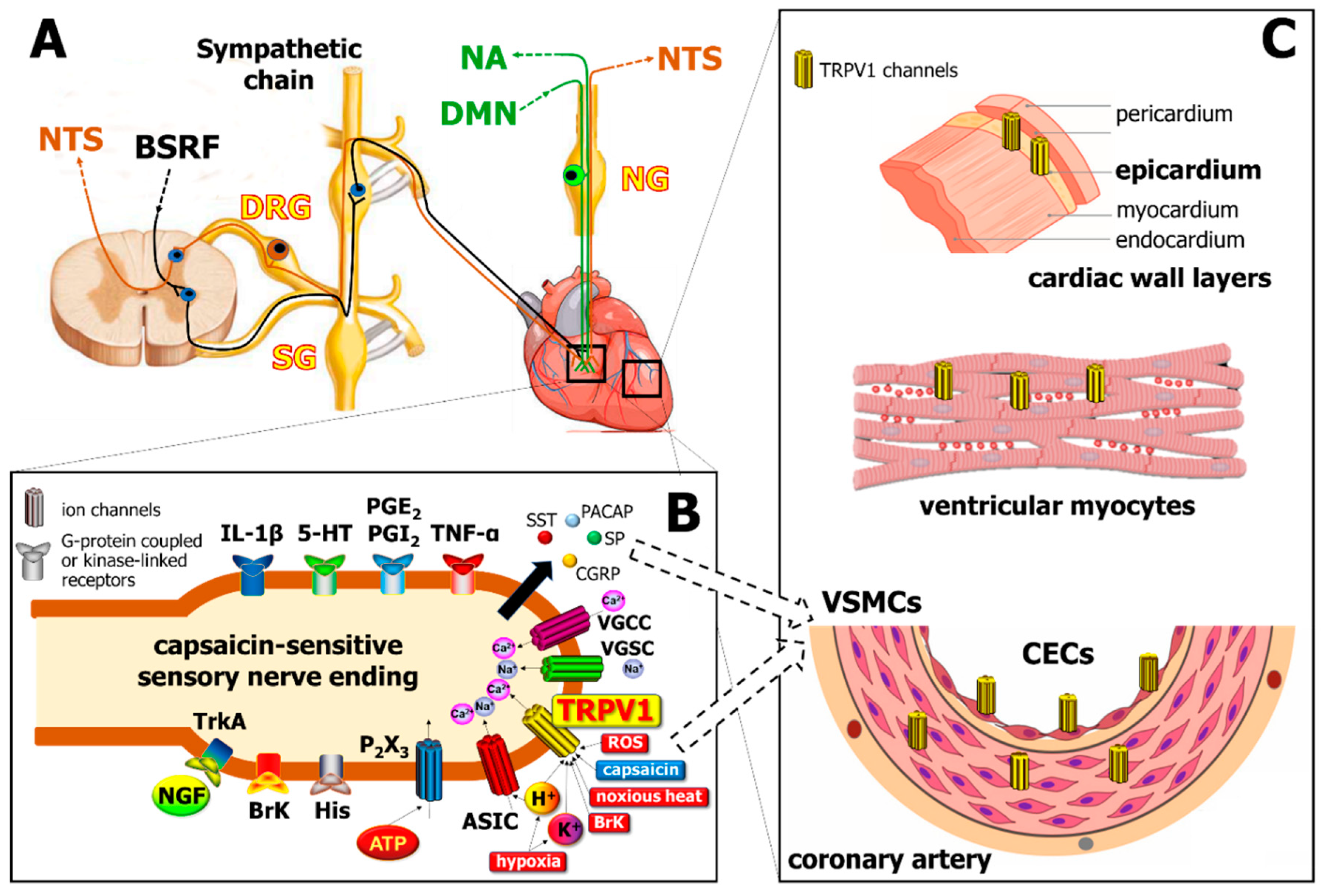
2. Capsaicin-Sensitive Sensory Nerves and TRPV1 Receptors in the Heart
3. Experimental Tools to Investigate the Function of Capsaicin-Sensitive Sensory Nerves and TRPV1 Receptors
4. The Capsaicin-Sensitive Sensory Nerves and the TRPV1 Receptor in Cardiac Pathologies
4.1. Coronary Heart Disease
4.1.1. In Vitro Studies
4.1.2. In Vivo Studies
Dietary Models
Non-Pharmacological TRPV1 Activation

{kind=link}
| TRPV1 Activation | Experimental Model | Beneficial Effects of TRPV1 Activation | Reference(s) | |
|---|---|---|---|---|
| capsaicin | in vitro | HUVEC cell culture | reduced oxLDL induced ROS generation | [66] |
| in vivo by dietary capsaicin | VSMC of WT or TRPV1 KO mice on high-fat diet | reduced foam cell formation | [67] | |
| dihydrocapsaicin | human monocytic THP-1 macrophage cell culture | downregulated LPS induced proinflammatory cytokines (TNF-α, IL-1β and IL-6) | [58] | |
| capsaicin | VSMC of ApoE, TRPV1 double KO mouse | reduced lipid accumulation | [57] | |
| CuS-TRPV1 antibody nanoparticles excited by near infrared light | VSMC of ApoE KO mouse | inhibited foam cell formation | [72] | |
| dietary capsaicin (24 weeks) | in vivo | Apo E, TRPV1 double KO mouse on high-fat diet | reduced lipid storage and atherosclerotic lesions | [57] |
| daily oral gavage of dihydrocapsaicin (12 weeks) | Apo E KO mouse on high fat diet | downregulated proinflammatory cytokines (TNF-α, IL-1β and IL-6) | [58] | |
| dietary evodiamine activation | Apo E, TRPV1 double KO mouse | alleviation of hyperlipidemia, inflammation and hepatic macrovesicular steatosis | [71] | |
| dietary capsaicin (24 weeks) | ApoE, TRPV1 double KO mouse on high-fat diet | prolonged survival, upregulated uncoupling protein 2 expression | [56] | |
| CuS-TRPV1 antibody nanoparticles excited by near infrared light | ApoE KO mouse | attenuated atherosclerotic lesion | [72] | |
4.1.3. Summary
4.2. Myocardial Infarction and Cardioprotection
4.2.1. Acute Infarction
Sensory Nerve Desensitization
TRPV1 Modulation
4.2.2. Ischemic Conditioning
Sensory Nerve Desensitization
TRPV1 Modulation
| Experimental Model | TRPV1 Modulation | Effects of TRPV1 on Major Study Endpoints | Role of TRPV1 Activation | Reference | |
|---|---|---|---|---|---|
| AMI + sensory desensitization | farm pig high dose capsaicin | ↓ | decreased infarct size and increased CGRP level | beneficial | [42] |
| in vivo rat neonatal capsaicin treatment | ↓ | decreased infarct | beneficial | [77] | |
| Ischemia reperfusion injury | H9C2 cell, capsazepine or TRPV1 siRNA | ↓ | decreased cell viability | detrimental | [79] |
| ex vivo TRPV1 KO mouse | ↓ | decreased infarct size | beneficial | [80] | |
| ex vivo TRPV1 KO mouse, capsazepine | ↓ | improved systolic and diastolic functions | beneficial | [81] | |
| in vivo rat Lentivirus-mediated spinal NGF gene knockdown-induced TRPV1 downregulation | ↓ | increased infarct size | detrimental | [84] | |
| in vivo rat Lentivirus-mediated spinal NGF gene knockdown-induced TRPV1 downregulation or capsazepine induced TRPV1 inhibition | ↓ | increased infarct size | detrimental | [85] | |
| ex vivo diabetic mouse Adenovirus-mediated NGF gene delivery induced TRPV1 upregulation | ↑ | Elevated cardiac CGRP level and improved systolic and diastolic functions | beneficial | [86] | |
| Sitagliptin-induced cardioprotection | ex vivo rat AMI, capsazepine and co-administration of sitagliptin | ↓ | capsazepine abolished infarct size limiting effects of sitagliptin | beneficial | [87] |
| Morphine-induced cardioprotection | in vivo rat AMI, capsazepine or P5 | ↓ | decreased infarct size | beneficial | [88] |
| Early or delayed preconditioning | in vivo/ex vivo rat AMI high dose capsaicin | ↓ | abolished preconditioning-induced cardioprotection | beneficial | [91,92,93,94] |
| Pacing-induced preconditioning | ex vivo rat AMI | ↓ | abolished preconditioning-induced cardioprotection | beneficial | [40] |
| Remote ischemic preconditioning | in vivo rat AMI, RIPC induced TRPV1 upregulation | ↑ | decreased infarct size | beneficial | [95] |
| Remote ischemic postconditioning | in vivo rat AMI, capsazepine | ↓ | decreased infarct size | beneficial | [96] |
| Ischemic postconditioning | type 1 diabetic rat ex vivo AMI, capsazepine | ↓ | abolished IPost-induced cardioprotection | beneficial | [97] |
4.2.3. Summary
4.3. Heart Failure
4.3.1. Sensory Desensitization in HF Studies
4.3.2. Studies on Pharmacological and Genetic Ablation of TRPV1
Post-Infarction HF
Chronic Hypertension-Induced HF
Pressure Overload-Induced HF
HF Models of Toxic Cardiac Injury
| Treatment to Modulate TRPV1 | TRPV1 up- or Downmodulation | Experimental Model | Effect of Treatment on Major Study Endpoints | Role of TRPV1 Activation on Cardiac Remodeling | Reference(s) | |
|---|---|---|---|---|---|---|
| Capsaicin-sensitive sensory nerve desensitization | neonatal capsaicin treatment | ↓ | neonatal capsaicin treatment, dilated cardiomyopathy (DCM) and control rat | enhanced EPR compared to control | detrimental | [101] |
| epicardial TRPV1 ablation by high dose RTX | epicardial ↓ | post-MI-induced HF with RTX treatment in rat | improved cardiac compliance | detrimental | [105] | |
| intrathecal RTX treatment | spinal cord ↓ | transverse aortic constriction (TAC)-induced HF | improved cardiac function | detrimental | [106] | |
| sc. capsaicin treatment for 3 days at increasing doses | ↓ | sensory neuropathy-induced HFpEF | impaired myocardial relaxation | beneficial | [103,104] | |
| Pharmacological or genetic modulation of the TRPV1 receptor | genetic deletion | ↓ | 7 days post-MI mouse | increased infarct size | beneficial | [108] |
| genetic deletion and dietary capsaicin for 24 weeks | ↑ | high-salt diet-induced cardiac hypertrophy, mouse | improved mitochondrial function | beneficial | [111] | |
| TRPV1 gene disruption | ↓ | TAC-induced HF | reduced cardiac hypertrophy | detrimental | [113] | |
| genetic deletion | ↓ | TAC-induced HF | decreased cardiac function and increased TNFα and IL-6 | beneficial | [112] | |
| genetic deletion and dietary capsaicin for 10 weeks | ↑ | TAC-induced HF | attenuated hypertrophy in WT | beneficial | [114] | |
| TRPV1 activation by eugenol, capsazepine | ↑ | acute doxorubicin cardiotoxicity | improved cardiac function | beneficial | [117] | |
| SA13353 TRPV1 agonist, and capsazepine | ↑ | doxorubicin-induced HF, ALDH2 transgene mouse | improved cardiac function | beneficial | [116] | |
| genetic deletion, AMG-9810 TRPV1 antagonist | ↓ | LPS-induced endotoxemia, mouse | cardiac dysfunction | beneficial | [120] |
4.3.3. Summary
4.4. Arrhythmias and Electrophysiology
4.4.1. Ventricular Arrhythmias
TRPV1-Independent Cardiac Actions of Capsaicin and RTX
Sensory Desensitization Studies on Ventricular Arrhythmias
Studies on Pharmacological and Genetic Ablation of TRPV1
4.4.2. Supraventricular Arrhythmias
4.4.3. Summary
4.5. Congenital Heart Diseases
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-HT | Serotonin |
| ABCA1 | ATP-binding cassette transporter A1 |
| ALDH2 | Aldehyde dehydrogenase 2 |
| AMI | Acute myocardial infarction |
| APD | Action potential duration |
| ASIC | Acid-sensitive ion channels |
| BAX | B-cell lymphoma 2 associated X protein |
| BCL2 | B-cell lymphoma 2 |
| BrK | Bradykinin |
| BSRF | Brain stem reticular formation |
| CAD | Coronary artery disease |
| CEC | Cardiac endothelial cell |
| CGRP | Calcitonin gene-related peptide |
| CHD | Congenital heart diseases |
| DCM | Dilated cardiomyopathy |
| DMN | Dorsal motor nucleus |
| DOAJ | Directory of open access journals |
| DRG | Dorsal root ganglia |
| EC | Endothelial cell |
| EF | Ejection fraction |
| eNOS | Endothelial nitric oxide synthase |
| EPR | Exercise pressor reflex |
| ERK1/2 | Extracellular signal-regulated protein kinase 1/2 |
| ERP | Effective refractory period |
| FS | Fractional shortening |
| H/R | Hypoxia/reoxygenation |
| HF | Heart failure |
| HFD | High-fat diet |
| HFmrEF | Heart failure with mid-range ejection fraction |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| HUVEC | Human umbilical vein endothelial cell |
| I/R | Ischemia/reperfusion |
| IPC | Ischemic preconditioning |
| LRP1 | LDL-related protein 1 |
| MDPI | Multidisciplinary Digital Publishing Institute |
| NA | Nucleus ambiguus |
| NG | Nodose ganglion |
| NGF | Nerve growth factor |
| NTS | Nucleus tractus solitarius |
| ONOO- | Peroxynitrite |
| PACAP | Pituitary adenylate cyclase-activating polypeptide |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PI3KI | Phosphatidylinositol 3-kinase inhibitor |
| ROS | Reactive oxygen species |
| RTX | Resiniferatoxin |
| SERCA2a | Sarco-endoplasmic reticulum Ca2+ ATPase 2a |
| SG | Sympathetic ganglion |
| SHU | Scoville Heat Unit |
| SP | Substance P |
| SST | Somatostatin |
| TAC | Transverse aortic constriction |
| TNF | Tumor necrosis factor alpha |
| TrkA | Tropomyosin receptor kinase A |
| TRPV1 | Transient receptor potential vanilloid type 1 |
| VGCC | Voltage-gated Ca2+ channel |
| VGSC | Voltage-gated Na+ channel |
| VMSC | Vascular smooth muscle cell |
References
- Jancsó, N. Desensitization with Capsaicin as a Tool for Studying the Function of Pain Receptors. In The Pharmacology of Pain; Pergamon Press: Oxford, UK, 1968; pp. 33–55. [Google Scholar]
- Julius, D. TRP channels and pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. [Google Scholar] [CrossRef] [Green Version]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Maggi, C.A.; Patacchini, R.; Tramontana, M.; Amann, R.; Giuliani, S.; Santicioli, P. Similarities and differences in the action of resiniferatoxin and capsaicin on central and peripheral endings of primary sensory neurons. Neuroscience 1990, 37, 531–539. [Google Scholar] [CrossRef]
- Jancsó, G.; Hökfelt, T.; Lundberg, J.M.; Kiraly, E.; Halász, N.; Nilsson, G.; Terenius, L.; Rehfeld, J.; Steinbusch, H.; Verhofstad, A.; et al. Immunohistochemical Studies on the Effect of Capsaicin on Spinal and Medullary Peptide and Monoamine Neurons Using Antisera to Substance P, gastrin/CCK, Somatostatin, VIP, Enkephalin, Neurotensin and 5-hydroxytryptamine. J. Neurocytol. 1981, 10, 963–980. [Google Scholar] [CrossRef] [PubMed]
- Jancso, G.; Such, G. Effects of capsaicin applied perineurally to the vagus nerve on cardiovascular and respiratory functions in the cat. J. Physiol. 1983, 341, 359–370. [Google Scholar] [CrossRef]
- Zahner, M.R.; Li, D.P.; Chen, S.R.; Pan, H.L. Cardiac vanilloid receptor 1-expressing afferent nerves and their role in the cardiogenic sympathetic reflex in rats. J. Physiol 2003, 551, 515–523. [Google Scholar] [CrossRef]
- Katona, M.; Boros, K.; Santha, P.; Ferdinandy, P.; Dux, M.; Jancso, G. Selective sensory denervation by capsaicin aggravates adriamycin-induced cardiomyopathy in rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 370, 436–443. [Google Scholar] [CrossRef]
- Mensah, G.A.; Roth, G.A.; Fuster, V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J. Am. Coll. Cardiol. 2019, 74, 2529–2532. [Google Scholar] [CrossRef]
- Hinton, W.; McGovern, A.; Coyle, R.; Han, T.S.; Sharma, P.; Correa, A.; Ferreira, F.; de Lusignan, S. Incidence and prevalence of cardiovascular disease in English primary care: A cross-sectional and follow-up study of the Royal College of General Practitioners (RCGP) Research and Surveillance Centre (RSC). BMJ Open 2018, 8, e020282. [Google Scholar] [CrossRef] [Green Version]
- Bencsik, P.; Gomori, K.; Szabados, T.; Santha, P.; Helyes, Z.; Jancso, G.; Ferdinandy, P.; Gorbe, A. Myocardial ischemia reperfusion injury and cardioprotection in the presence of sensory neuropathy: Therapeutic options. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Caterina, M.J.; Julius, D. The vanilloid receptor: A molecular gateway to the pain pathway. Annu. Rev. Neurosci. 2001, 24, 487–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Garcia-Martinez, C.; Morenilla-Palao, C.; Planells-Cases, R.; Merino, J.M.; Ferrer-Montiel, A. Identification of an aspartic residue in the P-loop of the vanilloid receptor that modulates pore properties. J. Biol. Chem. 2000, 275, 32552–32558. [Google Scholar] [CrossRef] [Green Version]
- Randhawa, P.K.; Jaggi, A.S. TRPV1 channels in cardiovascular system: A double edged sword? Int. J. Cardiol. 2017, 228, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Hwang, S.W.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Kim, W.B.; Kim, D.; Oh, U. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J. Neurosci. 1999, 19, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Activation of mitochondrial transient receptor potential vanilloid 1 channel contributes to microglial migration. Glia 2015, 63, 1870–1882. [Google Scholar] [CrossRef] [Green Version]
- Nagy, I.; Santha, P.; Jancso, G.; Urban, L. The role of the vanilloid (capsaicin) receptor (TRPV1) in physiology and pathology. Eur. J. Pharmacol. 2004, 500, 351–369. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Jaggi, A.S. TRPV1 and TRPV4 channels: Potential therapeutic targets for ischemic conditioning-induced cardioprotection. Eur. J. Pharmacol. 2015, 746, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Maggi, C.A.; Meli, A. The sensory-efferent function of capsaicin-sensitive sensory neurons. Gen. Pharmacol. 1988, 19, 1–43. [Google Scholar] [CrossRef]
- Holzer, P. Local effector functions of capsaicin-sensitive sensory nerve endings: Involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience 1988, 24, 739–768. [Google Scholar] [CrossRef]
- Jancso, G.; Kiraly, E.; Such, G.; Joo, F.; Nagy, A. Neurotoxic effect of capsaicin in mammals. Acta Physiol. Hung. 1987, 69, 295–313. [Google Scholar] [PubMed]
- Szolcsanyi, J.; Pinter, E.; Helyes, Z.; Oroszi, G.; Nemeth, J. Systemic anti-inflammatory effect induced by counter-irritation through a local release of somatostatin from nociceptors. Br. J. Pharmacol. 1998, 125, 916–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinter, E.; Helyes, Z.; Szolcsanyi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol. Ther. 2006, 112, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Szolcsanyi, J.; Pinter, E.; Helyes, Z.; Petho, G. Inhibition of the function of TRPV1-expressing nociceptive sensory neurons by somatostatin 4 receptor agonism: Mechanism and therapeutical implications. Curr. Top. Med. Chem. 2011, 11, 2253–2263. [Google Scholar] [CrossRef]
- Santha, P.; Pierau, F.K.; Jancso, G. Evidence for an inhibition by endogenous galanin of neurogenic cutaneous vasodilatation in the pigeon. Neurosci. Lett. 1998, 243, 101–104. [Google Scholar] [CrossRef]
- Chen, Y.; Lyga, J. Brain-skin connection: Stress, inflammation and skin aging. Inflamm. Allergy-Drug Targets 2014, 13, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Dux, M.; Santha, P.; Jancso, G. The role of chemosensitive afferent nerves and TRP ion channels in the pathomechanism of headaches. Pflug. Arch. Eur. J. Physiol. 2012, 464, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jancso, G.; Obal, F., Jr.; Toth-Kasa, I.; Katona, M.; Husz, S. The modulation of cutaneous inflammatory reactions by peptide-containing sensory nerves. Int. J. Tissue React. 1985, 7, 449–457. [Google Scholar]
- Jancsó, G.; Katona, M.; Horváth, V.; Sántha, P.; Nagy, J. Sensory Nerves as Modulators of Cutaneous Inflammatory Reactions in Health and Disease. In Neuroimmune Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 8, pp. 3–36. [Google Scholar]
- Andrei, S.R.; Sinharoy, P.; Bratz, I.N.; Damron, D.S. TRPA1 is functionally co-expressed with TRPV1 in cardiac muscle: Co-localization at z-discs, costameres and intercalated discs. Channels 2016, 10, 395–409. [Google Scholar] [CrossRef]
- Zurborg, S.; Yurgionas, B.; Jira, J.A.; Caspani, O.; Heppenstall, P.A. Direct activation of the ion channel TRPA1 by Ca2+. Nat. Neurosci. 2007, 10, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ye, D.; Ye, J.; Wang, M.; Liu, J.; Jiang, H.; Xu, Y.; Zhang, J.; Chen, J.; Wan, J. The TRPA1 Channel in the Cardiovascular System: Promising Features and Challenges. Front. Pharmacol. 2019, 10, 1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teofilo, T.M.; Duarte, G.P.; Borges, R.S.; Santos, A.A.; Magalhaes, P.J.C.; Lahlou, S. Stimulation of pulmonary vagal C-fibers by trans-4-methyl-beta-nitrostyrene induces bradycardiac and depressor reflex in rats: Role of vanilloid TRPV1 receptors. Eur. J. Pharmacol. 2019, 849, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Jancso, G.; Kiraly, E.; Jancso-Gabor, A. Direct evidence for an axonal site of action of capsaicin. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980, 313, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Tian, B.; Huang, Y.; Peng, X.Y.; Chen, L.H.; Li, J.C.; Liu, T. Hydrogen sulfide-induced itch requires activation of Cav3.2 T-type calcium channel in mice. Sci. Rep. 2015, 5, 16768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamse, R.; Petsche, U.; Lembeck, F.; Jancso, G. Capsaicin applied to peripheral nerve inhibits axoplasmic transport of substance P and somatostatin. Brain Res. 1982, 239, 447–462. [Google Scholar] [CrossRef]
- Ainsworth, A.; Hall, P.; Wall, P.D.; Allt, G.; MacKenzie, M.L.; Gibson, S.; Polak, J.M. Effects of capsaicin applied locally to adult peripheral nerve. II. Anatomy and enzyme and peptide chemistry of peripheral nerve and spinal cord. Pain 1981, 11, 379–388. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Csont, T.; Csonka, C.; Torok, M.; Dux, M.; Nemeth, J.; Horvath, L.I.; Dux, L.; Szilvassy, Z.; Jancso, G. Capsaicin-sensitive local sensory innervation is involved in pacing-induced preconditioning in rat hearts: Role of nitric oxide and CGRP? Naunyn-Schmiedeberg’s Arch. Pharmacol. 1997, 356, 356–363. [Google Scholar] [CrossRef]
- Wharton, J.; Gulbenkian, S.; Mulderry, P.K.; Ghatei, M.A.; McGregor, G.P.; Bloom, S.R.; Polak, J.M. Capsaicin induces a depletion of calcitonin gene-related peptide (CGRP)-immunoreactive nerves in the cardiovascular system of the guinea pig and rat. J. Auton. Nerv. Syst. 1986, 16, 289–309. [Google Scholar] [CrossRef]
- Kallner, G.; Franco-Cereceda, A. Aggravation of myocardial infarction in the porcine heart by capsaicin-induced depletion of calcitonin gene-related peptide (CGRP). J. Cardiovasc. Pharmacol. 1998, 32, 500–504. [Google Scholar] [CrossRef]
- Zvara, A.; Bencsik, P.; Fodor, G.; Csont, T.; Hackler, L., Jr.; Dux, M.; Furst, S.; Jancso, G.; Puskas, L.G.; Ferdinandy, P. Capsaicin-sensitive sensory neurons regulate myocardial function and gene expression pattern of rat hearts: A DNA microarray study. FASEB J. 2006, 20, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; He, E.Y.; Scott, G.I.; Ren, J. Alpha, beta-Unsaturated aldehyde pollutant acrolein suppresses cardiomyocyte contractile function: Role of TRPV1 and oxidative stress. Environ. Toxicol. 2015, 30, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Kun, J.; Helyes, Z.; Perkecz, A.; Ban, A.; Polgar, B.; Szolcsanyi, J.; Pinter, E. Effect of surgical and chemical sensory denervation on non-neural expression of the transient receptor potential vanilloid 1 (TRPV1) receptors in the rat. J. Mol. Neurosci. 2012, 48, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kark, T.; Bagi, Z.; Lizanecz, E.; Pasztor, E.T.; Erdei, N.; Czikora, A.; Papp, Z.; Edes, I.; Porszasz, R.; Toth, A. Tissue-specific regulation of microvascular diameter: Opposite functional roles of neuronal and smooth muscle located vanilloid receptor-1. Mol. Pharmacol. 2008, 73, 1405–1412. [Google Scholar] [CrossRef]
- Dux, M.; Santha, P.; Jancso, G. Capsaicin-sensitive neurogenic sensory vasodilatation in the dura mater of the rat. J. Physiol. 2003, 552, 859–867. [Google Scholar] [CrossRef]
- Porszasz, R.; Porkolab, A.; Ferencz, A.; Pataki, T.; Szilvassy, Z.; Szolcsanyi, J. Capsaicin-induced nonneural vasoconstriction in canine mesenteric arteries. Eur. J. Pharmacol. 2002, 441, 173–175. [Google Scholar] [CrossRef]
- Jancso, N.; Jancso-Gabor, A.; Szolcsanyi, J. The role of sensory nerve endings in neurogenic inflammation induced in human skin and in the eye and paw of the rat. Br. J. Pharmacol. Chemother. 1968, 33, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Jancso, G.; Kiraly, E.; Jancso-Gabor, A. Pharmacologically induced selective degeneration of chemosensitive primary sensory neurones. Nature 1977, 270, 741–743. [Google Scholar] [CrossRef]
- Rozsa, Z.; Jancso, G.; Varro, V. Possible involvement of capsaicin-sensitive sensory nerves in the regulation of intestinal blood flow in the dog. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1984, 326, 352–356. [Google Scholar] [CrossRef]
- Toth, A.; Czikora, A.; Pasztor, E.T.; Dienes, B.; Bai, P.; Csernoch, L.; Rutkai, I.; Csato, V.; Manyine, I.S.; Porszasz, R.; et al. Vanilloid receptor-1 (TRPV1) expression and function in the vasculature of the rat. J. Histochem. Cytochem. 2014, 62, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Domoki, F.; Santha, P.; Bari, F.; Jancso, G. Perineural capsaicin treatment attenuates reactive hyperaemia in the rat skin. Neurosci. Lett. 2003, 341, 127–130. [Google Scholar] [CrossRef]
- Mahmmoud, Y.A.; Shattock, M.; Cornelius, F.; Pavlovic, D. Inhibition of K+ transport through Na+, K+-ATPase by capsazepine: Role of membrane span 10 of the alpha-subunit in the modulation of ion gating. PLoS ONE 2014, 9, e96909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Docherty, R.J.; Yeats, J.C.; Piper, A.S. Capsazepine block of voltage-activated calcium channels in adult rat dorsal root ganglion neurones in culture. Br. J. Pharmacol. 1997, 121, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Wang, P.; Ma, L.; Gao, P.; Gong, L.; Li, L.; Li, Q.; Sun, F.; Zhou, X.; He, H.; et al. Ameliorating Endothelial Mitochondrial Dysfunction Restores Coronary Function via Transient Receptor Potential Vanilloid 1-Mediated Protein Kinase A/Uncoupling Protein 2 Pathway. Hypertension 2016, 67, 451–460. [Google Scholar] [CrossRef]
- Ma, L.; Zhong, J.; Zhao, Z.; Luo, Z.; Ma, S.; Sun, J.; He, H.; Zhu, T.; Liu, D.; Zhu, Z.; et al. Activation of TRPV1 reduces vascular lipid accumulation and attenuates atherosclerosis. Cardiovasc. Res. 2011, 92, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.J.; Hu, Y.W.; Huang, C.; Ma, X.; Kang, C.M.; Zhang, Y.; Guo, F.X.; Lu, J.B.; Xiu, J.C.; Qiu, Y.R.; et al. Dihydrocapsaicin suppresses proinflammatory cytokines expression by enhancing nuclear factor IA in a NF-kappaB-dependent manner. Arch. Biochem. Biophys. 2016, 604, 27–35. [Google Scholar] [CrossRef]
- Rollyson, W.D.; Stover, C.A.; Brown, K.C.; Perry, H.E.; Stevenson, C.D.; McNees, C.A.; Ball, J.G.; Valentovic, M.A.; Dasgupta, P. Bioavailability of capsaicin and its implications for drug delivery. J. Control. Release 2014, 196, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Kawada, T.; Suzuki, T.; Takahashi, M.; Iwai, K. Gastrointestinal absorption and metabolism of capsaicin and dihydrocapsaicin in rats. Toxicol. Appl. Pharmacol. 1984, 72, 449–456. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Capsaicin may have important potential for promoting vascular and metabolic health. Open Heart 2015, 2, e000262. [Google Scholar] [CrossRef] [Green Version]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Nazarian-Samani, Z.; Sewell, R.D.E.; Rafieian-Kopaei, M. Inflammasome Signaling and other Factors Implicated in Atherosclerosis Development and Progression. Curr. Pharm. Des. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bujak, J.K.; Kosmala, D.; Szopa, I.M.; Majchrzak, K.; Bednarczyk, P. Inflammation, Cancer and Immunity-Implication of TRPV1 Channel. Front. Oncol. 2019, 9, 1087. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, L.; Xu, H.; Liu, S.; Zhu, F.; Yan, F.; Shen, S.; Zhu, M. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 2017, 260, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; Chen, P.N.; Hsieh, Y.S.; Lin, C.Y.; Lee, Y.H.; Chu, S.C. Capsaicin protects endothelial cells and macrophage against oxidized low-density lipoprotein-induced injury by direct antioxidant action. Chem. Biol. Interact. 2015, 228, 35–45. [Google Scholar] [CrossRef]
- Li, B.H.; Yin, Y.W.; Liu, Y.; Pi, Y.; Guo, L.; Cao, X.J.; Gao, C.Y.; Zhang, L.L.; Li, J.C. TRPV1 activation impedes foam cell formation by inducing autophagy in oxLDL-treated vascular smooth muscle cells. Cell Death Dis. 2014, 5, e1182. [Google Scholar] [CrossRef] [Green Version]
- Poonyachoti, S.; Kulkarni-Narla, A.; Brown, D.R. Chemical coding of neurons expressing delta- and kappa-opioid receptor and type I vanilloid receptor immunoreactivities in the porcine ileum. Cell Tissue Res. 2002, 307, 23–33. [Google Scholar] [CrossRef]
- Rizopoulos, T.; Papadaki-Petrou, H.; Assimakopoulou, M. Expression Profiling of the Transient Receptor Potential Vanilloid (TRPV) Channels 1, 2, 3 and 4 in Mucosal Epithelium of Human Ulcerative Colitis. Cells 2018, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Wei, J.; Ching, L.C.; Zhao, J.F.; Shyue, S.K.; Lee, H.F.; Kou, Y.R.; Lee, T.S. Essential role of transient receptor potential vanilloid type 1 in evodiamine-mediated protection against atherosclerosis. Acta Physiol. 2013, 207, 299–307. [Google Scholar] [CrossRef]
- Gao, W.; Sun, Y.; Cai, M.; Zhao, Y.; Cao, W.; Liu, Z.; Cui, G.; Tang, B. Copper sulfide nanoparticles as a photothermal switch for TRPV1 signaling to attenuate atherosclerosis. Nat. Commun. 2018, 9, 231. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Schulz, R.; Baxter, G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef] [PubMed]
- Ustinova, E.E.; Bergren, D.; Schultz, H.D. Neuropeptide depletion impairs postischemic recovery of the isolated rat heart: Role of substance P. Cardiovasc. Res. 1995, 30, 55–63. [Google Scholar] [CrossRef]
- Zhang, R.L.; Guo, Z.; Wang, L.L.; Wu, J. Degeneration of capsaicin sensitive sensory nerves enhances myocardial injury in acute myocardial infarction in rats. Int. J. Cardiol. 2012, 160, 41–47. [Google Scholar] [CrossRef]
- Sogut, O.; Kaya, H.; Gokdemir, M.T.; Sezen, Y. Acute myocardial infarction and coronary vasospasm associated with the ingestion of cayenne pepper pills in a 25-year-old male. Int. J. Emerg. Med. 2012, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Sayin, M.R.; Karabag, T.; Dogan, S.M.; Akpinar, I.; Aydin, M. A case of acute myocardial infarction due to the use of cayenne pepper pills. Wien. Klin. Wochenschr. 2012, 124, 285–287. [Google Scholar] [CrossRef]
- Sun, Z.; Han, J.; Zhao, W.; Zhang, Y.; Wang, S.; Ye, L.; Liu, T.; Zheng, L. TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium overload and mitochondrial dysfunction. Int. J. Mol. Sci. 2014, 15, 18362–18380. [Google Scholar] [CrossRef]
- Jiang, X.X.; Liu, G.Y.; Lei, H.; Li, Z.L.; Feng, Q.P.; Huang, W. Activation of transient receptor potential vanilloid 1 protects the heart against apoptosis in ischemia/reperfusion injury through upregulating the PI3K/Akt signaling pathway. Int. J. Mol. Med. 2018, 41, 1724–1730. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, D.H. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation 2005, 112, 3617–3623. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Xiao, Z.S.; Peng, C.F.; Deng, H.W. Calcitonin gene-related peptide-induced preconditioning protects against ischemia-reperfusion injury in isolated rat hearts. Eur. J. Pharmacol. 1996, 311, 163–167. [Google Scholar] [CrossRef]
- Priestley, J.V.; Michael, G.J.; Averill, S.; Liu, M.; Willmott, N. Regulation of nociceptive neurons by nerve growth factor and glial cell line derived neurotrophic factor. Can. J. Physiol. Pharmacol. 2002, 80, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Y.; Chen, C.; He, S.F.; Huang, C.X.; Zhang, L.; Chen, Z.W.; Zhang, Y. Spinal NGF induces anti-intrathecal opioid-initiated cardioprotective effect via regulation of TRPV1 expression. Eur. J. Pharmacol. 2019, 844, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Ma, Z.; Cheng, X.; Zou, G.; Xu, Y.; Huang, C.; Xiong, W.; He, S.; Zhang, Y. Intrathecal lentivirus-mediated RNA interference targeting nerve growth factor attenuates myocardial ischaemia-reperfusion injury in rat. Br. J. Anaesth. 2019, 123, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.R.; Zhang, Y.Y.; Han, J.; Sun, Z.W.; Zhou, S.X.; Zhao, W.T.; Wang, L.H. Nerve growth factor rescues diabetic mice heart after ischemia/reperfusion injury via up-regulation of the TRPV1 receptor. J. Diabetes Complicat. 2015, 29, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Al-Awar, A.; Almasi, N.; Szabo, R.; Takacs, I.; Murlasits, Z.; Szucs, G.; Torok, S.; Posa, A.; Varga, C.; Kupai, K. Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model. Int. J. Mol. Sci. 2018, 19, 3226. [Google Scholar] [CrossRef] [Green Version]
- Heymann, H.M.; Wu, Y.; Lu, Y.; Qvit, N.; Gross, G.J.; Gross, E.R. Transient receptor potential vanilloid 1 inhibitors block laparotomy- and opioid-induced infarct size reduction in rats. Br. J. Pharmacol. 2017, 174, 4826–4835. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.J.; Li, Y.J.; Deng, H.W. Cardioprotective effect of bradykinin-induced preconditioning mediated by calcitonin gene-related peptide in isolated rat heart. Zhongguo Yao Li Xue Bao 1999, 20, 162–166. [Google Scholar]
- Peng, J.; Xiao, J.; Ye, F.; Deng, H.W.; Li, Y.J. Inhibition of cardiac tumor necrosis factor-alpha production by calcitonin gene-related peptide-mediated ischemic preconditioning in isolated rat hearts. Eur. J. Pharmacol. 2000, 407, 303–308. [Google Scholar] [CrossRef]
- Luo, D.; Deng, P.Y.; Ye, F.; Peng, W.J.; Deng, H.W.; Li, Y.J. Delayed preconditioning by cardiac ischemia involves endogenous calcitonin gene-related peptide via the nitric oxide pathway. Eur. J. Pharmacol. 2004, 502, 135–141. [Google Scholar] [CrossRef]
- He, S.Y.; Deng, H.W.; Li, Y.J. Monophosphoryl lipid A-induced delayed preconditioning is mediated by calcitonin gene-related peptide. Eur. J. Pharmacol. 2001, 420, 143–149. [Google Scholar] [CrossRef]
- Chen, K.; Yu, J.; Wang, Q.; Wu, L.; Liu, X.; Wong, G.T.C.; Lu, Y. The timing of propofol administration affects the effectiveness of remote ischemic preconditioning induced cardioprotection in rats. J. Cell Biochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Song, J.; Chen, H.; Cao, C.; Lee, C. TRPV1 activation is involved in the cardioprotection of remote limb ischemic postconditioning in ischemia-reperfusion injury rats. Biochem. Biophys. Res. Commun. 2015, 463, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.Y.; Song, J.X.; Lu, M.Y.; Chen, H. Cardioprotection by ischemic postconditioning is lost in isolated perfused heart from diabetic rats: Involvement of transient receptor potential vanilloid 1, calcitonin gene-related peptide and substance P. Regul. Pept. 2011, 169, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Wang, D.H. N-oleoyldopamine, a novel endogenous capsaicin-like lipid, protects the heart against ischemia-reperfusion injury via activation of TRPV1. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H728–H735. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Jancsó, G. Capsaicin-Sensitive Sensory Nerves in Myocardial Ischemia-Reperfusion Injury and Ischemic Stress Adaptation. Role of Nitric Oxide and Calcitonin Gene-Related Peptide. In Neurogenic Inflammation in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2009; pp. 267–288. [Google Scholar]
- Qi, Y.; Qi, Z.; Li, Z.; Wong, C.K.; So, C.; Lo, I.C.; Huang, Y.; Yao, X.; Tsang, S.Y. Role of TRPV1 in the Differentiation of Mouse Embryonic Stem Cells into Cardiomyocytes. PLoS ONE 2015, 10, e0133211. [Google Scholar] [CrossRef]
- Hajouli, S.; Ludhwani, D. Heart Failure and Ejection Fraction; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Smith, S.A.; Williams, M.A.; Mitchell, J.H.; Mammen, P.P.; Garry, M.G. The capsaicin-sensitive afferent neuron in skeletal muscle is abnormal in heart failure. Circulation 2005, 111, 2056–2065. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Wang, W.; Cornish, K.G.; Rozanski, G.J.; Zucker, I.H. Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension 2014, 64, 745–755. [Google Scholar] [CrossRef]
- Wang, D.; Wu, Y.; Chen, Y.; Wang, A.; Lv, K.; Kong, X.; He, Y.; Hu, N. Focal selective chemo-ablation of spinal cardiac afferent nerve by resiniferatoxin protects the heart from pressure overload-induced hypertrophy. Biomed. Pharmacother. 2019, 109, 377–385. [Google Scholar] [CrossRef]
- Bencsik, P.; Kiss, K.; Agg, B.; Baan, J.A.; Agoston, G.; Varga, A.; Gomori, K.; Mendler, L.; Farago, N.; Zvara, A.; et al. Sensory Neuropathy Affects Cardiac miRNA Expression Network Targeting IGF-1, SLC2a-12, EIF-4e, and ULK-2 mRNAs. Int. J. Mol. Sci. 2019, 20, 991. [Google Scholar] [CrossRef] [Green Version]
- Bencsik, P.; Kupai, K.; Giricz, Z.; Gorbe, A.; Huliak, I.; Furst, S.; Dux, L.; Csont, T.; Jancso, G.; Ferdinandy, P. Cardiac capsaicin-sensitive sensory nerves regulate myocardial relaxation via S-nitrosylation of SERCA: Role of peroxynitrite. Br. J. Pharmacol. 2008, 153, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Rubinstein, J.; Prieto, A.R.; Wang, D.H. Enhanced postmyocardial infarction fibrosis via stimulation of the transforming growth factor-beta-Smad2 signaling pathway: Role of transient receptor potential vanilloid type 1 channels. J. Hypertens. 2010, 28, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.; Li, Q.; Yu, H.; Li, P.; Lu, Z.; Xiong, S.; Yang, T.; Zhao, Y.; Huang, X.; Gao, P.; et al. Activation of TRPV1 attenuates high salt-induced cardiac hypertrophy through improvement of mitochondrial function. Br. J. Pharmacol. 2015, 172, 5548–5558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, C.L.; Stokes, A.J. Mice lacking functional TRPV1 are protected from pressure overload cardiac hypertrophy. Channels 2011, 5, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.; Rubinstein, J.; Ma, S.; Wang, D.H. Genetic ablation of TRPV1 exacerbates pressure overload-induced cardiac hypertrophy. Biomed. Pharmacother. 2018, 99, 261–270. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, S.; Li, D.; Zhang, Y.; Tang, B.; Qiu, C.; Yang, Y.; Yang, D. Dietary capsaicin ameliorates pressure overload-induced cardiac hypertrophy and fibrosis through the transient receptor potential vanilloid type 1. Am. J. Hypertens. 2014, 27, 1521–1529. [Google Scholar] [CrossRef] [Green Version]
- Fouad, A.A.; Yacoubi, M.T. Mechanisms underlying the protective effect of eugenol in rats with acute doxorubicin cardiotoxicity. Arch. Pharm. Res. 2011, 34, 821–828. [Google Scholar] [CrossRef]
- Ge, W.; Yuan, M.; Ceylan, A.F.; Wang, X.; Ren, J. Mitochondrial aldehyde dehydrogenase protects against doxorubicin cardiotoxicity through a transient receptor potential channel vanilloid 1-mediated mechanism. Biochim. Biophys. Acta 2016, 1862, 622–634. [Google Scholar] [CrossRef]
- Chen, J.; Hamers, A.J.P.; Finsterbusch, M.; Massimo, G.; Zafar, M.; Corder, R.; Colas, R.A.; Dalli, J.; Thiemermann, C.; Ahluwalia, A. Endogenously generated arachidonate-derived ligands for TRPV1 induce cardiac protection in sepsis. FASEB J. 2018, 32, 3816–3831. [Google Scholar] [CrossRef] [Green Version]
- Csont, T.; Csonka, C.; Kovacs, P.; Jancso, G.; Ferdinandy, P. Capsaicin-sensitive sensory neurons regulate myocardial nitric oxide and cGMP signaling. Eur. J. Pharmacol. 2003, 476, 107–113. [Google Scholar] [CrossRef]
- Wang, H.J.; Li, Y.L.; Gao, L.; Zucker, I.H.; Wang, W. Alteration in skeletal muscle afferents in rats with chronic heart failure. J. Physiol. 2010, 588, 5033–5047. [Google Scholar] [CrossRef] [PubMed]
- Dragun, M.; Gazova, A.; Kyselovic, J.; Hulman, M.; Matus, M. TRP Channels Expression Profile in Human End-Stage Heart Failure. Medicina 2019, 55, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, D.H. A novel mechanism contributing to development of Dahl salt-sensitive hypertension: Role of the transient receptor potential vanilloid type 1. Hypertension 2006, 47, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boros, K.; Jancso, G.; Dux, M.; Fekecs, Z.; Bencsik, P.; Oszlacs, O.; Katona, M.; Ferdinandy, P.; Nogradi, A.; Santha, P. Multiple impairments of cutaneous nociceptor function induced by cardiotoxic doses of Adriamycin in the rat. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016, 389, 1009–1020. [Google Scholar] [CrossRef]
- Choudhary, R.; Mishra, K.P.; Subramanyam, C. Interrelations between oxidative stress and calcineurin in the attenuation of cardiac apoptosis by eugenol. Mol. Cell Biochem. 2006, 283, 115–122. [Google Scholar] [CrossRef]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Janse, M.J.; Wit, A.L. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989, 69, 1049–1169. [Google Scholar] [CrossRef]
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456. [Google Scholar] [CrossRef]
- Lip, G.Y.; Heinzel, F.R.; Gaita, F.; Juanatey, J.R.; Le Heuzey, J.Y.; Potpara, T.; Svendsen, J.H.; Vos, M.A.; Anker, S.D.; Coats, A.J.; et al. European Heart Rhythm Association/Heart Failure Association joint consensus document on arrhythmias in heart failure, endorsed by the Heart Rhythm Society and the Asia Pacific Heart Rhythm Society. Europace 2016, 18, 12–36. [Google Scholar] [CrossRef]
- Sewell, W.H.; Koth, D.R.; Huggins, C.E. Ventricular fibrillation in dogs after sudden return of flow to the coronary artery. Surgery 1955, 38, 1050–1053. [Google Scholar]
- Krumholz, H.M.; Goldberger, A.L. Reperfusion arrhythmias after thrombolysis. Electrophysiologic tempest, or much ado about nothing. Chest 1991, 99, 135S–140S. [Google Scholar] [CrossRef] [PubMed]
- Castle, N.A. Differential inhibition of potassium currents in rat ventricular myocytes by capsaicin. Cardiovasc. Res. 1992, 26, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- D’Alonzo, A.J.; Grover, G.J.; Darbenzio, R.B.; Hess, T.A.; Sleph, P.G.; Dzwonczyk, S.; Zhu, J.L.; Sewter, J.C. In vitro effects of capsaicin: Antiarrhythmic and antiischemic activity. Eur. J. Pharmacol. 1995, 272, 269–278. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, Z.; Wang, D.; Lv, K.; Hu, N. Resiniferatoxin reduces ventricular arrhythmias in heart failure via selectively blunting cardiac sympathetic afferent projection into spinal cord in rats. Eur. J. Pharmacol. 2020, 867, 172836. [Google Scholar] [CrossRef] [PubMed]
- Yoshie, K.; Rajendran, P.S.; Massoud, L.; Mistry, J.; Swid, M.A.; Wu, X.; Sallam, T.; Zhang, R.; Goldhaber, J.I.; Salavatian, S.; et al. Cardiac TRPV1 afferent signaling promotes arrhythmogenic ventricular remodeling after myocardial infarction. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Xu, Y.; Cheng, X.; Huang, C.; Pan, Y.; Jin, S.; Xiong, W.; Zhang, L.; He, S.; Zhang, Y. Inhibition of DRG-TRPV1 upregulation in myocardial ischemia contributes to exogenous cardioprotection. J. Mol. Cell Cardiol. 2020, 138, 175–184. [Google Scholar] [CrossRef]
- Horton, J.S.; Shiraishi, T.; Alfulaij, N.; Small-Howard, A.L.; Turner, H.C.; Kurokawa, T.; Mori, Y.; Stokes, A.J. TRPV1 is a component of the atrial natriuretic signaling complex, and using orally delivered antagonists, presents a valid therapeutic target in the longitudinal reversal and treatment of cardiac hypertrophy and heart failure. Channels 2019, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ambrosy, A.P.; Fonarow, G.C.; Butler, J.; Chioncel, O.; Greene, S.J.; Vaduganathan, M.; Nodari, S.; Lam, C.S.P.; Sato, N.; Shah, A.N.; et al. The global health and economic burden of hospitalizations for heart failure: Lessons learned from hospitalized heart failure registries. J. Am. Coll. Cardiol. 2014, 63, 1123–1133. [Google Scholar] [CrossRef]
- Linz, D.; Elliott, A.D.; Hohl, M.; Malik, V.; Schotten, U.; Dobrev, D.; Nattel, S.; Bohm, M.; Floras, J.; Lau, D.H.; et al. Role of autonomic nervous system in atrial fibrillation. Int. J. Cardiol. 2019, 287, 181–188. [Google Scholar] [CrossRef]
- Ghias, M.; Scherlag, B.J.; Lu, Z.; Niu, G.; Moers, A.; Jackman, W.M.; Lazzara, R.; Po, S.S. The role of ganglionated plexi in apnea-related atrial fibrillation. J. Am. Coll. Cardiol. 2009, 54, 2075–2083. [Google Scholar] [CrossRef] [Green Version]
- Tavares, L.; Rodriguez-Manero, M.; Kreidieh, B.; Ibarra-Cortez, S.H.; Chen, J.; Wang, S.; Markovits, J.; Barrios, R.; Valderrabano, M. Cardiac Afferent Denervation Abolishes Ganglionated Plexi and Sympathetic Responses to Apnea: Implications for Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2019, 12, e006942. [Google Scholar] [CrossRef] [PubMed]
- Duzen, I.V.; Yavuz, F.; Vuruskan, E.; Saracoglu, E.; Poyraz, F.; Goksuluk, H.; Candemir, B.; Demiryurek, S. Leukocyte TRP channel gene expressions in patients with non-valvular atrial fibrillation. Sci. Rep. 2017, 7, 9272. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Van Wagoner, D.R. Oxidant and Inflammatory Mechanisms and Targeted Therapy in Atrial Fibrillation: An Update. J. Cardiovasc. Pharmacol. 2015, 66, 523–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varro, A.; Lathrop, D.A.; Hester, S.B.; Nanasi, P.P.; Papp, J.G. Ionic currents and action potentials in rabbit, rat, and guinea pig ventricular myocytes. Basic Res. Cardiol. 1993, 88, 93–102. [Google Scholar] [CrossRef]
- Baczko, I.; Jost, N.; Virag, L.; Bosze, Z.; Varro, A. Rabbit models as tools for preclinical cardiac electrophysiological safety testing: Importance of repolarization reserve. Prog. Biophys. Mol. Biol. 2016, 121, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Carson, J.; Lo, C. Genetics of Congenital Heart Disease. Biomolecules 2019, 9, 879. [Google Scholar] [CrossRef] [Green Version]
- Botto, L.D.; Panichello, J.D.; Browne, M.L.; Krikov, S.; Feldkamp, M.L.; Lammer, E.; Shaw, G.M.; National Birth Defects Prevention, S. Congenital heart defects after maternal fever. Am. J. Obstet. Gynecol. 2014, 210, 359.e1. [Google Scholar] [CrossRef]
- Hutson, M.R.; Keyte, A.L.; Hernandez-Morales, M.; Gibbs, E.; Kupchinsky, Z.A.; Argyridis, I.; Erwin, K.N.; Pegram, K.; Kneifel, M.; Rosenberg, P.B.; et al. Temperature-activated ion channels in neural crest cells confer maternal fever-associated birth defects. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabados, T.; Gömöri, K.; Pálvölgyi, L.; Görbe, A.; Baczkó, I.; Helyes, Z.; Jancsó, G.; Ferdinandy, P.; Bencsik, P. Capsaicin-Sensitive Sensory Nerves and the TRPV1 Ion Channel in Cardiac Physiology and Pathologies. Int. J. Mol. Sci. 2020, 21, 4472. https://doi.org/10.3390/ijms21124472
Szabados T, Gömöri K, Pálvölgyi L, Görbe A, Baczkó I, Helyes Z, Jancsó G, Ferdinandy P, Bencsik P. Capsaicin-Sensitive Sensory Nerves and the TRPV1 Ion Channel in Cardiac Physiology and Pathologies. International Journal of Molecular Sciences. 2020; 21(12):4472. https://doi.org/10.3390/ijms21124472
Chicago/Turabian StyleSzabados, Tamara, Kamilla Gömöri, Laura Pálvölgyi, Anikó Görbe, István Baczkó, Zsuzsanna Helyes, Gábor Jancsó, Péter Ferdinandy, and Péter Bencsik. 2020. "Capsaicin-Sensitive Sensory Nerves and the TRPV1 Ion Channel in Cardiac Physiology and Pathologies" International Journal of Molecular Sciences 21, no. 12: 4472. https://doi.org/10.3390/ijms21124472