Molecular Basis of the Pathogenic Mechanism Induced by the m.9191T>C Mutation in Mitochondrial ATP6 Gene

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
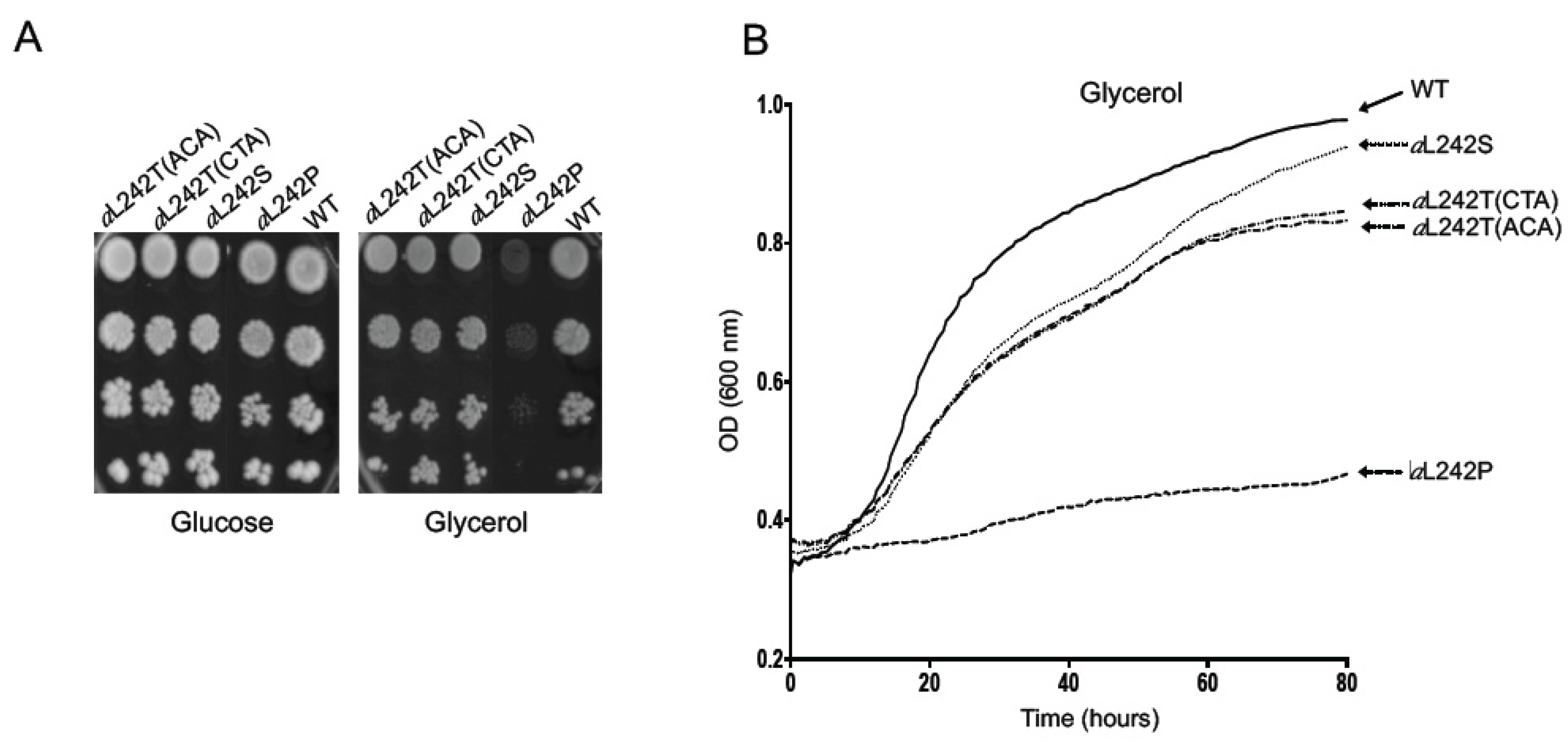
2.1. Isolation of Revertants from the Mutant aL242P
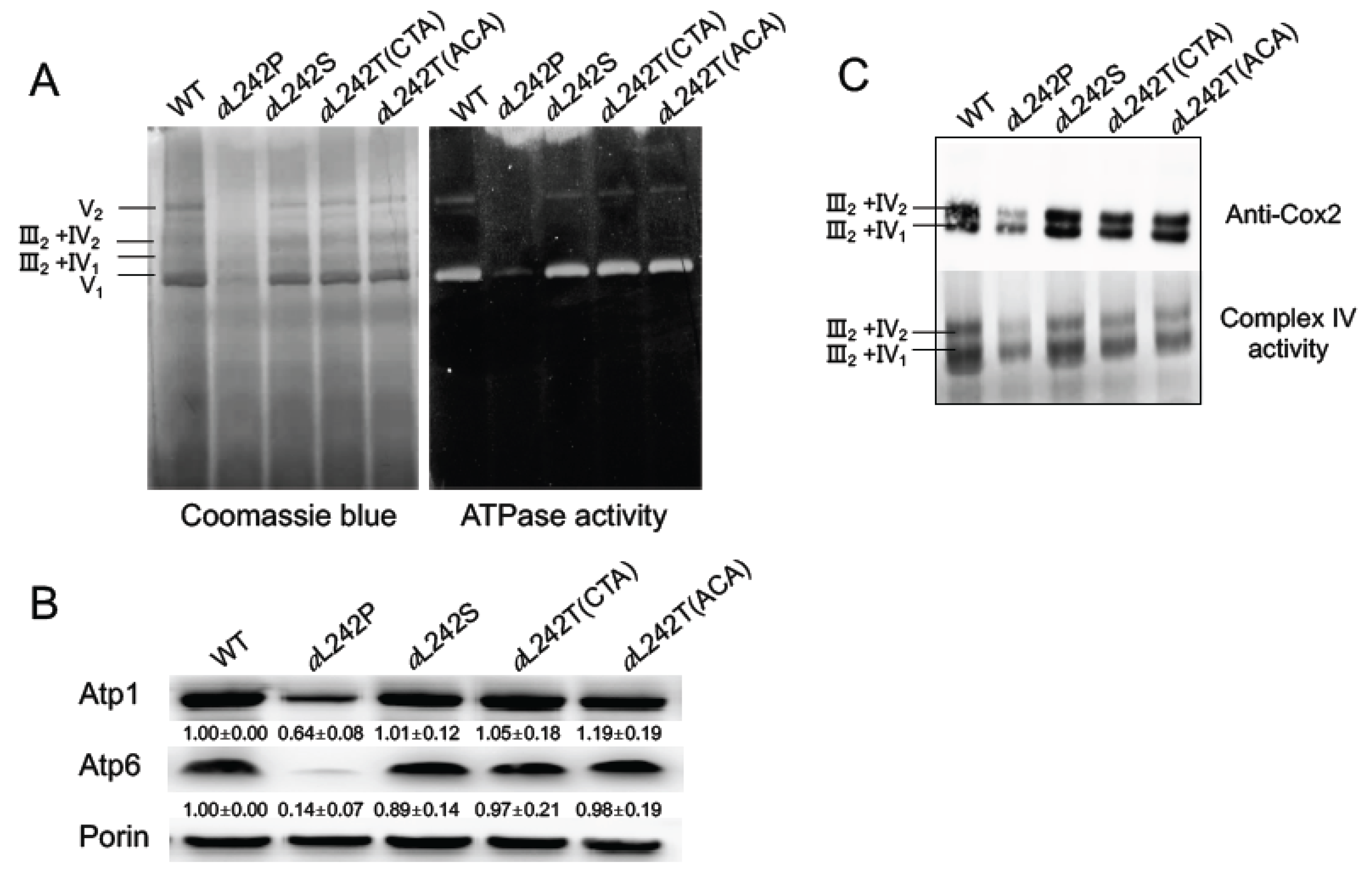
2.2. Assembly of OXPHOS Complexes
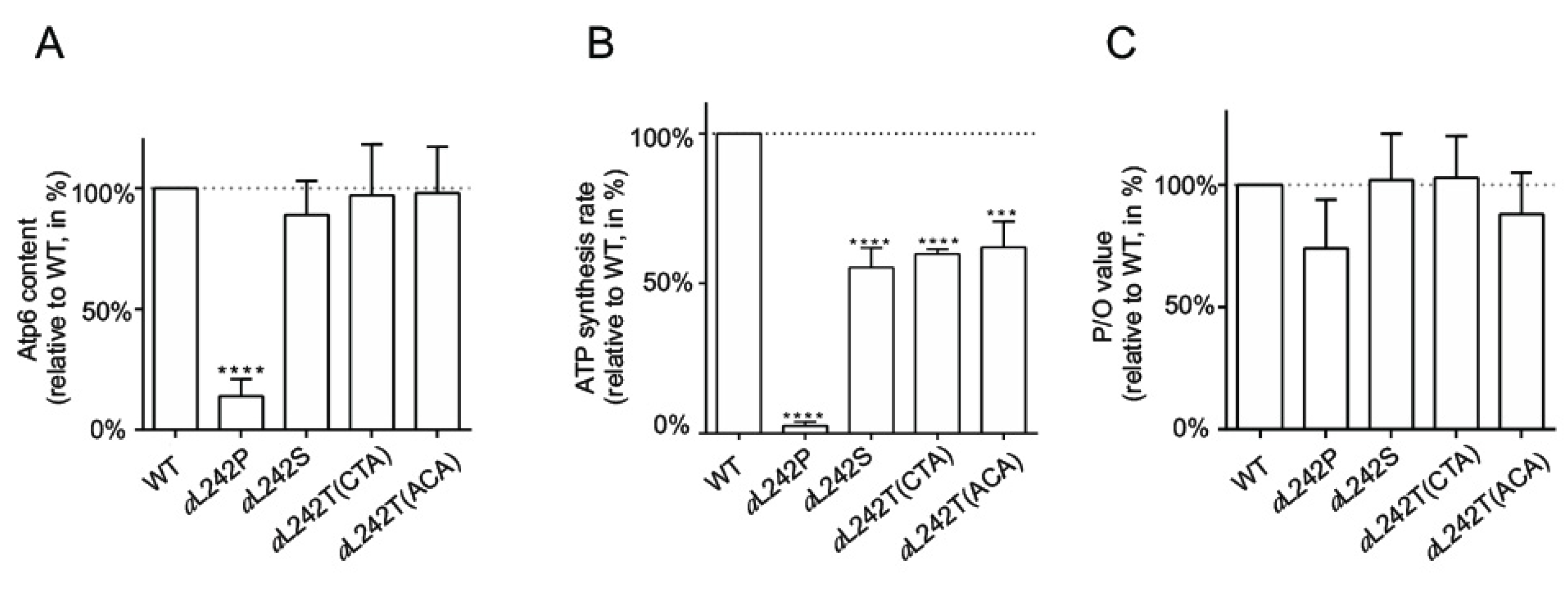
2.3. Mitochondrial Respiration and ATP Synthesis
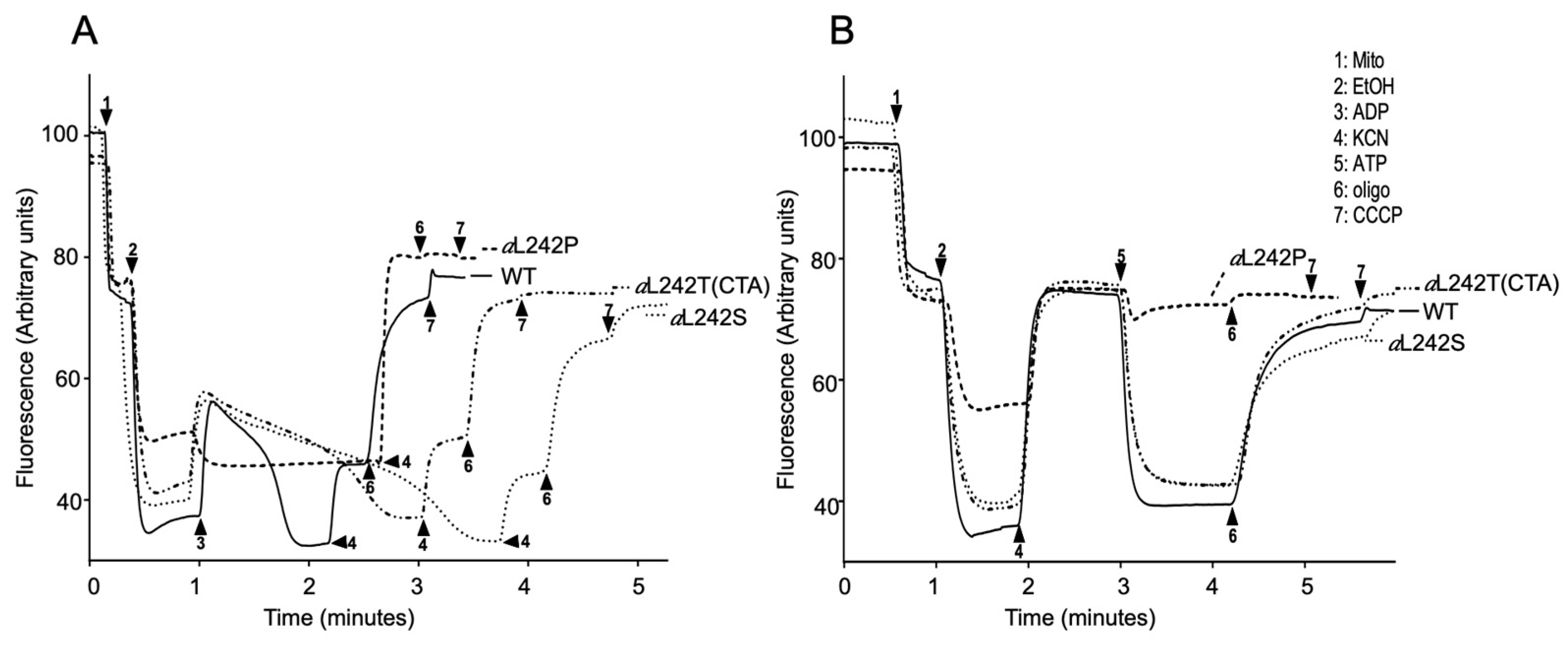
2.4. Mitochondrial ATP Hydrolysis
2.5. Mitochondrial Membrane Potential
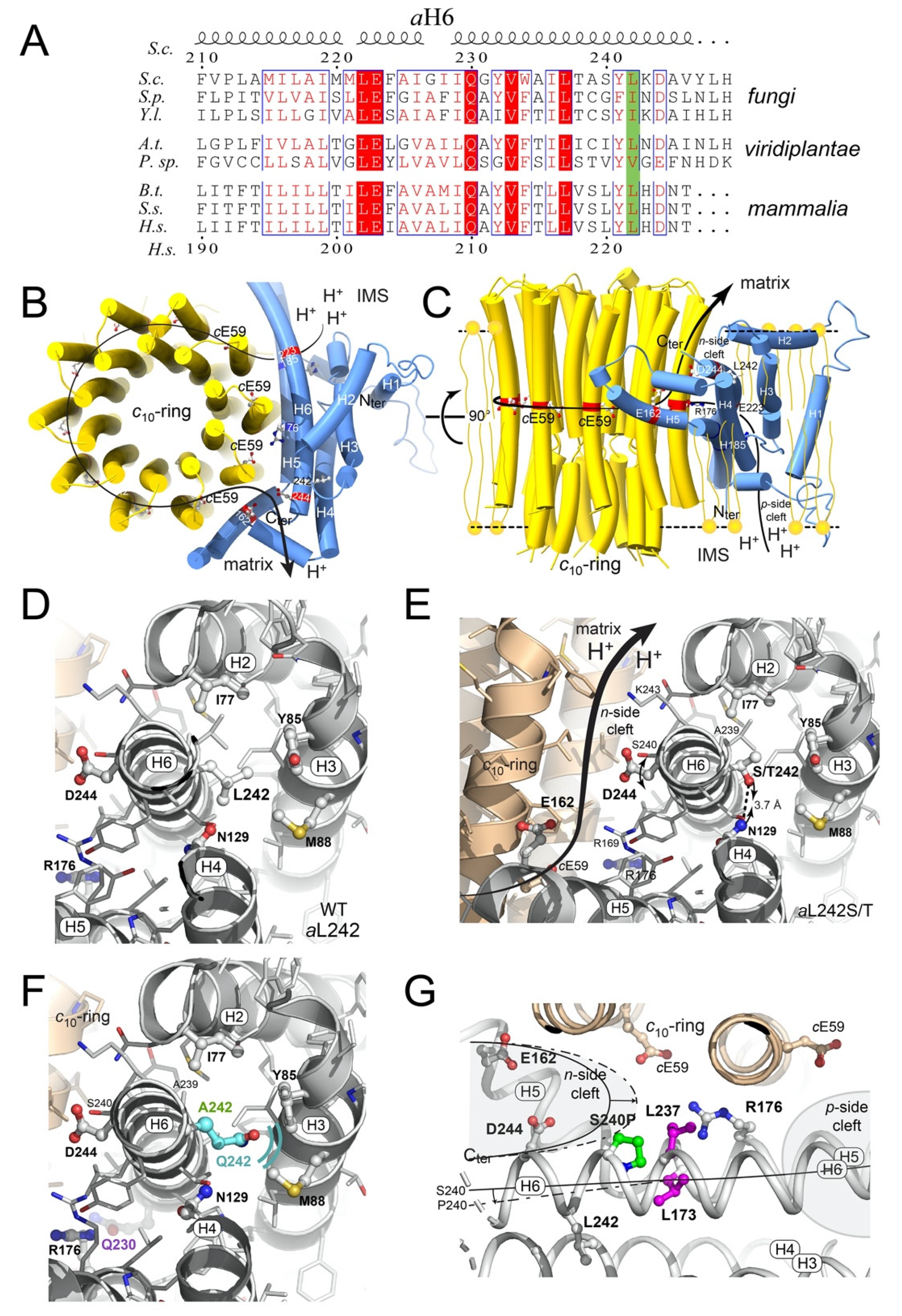
2.6. Structural Modeling
3. Discussion
4. Materials and Methods
4.1. Growth Media and Genotypes
4.2. Selection of Revertants from the Yeast Strain aL242P Mutant
4.3. Miscellaneous Procedures
4.4. Amino-Acid Alignments and Structural Modeling
4.5. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Asc | Ascorbate |
| BN-PAGE | Blue native polyacrylamide gel electrophoresis |
| CCCP | Carbonyl cyanide m-chlorophenylhydrazone |
| EtOH | Ethanol |
| IMS | Intermembrane space |
| KCN | Potassium cyanide |
| MILS | Maternally Inherited Leigh Syndrome |
| oligo | Oligomycin |
| OXPHOS | Oxidative phosphorylation |
| Pi | Inorganic phosphate |
| pmf | Proton-motive force |
| PVDF | Polyvinylidene difluoride |
| TMPD | N, N, N’, N’-tetramethyl-p-phenylenediamine |
| WT | Wild type |
| ΔΨ | Mitochondrial electrical potential |
References
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.D. The ATP synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Morales-Rios, E.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 A resolution. Proc. Natl. Acad Sci. USA 2015, 112, 13231–13236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.; Rohou, A.; Schep, D.G.; Bason, J.V.; Montgomery, M.G.; Walker, J.E.; Grigorieff, N.; Rubinstein, J.L. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife 2015, 4, e10180. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Klusch, N.; Mills, D.J.; Vonck, J.; Kuhlbrandt, W.; Davies, K.M. Horizontal membrane-intrinsic alpha-helices in the stator a-subunit of an F-type ATP synthase. Nature 2015, 521, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Bueler, S.A.; Rubinstein, J.L. Atomic model for the dimeric FO region of mitochondrial ATP synthase. Science 2017, 358, 936–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kuhlbrandt, W.; Meier, T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol Cell 2016, 63, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Morales-Rios, E.; Watt, I.N.; Zhang, Q.; Ding, S.; Fearnley, I.M.; Montgomery, M.G.; Wakelam, M.J.; Walker, J.E. Purification, characterization and crystallization of the F-ATPase from Paracoccus denitrificans. Open Biology 2015, 5, 150119. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.P.; Luo, M.; Zhou, W.; Symersky, J.; Bai, D.; Chambers, M.G.; Faraldo-Gómez, J.D.; Liao, M.; Mueller, D.M. High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 2018, 360. [Google Scholar] [CrossRef] [Green Version]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; di Rago, J.-P.; Kucharczyk, R. ATP synthase diseases of mitochondrial genetic origin. Front. Physiol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Houstek, J.; Pickova, A.; Vojtiskova, A.; Mracek, T.; Pecina, P.; Jesina, P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim. Biophys. Acta 2006, 1757, 1400–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; Santra, S.; Pallotti, F.; Girvin, M.E. Pathogenesis of primary defects in mitochondrial ATP synthesis. Semin. Cell Dev. Biol. 2001, 12, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Rak, M.; Tetaud, E.; Duvezin-Caubet, S.; Ezkurdia, N.; Bietenhader, M.; Rytka, J.; di Rago, J.P. A yeast model of the neurogenic ataxia retinitis pigmentosa (NARP) T8993G mutation in the mitochondrial ATP synthase-6 gene. J. Biol. Chem. 2007, 282, 34039–34047. [Google Scholar] [CrossRef] [Green Version]
- Kucharczyk, R.; Rak, M.; di Rago, J.-P. Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Salin, B.; di Rago, J.P. Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum. Mol. Genet. 2009, 18, 2889–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Ezkurdia, N.; Couplan, E.; Procaccio, V.; Ackerman, S.H.; Blondel, M.; di Rago, J.P. Consequences of the pathogenic T9176C mutation of human mitochondrial DNA on yeast mitochondrial ATP synthase. Biochim. Biophys. Acta 2010, 1797, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Giraud, M.F.; Brethes, D.; Wysocka-Kapcinska, M.; Ezkurdia, N.; Salin, B.; Velours, J.; Camougrand, N.; Haraux, F.; di Rago, J.P. Defining the pathogenesis of human mtDNA mutations using a yeast model: The case of T8851C. Int. J. Biochem. Cell Biol. 2013, 45, 130–140. [Google Scholar] [CrossRef]
- Kabala, A.M.; Lasserre, J.-P.; Ackerman, S.H.; di Rago, J.-P.; Kucharczyk, R. Defining the impact on yeast ATP synthase of two pathogenic human mitochondrial DNA mutations, T9185C and T9191C. Biochimie 2014, 100, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzwiecka, K.; Kabala, A.M.; Lasserre, J.P.; Tribouillard-Tanvier, D.; Golik, P.; Dautant, A.; di Rago, J.P.; Kucharczyk, R. Yeast models of mutations in the mitochondrial ATP6 gene found in human cancer cells. Mitochondrion 2016, 29, 7–17. [Google Scholar] [CrossRef]
- Wen, S.; Niedzwiecka, K.; Zhao, W.; Xu, S.; Liang, S.; Zhu, X.; Xie, H.; Tribouillard-Tanvier, D.; Giraud, M.F.; Zeng, C.; et al. Identification of G8969>A in mitochondrial ATP6 gene that severely compromises ATP synthase function in a patient with IgA nephropathy. Sci. Rep. 2016, 6, 36313. [Google Scholar] [CrossRef] [Green Version]
- Bonnefoy, N.; Fox, T.D. Genetic transformation of Saccharomyces cerevisiae mitochondria. Methods Cell Biol. 2001, 65, 381–396. [Google Scholar] [PubMed]
- Okamoto, K.; Perlman, P.S.; Butow, R.A. The sorting of mitochondrial DNA and mitochondrial proteins in zygotes: Preferential transmission of mitochondrial DNA to the medial bud. J. Cell Biol. 1998, 142, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Moslemi, A.-R.; Darin, N.; Tulinius, M.; Oldfors, A.; Holme, E. Two new mutations in the MTATP6 gene associated with Leigh syndrome. Neuropediatrics 2005, 36, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Michon, T.; Galante, M.; Velours, J. NH2-terminal sequence of the isolated yeast ATP synthase subunit 6 reveals post-translational cleavage. Eur. J. Biochem. 1988, 172, 621–625. [Google Scholar] [CrossRef] [PubMed]
- di Rago, J.P.; Netter, P.; Slonimski, P.P. Pseudo-wild type revertants from inactive apocytochrome b mutants as a tool for the analysis of the structure/function relationships of the mitochondrial ubiquinol-cytochrome c reductase of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 3332–3339. [Google Scholar]
- Mukhopadhyay, A.; Uh, M.; Mueller, D.M. Level of ATP synthase activity required for yeast Saccharomyces cerevisiae to grow on glycerol media. FEBS Lett. 1994, 343, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Bietenhader, M.; Martos, A.; Tetaud, E.; Aiyar, R.S.; Sellem, C.H.; Kucharczyk, R.; Clauder-Munster, S.; Giraud, M.F.; Godard, F.; Salin, B.; et al. Experimental relocation of the mitochondrial ATP9 gene to the nucleus reveals forces underlying mitochondrial genome evolution. PLoS Genet 2012, 8, e1002876. [Google Scholar] [CrossRef]
- Contamine, V.; Picard, M. Maintenance and integrity of the mitochondrial genome: A plethora of nuclear genes in the budding yeast. Microbiol. Mol. Biol. Rev. 2000, 64, 281–315. [Google Scholar] [CrossRef] [Green Version]
- Rak, M.; Tetaud, E.; Godard, F.; Sagot, I.; Salin, B.; Duvezin-Caubet, S.; Slonimski, P.P.; Rytka, J.; di Rago, J.P. Yeast cells lacking the mitochondrial gene encoding the ATP synthase subunit 6 exhibit a selective loss of complex IV and unusual mitochondrial morphology. J. Biol. Chem. 2007, 282, 10853–10864. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Rak, M.; Tetaud, E.; Godard, F.; Sardin, E.; Bouhier, M.; Gombeau, K.; Caetano-Anollés, D.; Salin, B.; Chen, H.; et al. Deregulating mitochondrial metabolite and ion transport has beneficial effects in yeast and human cellular models for NARP syndrome. Human Molecular Genetics 2019, 28, 3792–3804. [Google Scholar] [CrossRef]
- Venard, R.; Brèthes, D.; Giraud, M.F.; Vaillier, J.; Velours, J.; Haraux, F. Marie-France Giraud, Jacques Vaillier, Jean Velours, and Francis Haraux, Investigation of the Role and Mechanism of IF1 and STF1 Proteins, Twin Inhibitory Peptides Which Interact with the Yeast Mitochondrial ATP Synthase. Biochemistry 2003, 42, 7626–7636. [Google Scholar] [CrossRef]
- Guo, H.; Rubinstein, J.L. Cryo-EM of ATP synthases. Curr. Opin. Struct. Biol. 2018, 52, 71–79. [Google Scholar] [CrossRef]
- Rak, M.; Gokova, S.; Tzagoloff, A. Modular assembly of yeast mitochondrial ATP synthase. EMBO J. 2011, 30, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzagoloff, A.; Barrientos, A.; Neupert, W.; Herrmann, J.M. Atp10p assists assembly of Atp6p into the F0 unit of the yeast mitochondrial ATPase. J. Biol. Chem. 2004, 279, 19775–19780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, M.F.; Barrientos, A.; Tzagoloff, A. A single amino acid change in subunit 6 of the yeast mitochondrial ATPase suppresses a null mutation in ATP10. J. Biol. Chem. 2000, 275, 29238–29243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre-Legendre, L.; Vaillier, J.; Benabdelhak, H.; Velours, J.; Slonimski, P.P.; di Rago, J.P. Identification of a nuclear gene (FMC1) required for the assembly/stability of yeast mitochondrial F(1)-ATPase in heat stress conditions. J. Biol. Chem. 2001, 276, 6789–6796. [Google Scholar] [CrossRef] [Green Version]
- Elbaum, M.B.; Zondlo, N.J. OGlcNAcylation and phosphorylation have similar structural effects in alpha-helices: Post-translational modifications as inducible start and stop signals in alpha-helices, with greater structural effects on threonine modification. Biochemistry 2014, 53, 2242–2260. [Google Scholar] [CrossRef]
- Foury, F.; Roganti, T.; Lecrenier, N.; Purnelle, B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 1998, 440, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Bonitz, S.G.; Berlani, R.; Coruzzi, G.; Li, M.; Macino, G.; Nobrega, F.G.; Nobrega, M.P.; Thalenfeld, B.E.; Tzagoloff, A. Codon recognition rules in yeast mitochondria. Proc. Natl. Acad Sci. USA 1980, 77, 3167–3170. [Google Scholar] [CrossRef] [Green Version]
- Kucharczyk, R.; Dautant, A.; Gombeau, K.; Godard, F.; Tribouillard-Tanvier, D.; di Rago, J.P. The pathogenic MT-ATP6 m.8851T>C mutation prevents proton movements within the n-side hydrophilic cleft of the membrane domain of ATP synthase. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 562–572. [Google Scholar] [CrossRef]
- Skoczen, N.; Dautant, A.; Binko, K.; Godard, F.; Bouhier, M.; Su, X.; Lasserre, J.P.; Giraud, M.F.; Tribouillard-Tanvier, D.; Chen, H.; et al. Molecular basis of diseases caused by the mtDNA mutation m.8969G>A in the subunit a of ATP synthase. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Guerin, B.; Labbe, P.; Somlo, M. Preparation of yeast mitochondria (Saccharomyces cerevisiae) with good P/O and respiratory control ratios. Methods Enzymol. 1979, 55, 149–159. [Google Scholar] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Rigoulet, M.; Guerin, B. Phosphate transport and ATP synthesis in yeast mitochondria: Effect of a new inhibitor: The tribenzylphosphate. FEBS Lett. 1979, 102, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Emaus, R.K.; Grunwald, R.; Lemasters, J.J. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: Spectral and metabolic properties. Biochim. Biophys. Acta 1986, 850, 436–448. [Google Scholar] [CrossRef]
- Somlo, M. Induction and repression of mitochondrial ATPase in yeast. Eur. J. Biochem. 1968, 5, 276–284. [Google Scholar] [CrossRef]
- Lanzetta, P.A.; Alvarez, L.J.; Reinach, P.S.; Candia, O.A. An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 1979, 100, 95–97. [Google Scholar] [CrossRef]
- Schagger, H.; von Jagow, G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991, 199, 223–231. [Google Scholar] [CrossRef]
- Grandier-Vazeille, X.; Guerin, M. Separation by blue native and colorless native polyacrylamide gel electrophoresis of the oxidative phosphorylation complexes of yeast mitochondria solubilized by different detergents: Specific staining of the different complexes. Anal. Biochem. 1996, 242, 248–254. [Google Scholar] [CrossRef]
- Arselin, G.; Vaillier, J.; Graves, P.V.; Velours, J. ATP synthase of yeast mitochondria. Isolation of the subunit h and disruption of the ATP14 gene. J. Biol. Chem. 1996, 271, 20284–20290. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Grenacher, B.; Rohde, B.; Vogel, J. Quantifying Western blots: Pitfalls of densitometry. Electrophoresis 2009, 30, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. PyMOL Molecular Graphics System. 2002. The PyMOL Molecular Graphics System, Version 0.99, Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 17 July 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Codon Change | Amino Acid Change | Number |
|---|---|---|
| Original mutant | ||
| TTA252CCA | aL242P | - |
| Intragenic suppressors | ||
| CCA252TCA | aL242S | 31 |
| CCA252ACA | aL242T | 11 |
| CCA252CTA | aL242T | 3 |
| Other possible single nucleotide substitutions not obtained | ||
| CCA252CAA | aL242Q | 0 |
| CCA252CGA | aL242R | 0 |
| CCA252GCA | aL242A | 0 |
| Strain | Respiration Rates nmol O/min/mg | ATP Synthesis Rate nmol Pi/min/mg | ATPase Activity µmol Pi/min/mg | P/O | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NADH | NADH + ADP | NADH + CCCP | Asc/TMPD + CCCP | -oligo | +oligo | -oligo | +oligo | Inhib % | ||
| WT | 329 ± 3 | 619 ± 99 | 1548 ± 113 | 2865 ± 325 | 1531 ± 168 | 151 ± 30 | 4.7 ± 0.2 | 0.5 ± 0.1 | 88 | 1.21 ± 0.18 |
| aL242P | 96 ± 8 | 79 ± 11 | 242 ± 48 | 440 ± 26 | 142 ± 43 | 96 ± 34 | 3.3 ± 0.4 | 3.2 ± 0.2 | 4 | 0.89 ± 0.20 |
| aL242S | 192 ± 21 | 353 ± 23 | 793 ± 60 | 1443 ± 264 | 866 ± 170 | 170 ± 31 | 4.2 ± 0.1 | 0.6 ± 0.2 | 85 | 1.24 ± 0.19 |
| aL242T(CTA) | 220 ± 13 | 410 ± 22 | 949 ± 47 | 1704 ± 75 | 1056 ± 90 | 164 ± 32 | 4.2 ± 0.5 | 0.5 ± 0.2 | 88 | 1.25 ± 0.17 |
| aL242T(ACA) | 224 ± 28 | 462 ± 84 | 1133 ± 127 | 2101 ± 316 | 1066 ± 89 | 160 ± 18 | 4.0 ± 0.2 | 0.5 ± 0.1 | 87 | 1.06 ± 0.17 |
| Strain | Nuclear Genotype | mtDNA | Source |
|---|---|---|---|
| MR6 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ + | [29] |
| RKY66 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+ ATP6-aL242P | [18] |
| RRKY66/1 | Revertant isolated from RKY66 | ρ+ ATP6-aL242S | This study |
| RRKY66/2 | Revertant isolated from RKY66 | ρ+ ATP6-aL242T (CTA) | This study |
| RRKY66/3 | Revertant isolated from RKY66 | ρ+ ATP6-aL242T (ACA) | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.; Dautant, A.; Godard, F.; Bouhier, M.; Zoladek, T.; Kucharczyk, R.; di Rago, J.-P.; Tribouillard-Tanvier, D. Molecular Basis of the Pathogenic Mechanism Induced by the m.9191T>C Mutation in Mitochondrial ATP6 Gene. Int. J. Mol. Sci. 2020, 21, 5083. https://doi.org/10.3390/ijms21145083
Su X, Dautant A, Godard F, Bouhier M, Zoladek T, Kucharczyk R, di Rago J-P, Tribouillard-Tanvier D. Molecular Basis of the Pathogenic Mechanism Induced by the m.9191T>C Mutation in Mitochondrial ATP6 Gene. International Journal of Molecular Sciences. 2020; 21(14):5083. https://doi.org/10.3390/ijms21145083
Chicago/Turabian StyleSu, Xin, Alain Dautant, François Godard, Marine Bouhier, Teresa Zoladek, Roza Kucharczyk, Jean-Paul di Rago, and Déborah Tribouillard-Tanvier. 2020. "Molecular Basis of the Pathogenic Mechanism Induced by the m.9191T>C Mutation in Mitochondrial ATP6 Gene" International Journal of Molecular Sciences 21, no. 14: 5083. https://doi.org/10.3390/ijms21145083