Betacoronavirus Genomes: How Genomic Information has been Used to Deal with Past Outbreaks and the COVID-19 Pandemic

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Genome Structure and Protein-Coding Genes

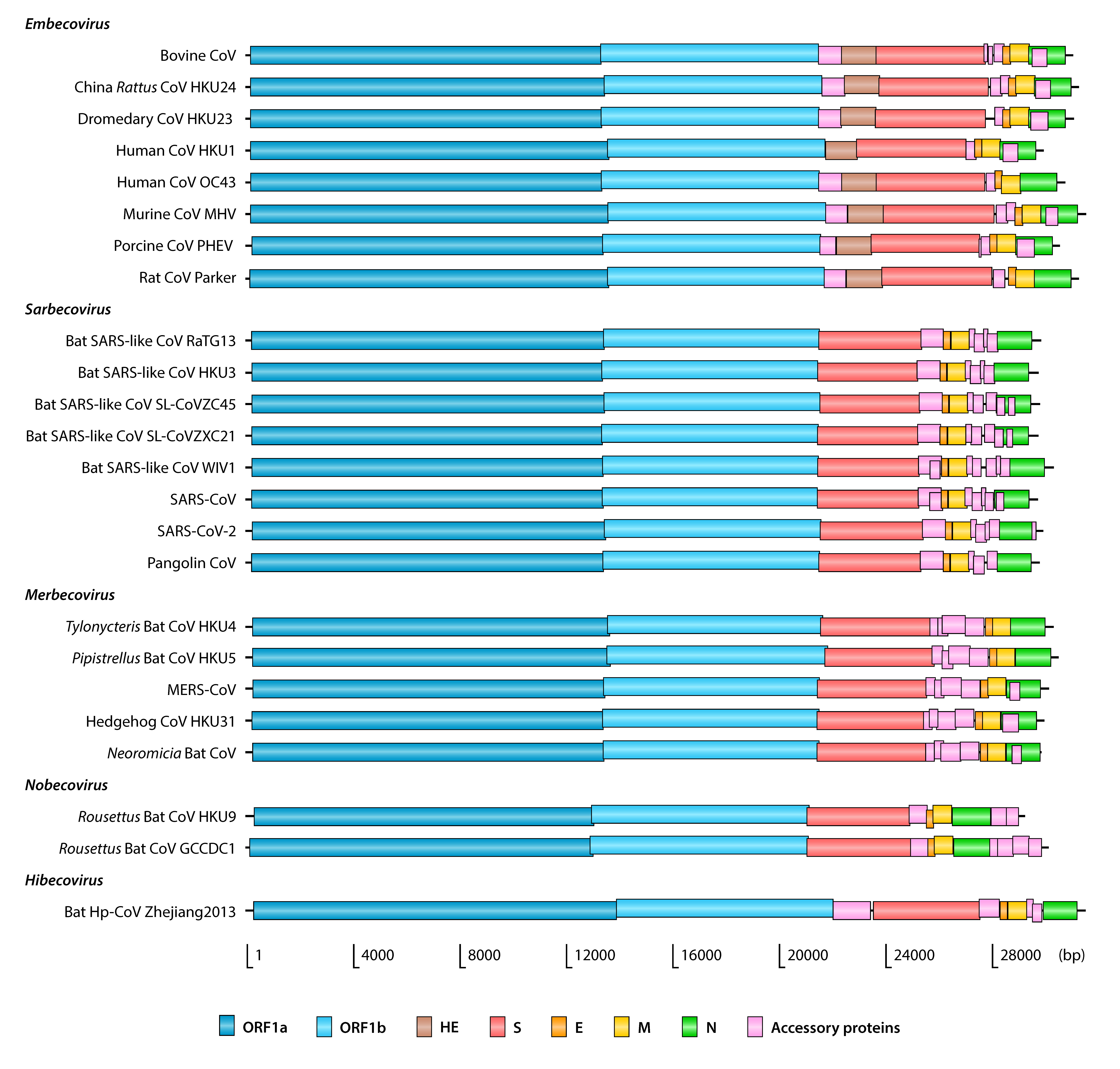{kind=link}
{kind=link}
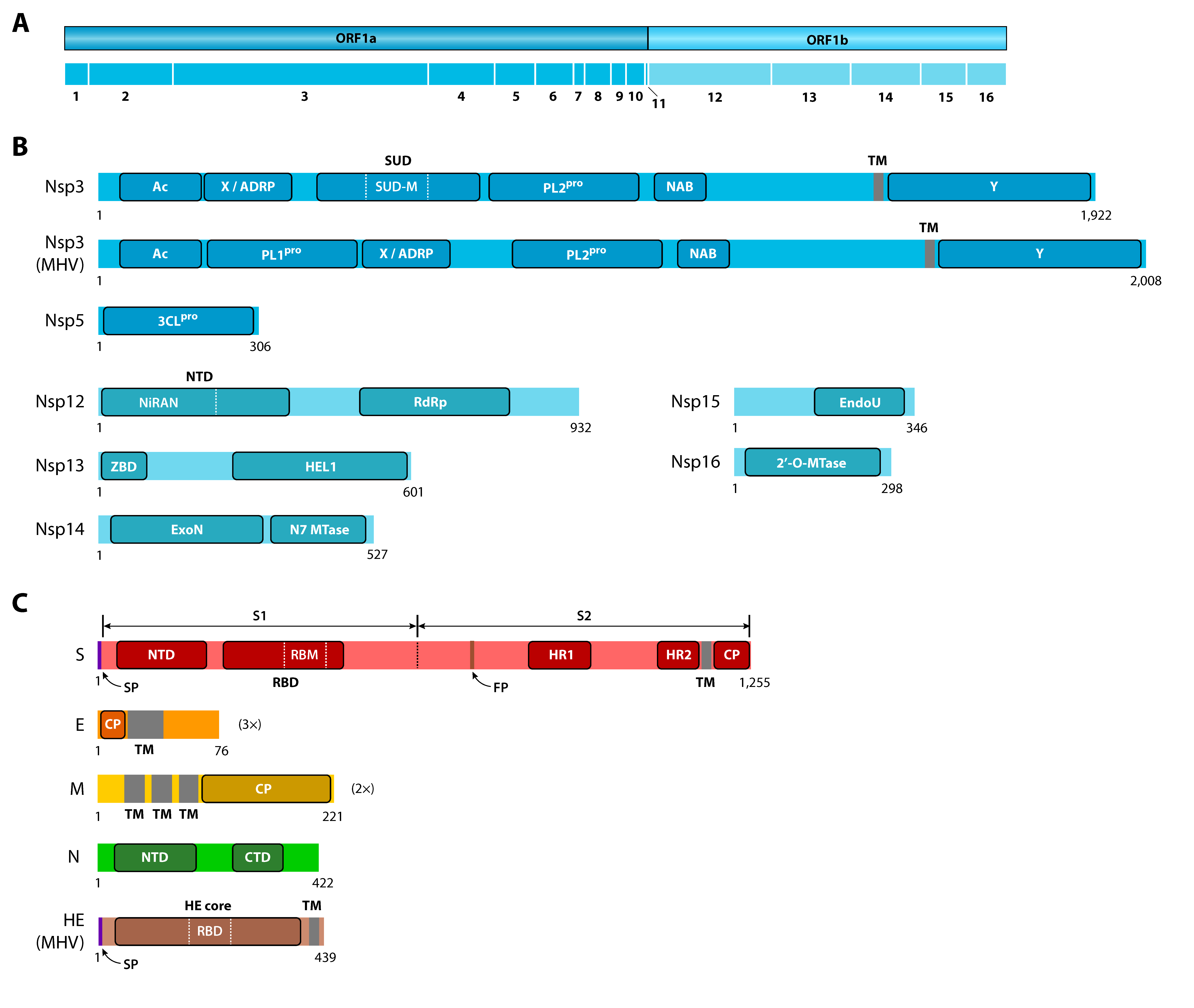{kind=link}
{kind=link}
| Virus 1 | GenBank Accession | Size (bp) | GC% | ORFs/Accessory Proteins 2 |
|---|---|---|---|---|
| Embecovirus | ||||
| Bovine CoV | NC_003045 | 31,028 | 37.12 | 12/5 |
| China Rattus CoV HKU24 | NC_026011 | 31,249 | 40.07 | 11/4 |
| Dromedary CoV HKU23 | KF906249 | 31,052 | 36.95 | 9/2 |
| Human CoV HKU1 | NC_006577 | 29,926 | 32.06 | 9/2 |
| Human CoV OC43 | NC_006213 | 30,741 | 36.79 | 9/2 |
| Mouse hepatitis virus (MHV) | NC_001846 | 31,526 | 42.03 | 11/4 |
| Porcine hemagglutinating encephalomyelitis virus (PHEV) | DQ011855 | 30,480 | 37.25 | 12/5 |
| Rat CoV Parker | NC_012936 | 31,250 | 41.26 | 10/3 |
| Sarbecovirus | ||||
| Bat SARS-like CoV RaTG13 | MN996532 | 29,855 | 38.04 | 11/5 |
| Bat SARS-like CoV HKU3 | DQ022305 | 29,728 | 41.12 | 12/6 |
| Bat SARS-like CoV SL-CoVZC45 | MG772933 | 29,802 | 38.90 | 12/6 |
| Bat SARS-like CoV SL-CoVZXC21 | MG772934 | 29,732 | 38.82 | 12/6 |
| Bat SARS-like CoV WIV1 | KF367457 | 30,309 | 40.77 | 13/7 |
| SARS-CoV (Human) | NC_004718 | 29,751 | 40.76 | 14/8 |
| SARS-CoV (Civet) | AY686863 | 29,499 | 40.85 | 13/7 |
| SARS-CoV-2 (Human) | NC_045512 | 29,903 | 37.97 | 12/6 |
| SARS-CoV-2 (Tiger) | MT365033 | 29,897 | 37.97 | 11/5 |
| Pangolin CoV | MT040333 | 29,805 | 38.52 | 10/4 |
| Merbecovirus | ||||
| Hedgehog CoV HKU31 | MK907286 | 29,951 | 37.69 | 10/4 |
| MERS-CoV (Human) | NC_019843 | 30,119 | 41.24 | 11/5 |
| MERS-CoV (Dromedary camel) | KF917527 | 29,851 | 41.19 | 10/4 |
| Neoromicia bat CoV | MF593268 | 30,009 | 40.21 | 10/4 |
| Pipistrellus bat CoV HKU5 | NC_009020 | 30,482 | 43.19 | 10/4 |
| Tylonycteris bat CoV HKU4 | NC_009019 | 30,286 | 37.82 | 10/4 |
| Nobecovirus | ||||
| Rousettus bat CoV GCCDC1 | NC_030886 | 30,161 | 45.30 | 11/5 |
| Rousettus bat CoV HKU9 | NC_009021 | 29,114 | 41.05 | 9/3 |
| Hibecovirus | ||||
| Bat Hp-BetaCoV Zhejiang2013 | NC_025217 | 31,491 | 41.28 | 10/4 |

2.1. Spike (S) Protein
2.2. Replicase/Transcriptase and Nonstructural Proteins
2.3. Envelope (E) and Membrane (M) Proteins
2.4. Nucleocapsid (N) Protein
2.5. Accessory Proteins
2.6. Haemagglutinin Esterase (HE)
3. Basic Phylogenetic Relationships
4. Molecular Epidemiology
4.1. Evolutionary Rates and Divergence
4.2. Recombination, RBD Mutations and Host/Tissue Tropism
4.3. Genetic Variation and Transmission in Human Populations
5. Diagnostics, Drug Design and Vaccine Candidates
5.1. Reverse Genetic Systems
5.2. Diagnostics
5.3. Drug Design
5.4. Vaccine Candidates
6. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moore, M.J.; Vasllieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greeneugh, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.K.; Jiang, G.S.; Holmes, K.V. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. USA 1991, 88, 5533–5536. [Google Scholar] [CrossRef] [Green Version]
- Nédellec, P.; Dveksler, G.S.; Daniels, E.; Turbide, C.; Chow, B.; Basile, A.A.; Holmes, K.V.; Beauchemin, N. Bgp2, a new member of the carcinoembryonic antigen-related gene family, encodes an alternative receptor for mouse hepatitis viruses. J. Virol. 1994, 68, 4525–4537. [Google Scholar] [CrossRef] [Green Version]
- Schultze, B.; Cavanagh, D.; Herrler, G. Neuraminidase treatment of avian infectious bronchitis coronavirus reveals a hemagglutinating activity that is dependent on sialic acid-containing receptors on erythrocytes. Virology 1992, 189, 792–794. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structural biology: Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science (80) 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Rajashankar, K.R.; Yang, Y.; Agnihothram, S.S.; Liu, C.; Lin, Y.-L.; Baric, R.S.; Li, F. Crystal Structure of the Receptor-Binding Domain from Newly Emerged Middle East Respiratory Syndrome Coronavirus. J. Virol. 2013, 87, 10777–10783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Sun, D.; Rajashankar, K.R.; Qian, Z.; Holmes, K.V.; Li, F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 10696–10701. [Google Scholar] [CrossRef] [Green Version]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.M.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B.; Rausch, J.M.; Gallaher, W.R.; Garry, R.F.; Wimley, W.C. Identification and Characterization of the Putative Fusion Peptide of the Severe Acute Respiratory Syndrome-Associated Coronavirus Spike Protein. J. Virol. 2005, 79, 7195–7206. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.M.; Guan, Y.; Rozanov, M.; Spaan, W.J.M.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Chatterjee, A.; Johnson, M.A.; Serrano, P.; Pedrini, B.; Joseph, J.S.; Neuman, B.W.; Saikatendu, K.; Buchmeier, M.J.; Kuhn, P.; Wüthrich, K. Nuclear Magnetic Resonance Structure Shows that the Severe Acute Respiratory Syndrome Coronavirus-Unique Domain Contains a Macrodomain Fold. J. Virol. 2009, 83, 1823–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, P.; Johnson, M.A.; Chatterjee, A.; Neuman, B.W.; Joseph, J.S.; Buchmeier, M.J.; Kuhn, P.; Wüthrich, K. Nuclear Magnetic Resonance Structure of the Nucleic Acid-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein 3. J. Virol. 2009, 83, 12998–13008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.C.; Ruben, M.; Overkleeft, H.S.; Van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogando, N.S.; Ferron, F.; Decroly, E.; Canard, B.; Posthuma, C.C.; Snijder, E.J. The Curious Case of the Nidovirus Exoribonuclease: Its Role in RNA Synthesis and Replication Fidelity. Front. Microbiol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Seybert, A.; Posthuma, C.C.; van Dinten, L.C.; Snijder, E.J.; Gorbalenya, A.E.; Ziebuhr, J. A Complex Zinc Finger Controls the Enzymatic Activities of Nidovirus Helicases. J. Virol. 2005, 79, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Hurst, K.R.; Kuo, L.; Koetzner, C.A.; Ye, R.; Hsue, B.; Masters, P.S. A Major Determinant for Membrane Protein Interaction Localizes to the Carboxy-Terminal Domain of the Mouse Coronavirus Nucleocapsid Protein. J. Virol. 2005, 79, 13285–13297. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Hogue, B.G. Role of the Coronavirus E Viroporin Protein Transmembrane Domain in Virus Assembly. J. Virol. 2007, 81, 3597–3607. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Kuo, L.; Hurst-Hess, K.R.; Koetzner, C.A.; Masters, P.S. Analyses of Coronavirus Assembly Interactions with Interspecies Membrane and Nucleocapsid Protein Chimeras. J. Virol. 2016, 90, 4357–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.; Mckinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madan, V.; García, M.D.J.; Sanz, M.A.; Carrasco, L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 2005, 579, 3607–3612. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; De Diego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Koetzner, C.A.; Masters, P.S. A key role for the carboxy-terminal tail of the murine coronavirus nucleocapsid protein in coordination of genome packaging. Virology 2016, 494, 100–107. [Google Scholar] [CrossRef]
- Jayaram, H.; Fan, H.; Bowman, B.R.; Ooi, A.; Jayaram, J.; Collisson, E.W.; Lescar, J.; Prasad, B.V.V. X-ray Structures of the N- and C-Terminal Domains of a Coronavirus Nucleocapsid Protein: Implications for Nucleocapsid Formation. J. Virol. 2006, 80, 6612–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.X.; Fung, T.S.; Chong, K.K.L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef]
- Pewe, L.; Zhou, H.; Netland, J.; Tangudu, C.; Olivares, H.; Shi, L.; Look, D.; Gallagher, T.; Perlman, S. A Severe Acute Respiratory Protein Enhances Virulence of an Attenuated Murine Coronavirus. J. Virol. 2005, 79, 11335–11342. [Google Scholar] [CrossRef] [Green Version]
- Pfefferle, S.; Krähling, V.; Ditt, V.; Grywna, K.; Mühlberger, E.; Drosten, C. Reverse genetic characterization of the natural genomic deletion in SARS-Coronavirus strain Frankfurt-1 open reading frame 7b reveals an attenuating function of the 7b protein in-vitro and in-vivo. Virol. J. 2009, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.Y.; Yuen, K.S.; Ye, Z.W.; Chan, C.P.; Jin, D.Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg. Microbes Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef]
- Fischer, F.; Peng, D.; Hingley, S.T.; Weiss, S.R.; Masters, P.S. The internal open reading frame within the nucleocapsid gene of mouse hepatitis virus encodes a structural protein that is not essential for viral replication. J. Virol. 1997, 71, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senanayake, S.D.; Brian, D.A. Bovine coronavirus I protein synthesis follows ribosomal scanning on the bicistronic N mRNA. Virus Res. 1997, 48, 101–105. [Google Scholar] [CrossRef]
- Xu, K.; Zheng, B.J.; Zeng, R.; Lu, W.; Lin, Y.P.; Xue, L.; Li, L.; Yang, L.L.; Xu, C.; Dai, J.; et al. Severe acute respiratory syndrome coronavirus accessory protein 9b is a virion-associated protein. Virology 2009, 388, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.Y. Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1805–1820. [Google Scholar] [CrossRef] [Green Version]
- Vlasak, R.; Luytjes, W.; Leider, J.; Spaan, W.; Palese, P. The E3 protein of bovine coronavirus is a receptor-destroying enzyme with acetylesterase activity. J. Virol. 1988, 62, 4686–4690. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Langereis, M.A.; Van Vliet, A.L.W.; Huizinga, E.G.; De Groot, R.J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 9065–9069. [Google Scholar] [CrossRef] [Green Version]
- Luytjes, W.; Bredenbeek, P.J.; Noten, A.F.H.; Horzinek, M.C.; Spaan, W.J.M. Sequence of mouse hepatitis virus A59 mRNA 2: Indications for RNA recombination between coronaviruses and influenza C virus. Virology 1988, 166, 415–422. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Tsoi, H.; Huang, Y.; Poon, R.W.S.; Chu, C.; Lee, R.A.; Luk, W.; Wong, G.K.M.; Wong, B.H.L.; et al. Clinical and Molecular Epidemiological Features of Coronavirus HKU1–Associated Community-Acquired Pneumonia. J. Infect. Dis. 2005, 192, 1898–1907. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Tse, H.; Tsoi, H.W.; Cheng, V.C.C.; Lee, P.; Tang, B.S.F.; Cheung, C.H.Y.; Lee, R.A.; et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J. Clin. Microbiol. 2006, 44, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Tsoi, H.-W.; Chan, K.-H.; Yuen, K.-Y. Comparative Analysis of 22 Coronavirus HKU1 Genomes Reveals a Novel Genotype and Evidence of Natural Recombination in Coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Saberi, A.; Gulyaeva, A.A.; Brubacher, J.L.; Newmark, P.A.; Gorbalenya, A.E. A planarian nidovirus expands the limits of RNA genome size. PLoS Pathog. 2018, 14, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ge, X.; Wang, L.F.; Shi, Z. Bat origin of human coronaviruses Coronaviruses: Emerging and re-emerging pathogens in humans and animals Susanna Lau Positive-strand RNA viruses. Virol. J. 2015, 12, 1–10. [Google Scholar]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science (80) 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Tsoi, H.W.; Wong, B.H.L.; Wong, S.S.Y.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [Green Version]
- Luk, H.K.H.; Li, X.; Fung, J.; Lau, S.K.P.; Woo, P.C.Y. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Van Boheemen, S.; De Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.; et al. Genomic Characterization of a Newly Discovered Coronavirus. MBio 2012, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldenhuys, M.; Mortlock, M.; Weyer, J.; Bezuidt, O.; Seamark, E.C.J.; Kearney, T.; Gleasner, C.; Erkkila, T.H.; Cui, H.; Markotter, W. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE 2018, 13, 1–27. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science (80) 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Kan, B.; Wang, M.; Jing, H.; Xu, H.; Jiang, X.; Yan, M.; Liang, W.; Zheng, H.; Wan, K.; Liu, Q.; et al. Molecular Evolution Analysis and Geographic Investigation of Severe Acute Respiratory Syndrome Coronavirus-Like Virus in Palm Civets at an Animal Market and on Farms. J. Virol. 2005, 79, 11892–11900. [Google Scholar] [CrossRef] [Green Version]
- Haagmans, B.L.; Al Dhahiry, S.H.S.; Reusken, C.B.E.M.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2014, 14, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Stalin Raj, V.; Farag, E.A.B.A.; Reusken, C.B.E.M.; Lamers, M.M.; Pas, S.D.; Voermans, J.; Smits, S.L.; Osterhaus, A.D.M.E.; Al-Mawlawi, N.; Al-Romaihi, H.E.; et al. Isolation of MERS coronavirus from dromedary camel, Qatar, 2014. Emerg. Infect. Dis. 2014, 20, 1339–1342. [Google Scholar]
- Woo, P.C.Y.; Lau, S.K.P.; Wernery, U.; Wong, E.Y.M.; Tsang, A.K.L.; Johnson, B.; Yip, C.C.Y.; Lau, C.C.Y.; Sivakumar, S.; Cai, J.P.; et al. Novel betacoronavirus in dromedaries of the Middle East, 2013. Emerg. Infect. Dis. 2014, 20, 560–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuto, D.; Giovanetti, M.; Ciccozzi, A.; Spoto, S.; Angeletti, S.; Ciccozzi, M. The 2019-new coronavirus epidemic: Evidence for virus evolution. J. Med. Virol. 2020, 92, 455–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.T.Y.; Shum, M.H.H.; Zhu, H.C.; Tong, Y.G.; Ni, X.B.; Liao, Y.S.; Wei, W.; Cheung, W.Y.M.; Li, W.J.; Li, L.F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020, in press. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, C.M.; Gebauer, F.; Suñé, C.; Mendez, A.; Dopazo, J.; Enjuanes, L. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 1992, 190, 92–105. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Van Ranst, M. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotten, M.; Watson, S.J.; Zumla, A.I.; Makhdoom, H.Q.; Palser, A.L.; Ong, S.H.; Al Rabeeah, A.A.; Alhakeem, R.F.; Assiri, A.; Al-Tawfiq, J.A.; et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. MBio 2014, 5, e01062-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.D.; Zhang, G.W.; Cheng, F.; Pan, C.M.; Chen, S.J.; Tu, C.C.; Lei, L.C.; Gao, Y.W.; Xiang, H.; Xuan, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, M.; Fitch, W.M.; Ciccozzi, M.; Ruiz-Alvarez, M.J.; Rezza, G.; Lewis, M.J. Severe Acute Respiratory Syndrome Coronavirus Sequence Characteristics and Evolutionary Rate Estimate from Maximum Likelihood Analysis. J. Virol. 2004, 78, 1602–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijaykrishna, D.; Smith, G.J.D.; Zhang, J.X.; Peiris, J.S.M.; Chen, H.; Guan, Y. Evolutionary Insights into the Ecology of Coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [Green Version]
- Hon, C.-C.; Lam, T.-Y.; Shi, Z.-L.; Drummond, A.J.; Yip, C.-W.; Zeng, F.; Lam, P.-Y.; Leung, F.C.-C. Evidence of the Recombinant Origin of a Bat Severe Acute Respiratory Syndrome (SARS)-Like Coronavirus and Its Implications on the Direct Ancestor of SARS Coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Li, K.S.M.; Huang, Y.; Shek, C.-T.; Tse, H.; Wang, M.; Choi, G.K.Y.; Xu, H.; Lam, C.S.F.; Guo, R.; et al. Ecoepidemiology and Complete Genome Comparison of Different Strains of Severe Acute Respiratory Syndrome-Related Rhinolophus Bat Coronavirus in China Reveal Bats as a Reservoir for Acute, Self-Limiting Infection That Allows Recombination Events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.; Castoe, T.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. bioRxiv 2020. [Google Scholar] [CrossRef]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Lee, P.; Tsang, A.K.L.; Yip, C.C.Y.; Tse, H.; Lee, R.A.; So, L.-Y.; Lau, Y.-L.; Chan, K.-H.; Woo, P.C.Y.; et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Feng, Y.; Chen, H.; Luk, H.K.H.; Yang, W.-H.; Li, K.S.M.; Zhang, Y.-Z.; Huang, Y.; Song, Z.-Z.; Chow, W.-N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; Zhang, J.; Yang, F.; Zhang, S.; Jin, Q. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2016, 213, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Hu, B.; Wang, B.; Wang, M.-N.; Zhang, Q.; Zhang, W.; Wu, L.-J.; Ge, X.-Y.; Zhang, Y.-Z.; Daszak, P.; et al. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2015, 90, 3253–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 2020, nwaa036. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.F.; Peng, G.W.; Min, J.; Yu, D.W.; Liang, W.J.; Zhang, S.Y.; Xu, R.H.; Zheng, H.Y.; Wu, X.W.; Xu, J.; et al. Molecular Evolution of the SARS Coronavirus, during the Course of the SARS Epidemic in China. Science (80) 2004, 303, 1666–1669. [Google Scholar]
- Zhang, C.Y.; Wei, J.F.; He, S.N. Adaptive evolution of the spike gene of SARS coronavirus: Changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006, 6, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Shen, L.; Gu, X. Evolutionary Dynamics of MERS-CoV: Potential Recombination, Positive Selection and Transmission. Sci. Rep. 2016, 6, 25049. [Google Scholar] [CrossRef] [Green Version]
- Bolles, M.; Donaldson, E.; Baric, R. SARS-CoV and emergent coronaviruses: Viral determinants of interspecies transmission. Curr. Opin. Virol. 2011, 1, 624–634. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Computational inference of selection underlying the evolution of the novel coronavirus, SARS-CoV-2. J. Virol. 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.T.Y.; Hon, C.C.; Tang, J.W. Use of Phylogenetics in the Molecular Epidemiology and Evolutionary Studies of Viral Infections. Crit. Rev. Clin. Lab. Sci. 2010, 47, 5–49. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Gardy, J.; Colijn, C. Bayesian inference of infectious disease transmission from whole-genome sequence data. Mol. Biol. Evol. 2014, 31, 1869–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinkenberg, D.; Backer, J.A.; Didelot, X.; Colijn, C.; Wallinga, J. Simultaneous Inference of Phylogenetic and Transmission Trees in Infectious Disease Outbreaks. PLoS Comput. Biol. 2017, 13, e1005495. [Google Scholar] [CrossRef] [Green Version]
- Wymant, C.; Hall, M.; Ratmann, O.; Bonsall, D.; Golubchik, T.; De Cesare, M.; Gall, A.; Cornelissen, M.; Fraser, C. Phyloscanner: Inferring transmission from within- and between-host pathogen genetic diversity. Mol. Biol. Evol. 2018, 35, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Didelot, X.; Fitzjohn, R.; Ferguson, N.; Cori, A.; Jombart, T. Outbreaker2: A modular platform for outbreak reconstruction. BMC Bioinform. 2018, 19, 363. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Cori, A.; Didelot, X.; Cauchemez, S.; Fraser, C.; Ferguson, N. Bayesian reconstruction of disease outbreaks by combining epidemiologic and genomic data. PLoS Comput. Biol. 2014, 10, e1003457. [Google Scholar] [CrossRef]
- Fraser, C.; Donnelly, C.A.; Cauchemez, S.; Hanage, W.P.; Van Kerkhove, M.D.; Hollingsworth, T.D.; Griffin, J.; Baggaley, R.F.; Jenkins, H.E.; Lyons, E.J.; et al. Pandemic potential of a strain of influenza A (H1N1): Early findings. Science (80) 2009, 324, 1557–1561. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Singer, J.B.; Thomson, E.C.; McLauchlan, J.; Hughes, J.; Gifford, R.J. GLUE: A flexible software system for virus sequence data. BMC Bioinform. 2018, 19, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, P.; Forster, L.; Renfrew, C.; Forster, M. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 9241–9243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, D. Coronavirus SARS-CoV-2: Filtering fact from fiction in the infodemic: Q&A with virologist Professor Urs Greber. FEBS Lett. 2020, 594, 1127–1131. [Google Scholar] [PubMed] [Green Version]
- Sánchez-Pacheco, S.J.; Kong, S.; Pulido-Santacruz, P.; Murphy, R.W.; Kubatko, L. Median-joining network analysis of SARS-CoV-2 genomes is neither phylogenetic nor evolutionary. Proc. Natl. Acad. Sci. USA 2020, 117, 12518–12519. [Google Scholar] [CrossRef]
- Mavian, C.; Pond, S.K.; Marini, S.; Magalis, B.R.; Vandamme, A.-M.; Dellicour, S.; Scarpino, S.V.; Houldcroft, C.; Villabona-Arenas, J.; Paisie, T.K.; et al. Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable. Proc. Natl. Acad. Sci. USA 2020, 117, 12522–12523. [Google Scholar] [CrossRef]
- Chookajorn, T. Evolving COVID-19 conundrum and its impact. Proc. Natl. Acad. Sci. USA 2020, 117, 12520. [Google Scholar] [CrossRef]
- Cockrell, A.S.; Beall, A.; Yount, B.; Baric, R. Efficient Reverse Genetic Systems for Rapid Genetic Manipulation of Emergent and Preemergent Infectious Coronaviruses. Methods Mol. Biol. 2017, 1602, 59–81. [Google Scholar]
- Totura, A.L.; Bavari, S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin. Drug Discov. 2019, 14, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Almazán, F.; Sola, I.; Zuñiga, S.; Marquez-Jurado, S.; Morales, L.; Becares, M.; Enjuanes, L. Coronavirus reverse genetic systems: Infectious clones and replicons. Virus Res. 2014, 194, 67–75. [Google Scholar] [CrossRef]
- Yount, B.; Curtis, K.M.; Baric, R.S. Strategy for Systematic Assembly of Large RNA and DNA Genomes: Transmissible Gastroenteritis Virus Model. J. Virol. 2000, 74, 10600–10611. [Google Scholar] [CrossRef] [Green Version]
- Scobey, T.; Yount, B.L.; Sims, A.C.; Donaldson, E.F.; Agnihothram, S.S.; Menachery, V.D.; Graham, R.L.; Swanstrom, J.; Bove, P.F.; Kim, J.D.; et al. Reverse genetics with a full-length infectious cDNA of the Middle East respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 16157–16162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Muruato, A.; Lokugamage, K.G.; Narayanan, K.; Zhang, X.; Zou, J.; Liu, J.; Schindewolf, C.; Bopp, N.E.; Aguilar, P.V.; et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 2020, 27, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Yount, B.; Curtis, K.M.; Fritz, E.A.; Hensley, L.E.; Jahrling, P.B.; Prentice, E.; Denison, M.R.; Geisbert, T.W.; Baric, R.S. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2003, 100, 12995–13000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, A.C.; Baric, R.S.; Yount, B.; Burkett, S.E.; Collins, P.L.; Pickles, R.J. Severe Acute Respiratory Syndrome Coronavirus Infection of Human Ciliated Airway Epithelia: Role of Ciliated Cells in Viral Spread in the Conducting Airways of the Lungs. J. Virol. 2005, 79, 15511–15524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yount, B.; Roberts, R.S.; Lindesmith, L.; Baric, R.S. Rewiring the severe acute respiratory syndrome coronavirus (SARS-CoV) transcription circuit: Engineering a recombination-resistant genome. Proc. Natl. Acad. Sci. USA 2006, 103, 12546–12551. [Google Scholar] [CrossRef] [Green Version]
- Frieman, M.; Yount, B.; Agnihothram, S.; Page, C.; Donaldson, E.; Roberts, A.; Vogel, L.; Woodruff, B.; Scorpio, D.; Subbarao, K.; et al. Molecular Determinants of Severe Acute Respiratory Syndrome Coronavirus Pathogenesis and Virulence in Young and Aged Mouse Models of Human Disease. J. Virol. 2012, 86, 884–897. [Google Scholar] [CrossRef] [Green Version]
- Cockrell, A.S.; Yount, B.L.; Scobey, T.; Jensen, K.; Douglas, M.; Beall, A.; Tang, X.C.; Marasco, W.A.; Heise, M.T.; Baric, R.S. A mouse model for MERS coronavirus-induced acute respiratory distress syndrome. Nat. Microbiol. 2016, 2, 1–11. [Google Scholar] [CrossRef]
- Roberts, A.; Deming, D.; Paddock, C.D.; Cheng, A.; Yount, B.; Vogel, L.; Herman, B.D.; Sheahan, T.; Heise, M.; Genrich, G.L.; et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007, 3, 0023–0037. [Google Scholar] [CrossRef]
- Day, C.W.; Baric, R.; Cai, S.X.; Frieman, M.; Kumaki, Y.; Morrey, J.D.; Smee, D.F.; Barnard, D.L. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology 2009, 395, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, T.; Rockx, B.; Donaldson, E.; Sims, A.; Pickles, R.; Corti, D.; Baric, R. Mechanisms of Zoonotic Severe Acute Respiratory Syndrome Coronavirus Host Range Expansion in Human Airway Epithelium. J. Virol. 2008, 82, 2274–2285. [Google Scholar] [CrossRef] [Green Version]
- Rockx, B.; Sheahan, T.; Donaldson, E.; Harkema, J.; Sims, A.; Heise, M.; Pickles, R.; Cameron, M.; Kelvin, D.; Baric, R. Synthetic Reconstruction of Zoonotic and Early Human Severe Acute Respiratory Syndrome Coronavirus Isolates That Produce Fatal Disease in Aged Mice. J. Virol. 2007, 81, 7410–7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockx, B.; Feldmann, F.; Brining, D.; Gardner, D.; LaCasse, R.; Kercher, L.; Long, D.; Rosenke, R.; Virtaneva, K.; Sturdevant, D.E.; et al. Comparative pathogenesis of three human and zoonotic sars-cov strains in cynomolgus macaques. PLoS ONE 2011, 6, e18558. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.M.; Graham, R.L.; Donaldson, E.F.; Rockx, B.; Sims, A.C.; Sheahan, T.; Pickles, R.J.; Corti, D.; Johnston, R.E.; Baric, R.S.; et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19944–19949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihothram, S.; Yount, B.L.; Donaldson, E.F. A Mouse Model for Betacoronavirus Subgroup 2c Using a Bat. MBio 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Menachery, V.D.; Yount, B.L.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. USA 2016, 113, 3048–3053. [Google Scholar] [CrossRef] [Green Version]
- Loeffelholz, M.J.; Tang, Y.W. Laboratory diagnosis of emerging human coronavirus infections–the state of the art. Emerg. Microbes Infect. 2020, 9, 747–756. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Briese, T.; Mishra, N.; Jain, K.; East, M.; Syndrome, R.; Quasispecies, C.; Include, T.; Revealed, H.I.; Analysis, W.; Cultured, V.; et al. Dromedary Camels in Saudi Arabia Include Homologues of Human Isolates Revealed through Whole-Genome analysis etc. MBio 2014, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Hui, R.K.H.; Zeng, F.; Chan, C.M.N.; Yuen, K.Y.; Peiris, J.S.M.; Leung, F.C.C. Reverse Transcriptase PCR Diagnostic Assay for the Coronavirus Associated with Severe Acute Respiratory Syndrome. J. Clin. Microbiol. 2004, 42, 1994–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.M.; Eckerle, I.; Bleicker, T.; Zaki, A.; Landt, O.; Eschbach-Bludau, M.; van Boheemen, S.; Gopal, R.; Ballhause, M.; Bestebroer, T.M.; et al. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Eurosurveillance 2012, 17, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Whitaker, B.; Sakthivel, S.K.K.; Kamili, S.; Rose, L.E.; Lowe, L.; Mohareb, E.; Elassal, E.M.; Al-sanouri, T.; Haddadin, A.; et al. Real-time reverse transcription-pcr assay panel for middle east respiratory syndrome coronavirus. J. Clin. Microbiol. 2014, 52, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kang, H.; Liu, X.; Tong, Z. Combination of RT-qPCR testing and clinical features for diagnosis of COVID-19 facilitates management of SARS-CoV-2 outbreak. J. Med. Virol. 2020, 92, 538–539. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Wang, H.; Cao, Z.; Jin, H.; Chi, H.; Zhao, J.; Yu, B.; Yan, F.; Hu, X.; Wu, F.; et al. A rapid and specific assay for the detection of MERS-CoV. Front. Microbiol. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Irigoyen, N.; Firth, A.E.; Jones, J.D.; Chung, B.Y.W.; Siddell, S.G.; Brierley, I. High-Resolution Analysis of Coronavirus Gene Expression by RNA Sequencing and Ribosome Profiling. PLoS Pathog. 2016, 12, e1005473. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Woo, P.C.Y.; Wong, B.H.L.; Tsoi, H.W.; Woo, G.K.S.; Poon, R.W.S.; Chan, K.H.; Wei, W.I.; Malik Peiris, J.S.; Yuen, K.Y. Detection of severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein in SARS patients by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 2004, 42, 2884–2889. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.; Shi, Y.; Guo, Z.; Chen, Z.; He, R.; Chen, R.; Zhou, D.; Dai, E.; Wang, X.; Si, B.; et al. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microbes Infect. 2005, 7, 882–889. [Google Scholar] [CrossRef]
- Chen, Y.; Chan, K.H.; Kang, Y.; Chen, H.; Luk, H.K.H.; Poon, R.W.S.; Chan, J.F.W.; Yuen, K.Y.; Xia, N.; Lau, S.K.P.; et al. A sensitive and specific antigen detection assay for Middle East respiratory syndrome coronavirus. Emerg. Microbes Infect. 2015, 4, e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yi, Y.; Luo, X.; Xiong, N.; Liu, Y.; Li, S.; Sun, R.; Wang, Y.; Hu, B.; Chen, W.; et al. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J. Med. Virol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An Introduction to B-Cell Epitope Mapping and in Silico Epitope Prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef] [Green Version]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Abbott, W.M.; Damschroder, M.M.; Lowe, D.C. Current approaches to fine mapping of antigen-antibody interactions. Immunology 2014, 142, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.P.; Papenburg, J.; Desjardins, M.; Kanjilal, S.; Quach, C.; Libman, M.; Dittrich, S.; Yansouni, C.P. Diagnostic Testing for Severe Acute Respiratory Syndrome–Related Coronavirus-2: A Narrative Review. Ann. Intern. Med. 2020, 172, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.; Mattiuzzo, G.; Hassall, M.; Sieberg, A.; Müller, M.A.; Drosten, C.; Rigsby, P.; Oxenford, C.J.; Caly, L.; Li, C.; et al. Comparison of serologic assays for Middle East respiratory syndrome coronavirus. Emerg. Infect. Dis. 2019, 25, 1878–1883. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, X.S.; Agwanda, B.; Ommeh, S.; Zhao, K.; Lichoti, J.; Wang, N.; Chen, J.; Li, B.; Yang, X.L.; et al. Serological evidence of MERS-CoV and HKU8-related CoV co-infection in Kenyan camels. Emerg. Microbes Infect. 2019, 8, 1528–1534. [Google Scholar] [CrossRef]
- Hu, Z.; Song, C.; Xu, C.; Jin, G.; Chen, Y.; Xu, X.; Ma, H.; Chen, W.; Lin, Y.; Zheng, Y.; et al. Clinical characteristics of 24 asymptomatic infections with COVID-19 screened among close contacts in Nanjing, China. Sci. China Life Sci. 2020, 63, 706–711. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV2). Science 2020, 368, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Xu, S.; Rong, Z.; Xu, R.; Liu, X.; Deng, P.; Liu, H.; Xu, X. Delivery of infection from asymptomatic carriers of COVID-19 in a familial cluster. Int. J. Infect. Dis. 2020, 94, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.Y. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgenfeld, R.; Peiris, M. From SARS to MERS: 10 years of research on highly pathogenic human coronaviruses. Antivir. Res. 2013, 100, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Li, B.J.; Tang, Q.; Cheng, D.; Qin, C.; Xie, F.Y.; Wei, Q.; Xu, J.; Liu, Y.; Zheng, B.J.; Woodle, M.C.; et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat. Med. 2005, 11, 944–951. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Liu, K.; Wu, M.; Lin, H. Computational Identification of Small Interfering RNA Targets in SARS-CoV-2. Virol. Sin. 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Lee, J.; Yang, J.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 1–8. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, in press. [Google Scholar] [CrossRef]
- Roper, R.L.; Rehm, K.E. SARS vaccines: Where are we? Expert Rev. Vaccines 2009, 8, 887–898. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and Restoration of Severe Acute Respiratory Syndrome Coronavirus Mutant Lacking 2’-O-Methyltransferase Activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menachery, V.D.; Gralinski, L.E.; Mitchell, H.D.; Dinnon, K.H.; Leist, S.R.; Yount, B.L.; Graham, R.L.; McAnarney, E.T.; Stratton, K.G.; Cockrell, A.S.; et al. Middle East Respiratory Syndrome Coronavirus Nonstructural Protein 16 Is Necessary for Interferon Resistance and Viral Pathogenesis. mSphere 2017, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Diego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.-J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A Severe Acute Respiratory Syndrome Coronavirus That Lacks the E Gene Is Attenuated In Vitro and In Vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netland, J.; De Diego, M.L.; Zhao, J.; Fett, C.; Álvarez, E.; Nieto-Torres, J.L.; Enjuanes, L.; Perlman, S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010, 399, 120–128. [Google Scholar] [CrossRef]
- Almazán, F.; Dediego, M.L.; Sola, I.; Zuñiga, S.; Nieto-Torres, J.L.; Marquez-Jurado, S.; Andrés, G.; Enjuanes, L. Engineering a replication-competent, propagation-defective middle east respiratory syndrome coronavirus as a vaccine candidate. MBio 2013, 4, e00650-13. [Google Scholar] [CrossRef] [Green Version]
- Fett, C.; De Diego, M.L.; Regla-Nava, J.A.; Enjuanes, L.; Perlman, S. Complete Protection against Severe Acute Respiratory Syndrome Coronavirus-Mediated Lethal Respiratory Disease in Aged Mice by Immunization with a Mouse-Adapted Virus Lacking E Protein. J. Virol. 2013, 87, 6551–6559. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Tamin, A.; Soloff, A.; D’Aiuto, L.; Nwanegbo, E.; Robbins, P.D.; Bellini, W.J.; Barratt-Boyes, S.; Gambotto, A. Effects of a SARS-associated coronavirus vaccine in monkeys. Lancet 2003, 362, 1895–1896. [Google Scholar] [CrossRef] [Green Version]
- Hogan, R.J.; Gao, G.; Rowe, T.; Bell, P.; Flieder, D.; Paragas, J.; Kobinger, G.P.; Wivel, N.A.; Crystal, R.G.; Boyer, J.; et al. Resolution of Primary Severe Acute Respiratory Syndrome-Associated Coronavirus Infection Requires Stat1. J. Virol. 2004, 78, 11416–11421. [Google Scholar] [CrossRef] [Green Version]
- Zakhartchouk, A.N.; Viswanathan, S.; Mahony, J.B.; Glaudei, J.; Babiuk, L.A. Severe acute respiratory syndrome coronavirus nucleocapsid protein expressed by an adenovirus vector is phosphorylated and immunogenic in mice. J. Gen. Virol. 2005, 86, 211–215. [Google Scholar] [CrossRef]
- See, R.H.; Zakhartchouk, A.N.; Petric, M.; Lawrence, D.J.; Mok, C.P.Y.; Hogan, R.J.; Rowe, T.; Zitzow, L.A.; Karunakaran, K.P.; Hitt, M.M.; et al. Comparative evaluation of two severe acute respiratory syndrome (SARS) vaccine candidates in mice challenged with SARS coronavirus. J. Gen. Virol. 2006, 87, 641–650. [Google Scholar] [CrossRef] [PubMed]
- See, R.H.; Petric, M.; Lawrence, D.J.; Mok, C.P.Y.; Rowe, T.; Zitzow, L.A.; Karunakaran, K.P.; Voss, T.G.; Brunham, R.C.; Gauldie, J.; et al. Severe acute respiratory syndrome vaccine efficacy in ferrets: Whole killed virus and adenovirus-vectored vaccines. J. Gen. Virol. 2008, 89, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Okada, K.; Kenniston, T.; Raj, V.S.; AlHajri, M.M.; Farag, E.A.B.A.; AlHajri, F.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Gambotto, A. Immunogenicity of an adenoviral-based Middle East Respiratory Syndrome coronavirus vaccine in BALB/c mice. Vaccine 2014, 32, 5975–5982. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Deng, Y.; Chen, H.; Lan, J.; Wang, W.; Zou, X.; Hung, T.; Lu, Z.; Tan, W. Systemic and mucosal immunity in mice elicited by a single immunization with human adenovirus type 5 or 41 vector-based vaccines carrying the spike protein of Middle East respiratory syndrome coronavirus. Immunology 2015, 145, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukreyev, A.; Lamirande, E.W.; Buchholz, U.J.; Vogel, L.N.; Elkins, W.R.; St Claire, M.; Murphy, B.R.; Subbarao, K.; Collins, P.L. Mucosal immunisation of African green monkeys (Cercopithecus aethiops) with an attenuated parainfluenza virus expressing the SARS coronavirus spike protein for the prevention of SARS. Lancet 2004, 363, 2122–2127. [Google Scholar] [CrossRef] [Green Version]
- Kapadia, S.U.; Simon, I.D.; Rose, J.K. SARS vaccine based on a replication-defective recombinant vesicular stomatitis virus is more potent than one based on a replication-competent vector. Virology 2008, 376, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Liniger, M.; Zuniga, A.; Tamin, A.; Azzouz-Morin, T.N.; Knuchel, M.; Marty, R.R.; Wiegand, M.; Weibel, S.; Kelvin, D.; Rota, P.A.; et al. Induction of neutralising antibodies and cellular immune responses against SARS coronavirus by recombinant measles viruses. Vaccine 2008, 26, 2164–2174. [Google Scholar] [CrossRef]
- Bai, B.; Lu, X.; Meng, J.; Hu, Q.; Mao, P.; Lu, B.; Chen, Z.; Yuan, Z.; Wang, H. Vaccination of mice with recombinant baculovirus expressing spike or nucleocapsid protein of SARS-like coronavirus generates humoral and cellular immune responses. Mol. Immunol. 2008, 45, 868–875. [Google Scholar] [CrossRef]
- Czub, M.; Weingartl, H.; Czub, S.; He, R.; Cao, J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine 2005, 23, 2273–2279. [Google Scholar] [CrossRef]
- Song, F.; Fux, R.; Provacia, L.B.; Volz, A.; Eickmann, M.; Becker, S.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Sutter, G. Middle East Respiratory Syndrome Coronavirus Spike Protein Delivered by Modified Vaccinia Virus Ankara Efficiently Induces Virus-Neutralizing Antibodies. J. Virol. 2013, 87, 11950–11954. [Google Scholar] [CrossRef] [Green Version]
- Volz, A.; Kupke, A.; Song, F.; Jany, S.; Fux, R.; Shams-Eldin, H.; Schmidt, J.; Becker, C.; Eickmann, M.; Becker, S.; et al. Protective Efficacy of Recombinant Modified Vaccinia Virus Ankara Delivering Middle East Respiratory Syndrome Coronavirus Spike Glycoprotein. J. Virol. 2015, 89, 8651–8656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Feng, Y.; Liu, M.; Li, P.; Pan, Q.; Jeza, V.T.; Xia, B.; Wu, J.; Zhang, X.L. Type IVB pilus operon promoter controlling expression of the severe acute respiratory syndrome-associated coronavirus nucleocapsid gene in Salmonella enterica serovar Typhi elicits full immune response by intranasal vaccination. Clin. Vaccine Immunol. 2007, 14, 990–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.Y.; Kong, W.P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, F.; Chow, K.Y.C.; Hon, C.C.; Law, K.M.; Yip, C.W.; Chan, K.H.; Peiris, J.S.M.; Leung, F.C.C. Characterization of humoral responses in mice immunized with plasmid DNAs encoding SARS-CoV spike gene fragments. Biochem. Biophys. Res. Commun. 2004, 315, 1134–1139. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, Z.; Matsumoto, M.; Hengge, U.R.; Chang, Y.F. Immune responses with DNA vaccines encoded different gene fragments of severe acute respiratory syndrome coronavirus in BALB/c mice. Biochem. Biophys. Res. Commun. 2005, 327, 130–135. [Google Scholar] [CrossRef]
- Wang, L.; Shi, W.; Joyce, M.G.; Modjarrad, K.; Zhang, Y.; Leung, K.; Lees, C.R.; Zhou, T.; Yassine, H.M.; Kanekiyo, M.; et al. Evaluation of candidate vaccine approaches for MERS-CoV. Nat. Commun. 2015, 6, 7712. [Google Scholar] [CrossRef]
- Wang, X.; Xu, W.; Tong, D.; Ni, J.; Gao, H.; Wang, Y.; Chu, Y.; Li, P.; Yang, X.; Xiong, S. A chimeric multi-epitope DNA vaccine elicited specific antibody response against severe acute respiratory syndrome-associated coronavirus which attenuated the virulence of SARS-CoV in vitro. Immunol. Lett. 2008, 119, 71–77. [Google Scholar] [CrossRef]
- Zhu, M.S.; Pan, Y.; Chen, H.Q.; Shen, Y.; Wang, X.C.; Sun, Y.J.; Tao, K.H. Induction of SARS-nucleoprotein-specific immune response by use of DNA vaccine. Immunol. Lett. 2004, 92, 237–243. [Google Scholar] [CrossRef]
- Bolles, M.; Deming, D.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A Double-Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine Provides Incomplete Protection in Mice and Induces Increased Eosinophilic Proinflammatory Pulmonary Response upon Challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, T.; Whitmore, A.; Long, K.; Ferris, M.; Rockx, B.; Funkhouser, W.; Donaldson, E.; Gralinski, L.; Collier, M.; Heise, M.; et al. Successful Vaccination Strategies That Protect Aged Mice from Lethal Challenge from Influenza Virus and Heterologous Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2011, 85, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.S.; Tao, X.; Algaissi, A.; Garron, T.; Narayanan, K.; Peng, B.H.; Couch, R.B.; Tseng, C.T.K. Immunization with inactivated Middle East Respiratory Syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum. Vaccines Immunother. 2016, 12, 2351–2356. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, L.; Kuwahara, K.; Li, L.; Liu, Z.; Li, T.; Zhu, H.; Liu, J.; Xu, Y.; Xie, J.; et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect. Dis. 2016, 2, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Houser, K.V.; Broadbent, A.J.; Gretebeck, L.; Vogel, L.; Lamirande, E.W.; Sutton, T.; Bock, K.W.; Minai, M.; Orandle, M.; Moore, I.N.; et al. Enhanced inflammation in New Zealand white rabbits when MERS-CoV reinfection occurs in the absence of neutralizing antibody. PLoS Pathog. 2017, 13, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Cao, Y.; Du, J.; Bu, X.; Ma, R.; Wu, C. Priming with SARS CoV S DNA and boosting with SARS CoV S epitopes specific for CD4+ and CD8+ T cells promote cellular immune responses. Vaccine 2007, 25, 6981–6991. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Krammer, F. Perspective SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llanes, A.; Restrepo, C.M.; Caballero, Z.; Rajeev, S.; Kennedy, M.A.; Lleonart, R. Betacoronavirus Genomes: How Genomic Information has been Used to Deal with Past Outbreaks and the COVID-19 Pandemic. Int. J. Mol. Sci. 2020, 21, 4546. https://doi.org/10.3390/ijms21124546
Llanes A, Restrepo CM, Caballero Z, Rajeev S, Kennedy MA, Lleonart R. Betacoronavirus Genomes: How Genomic Information has been Used to Deal with Past Outbreaks and the COVID-19 Pandemic. International Journal of Molecular Sciences. 2020; 21(12):4546. https://doi.org/10.3390/ijms21124546
Chicago/Turabian StyleLlanes, Alejandro, Carlos M. Restrepo, Zuleima Caballero, Sreekumari Rajeev, Melissa A. Kennedy, and Ricardo Lleonart. 2020. "Betacoronavirus Genomes: How Genomic Information has been Used to Deal with Past Outbreaks and the COVID-19 Pandemic" International Journal of Molecular Sciences 21, no. 12: 4546. https://doi.org/10.3390/ijms21124546