Targeted Disruption of Mouse Dip2B Leads to Abnormal Lung Development and Prenatal Lethality

 , ,
, ,
Abstract
:1. Introduction
2. Results
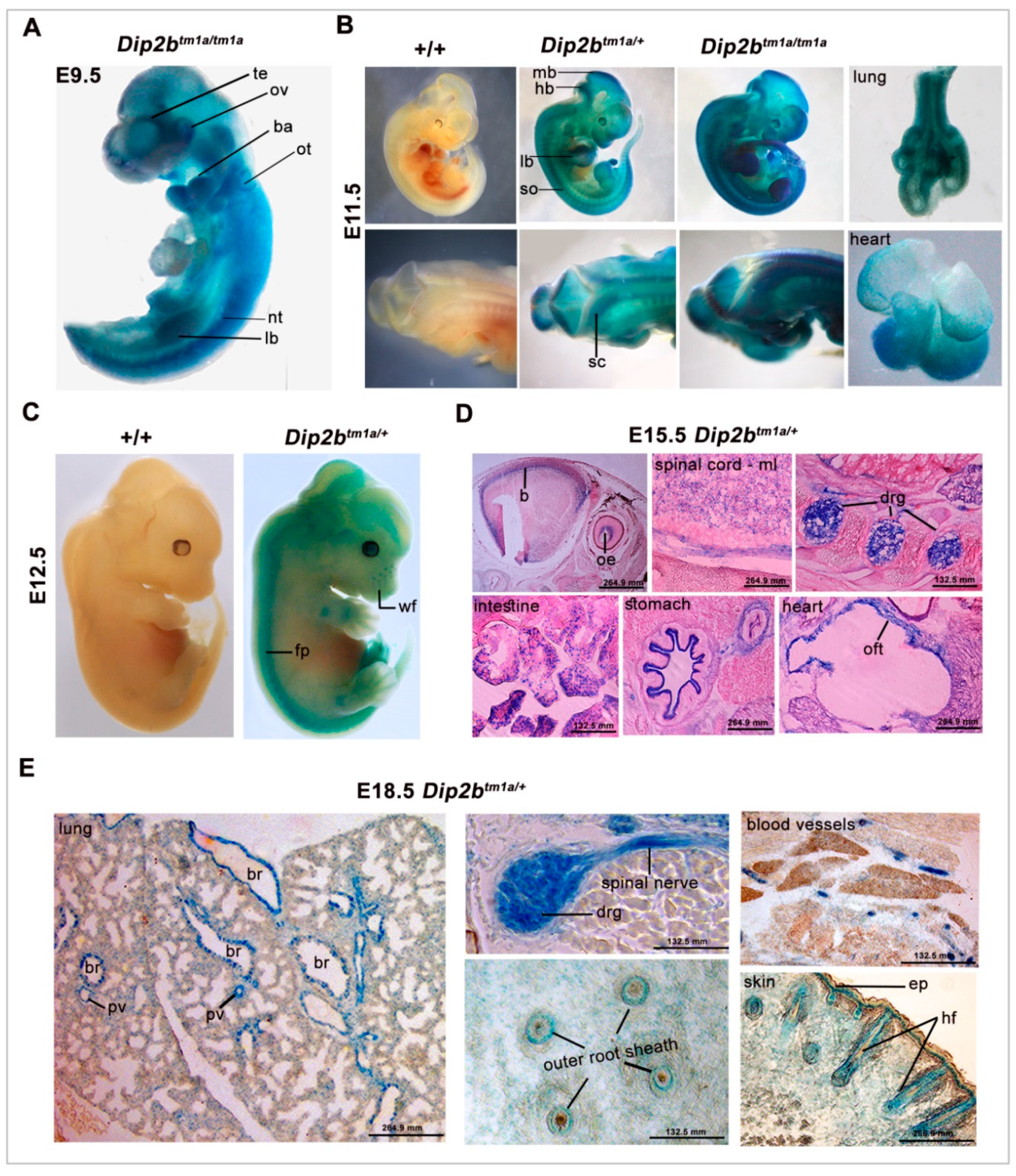
2.1. Dip2b Is Expressed in Multiple Organs during Development
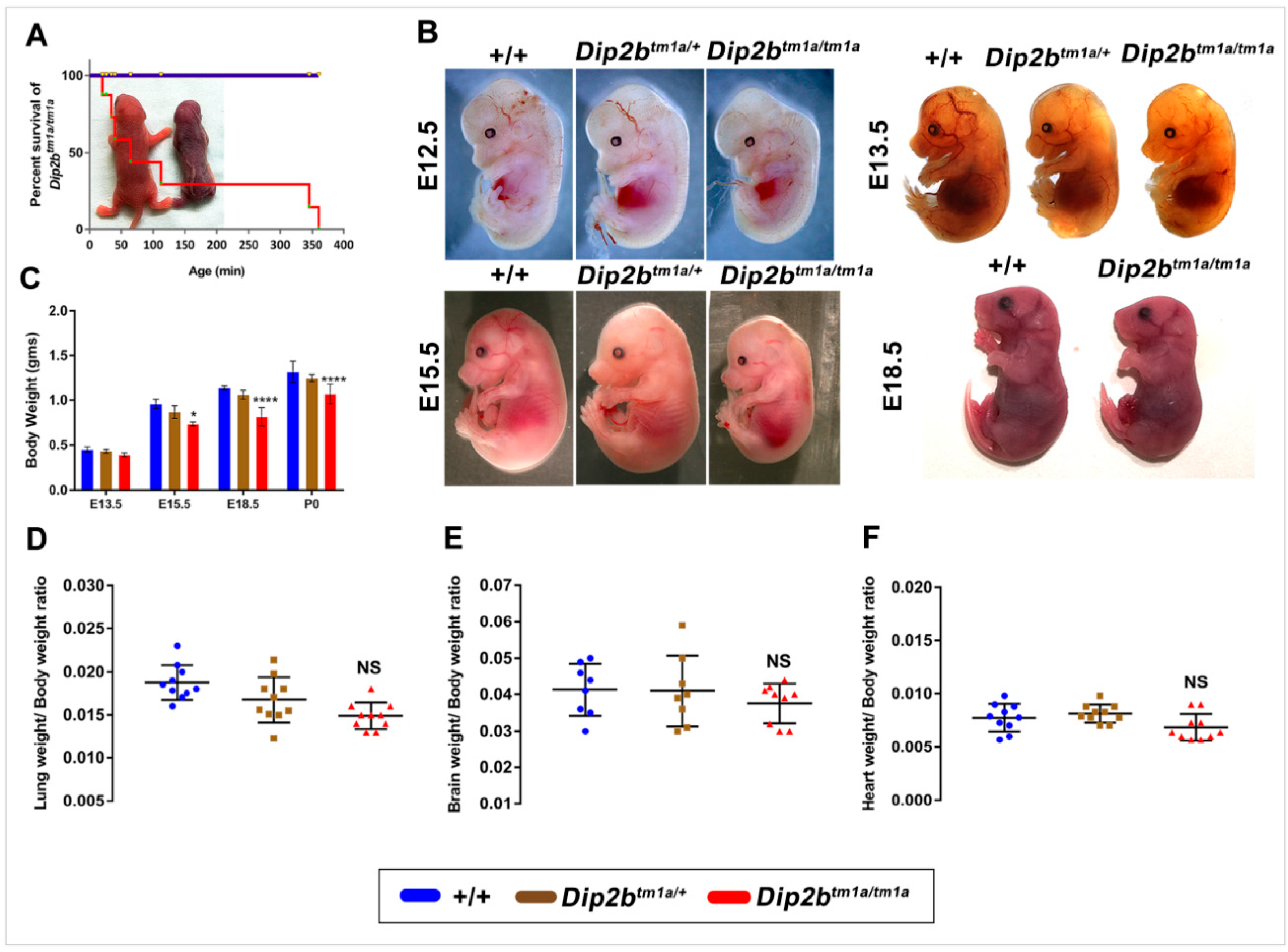
2.2. Dip2b Knockout Leads to Fetal Growth Restriction, Birth Weight Reduction and Perinatal Lethality
2.3. Dip2b Inactivation Causes Respiratory Distress and Pathologic Lung Development
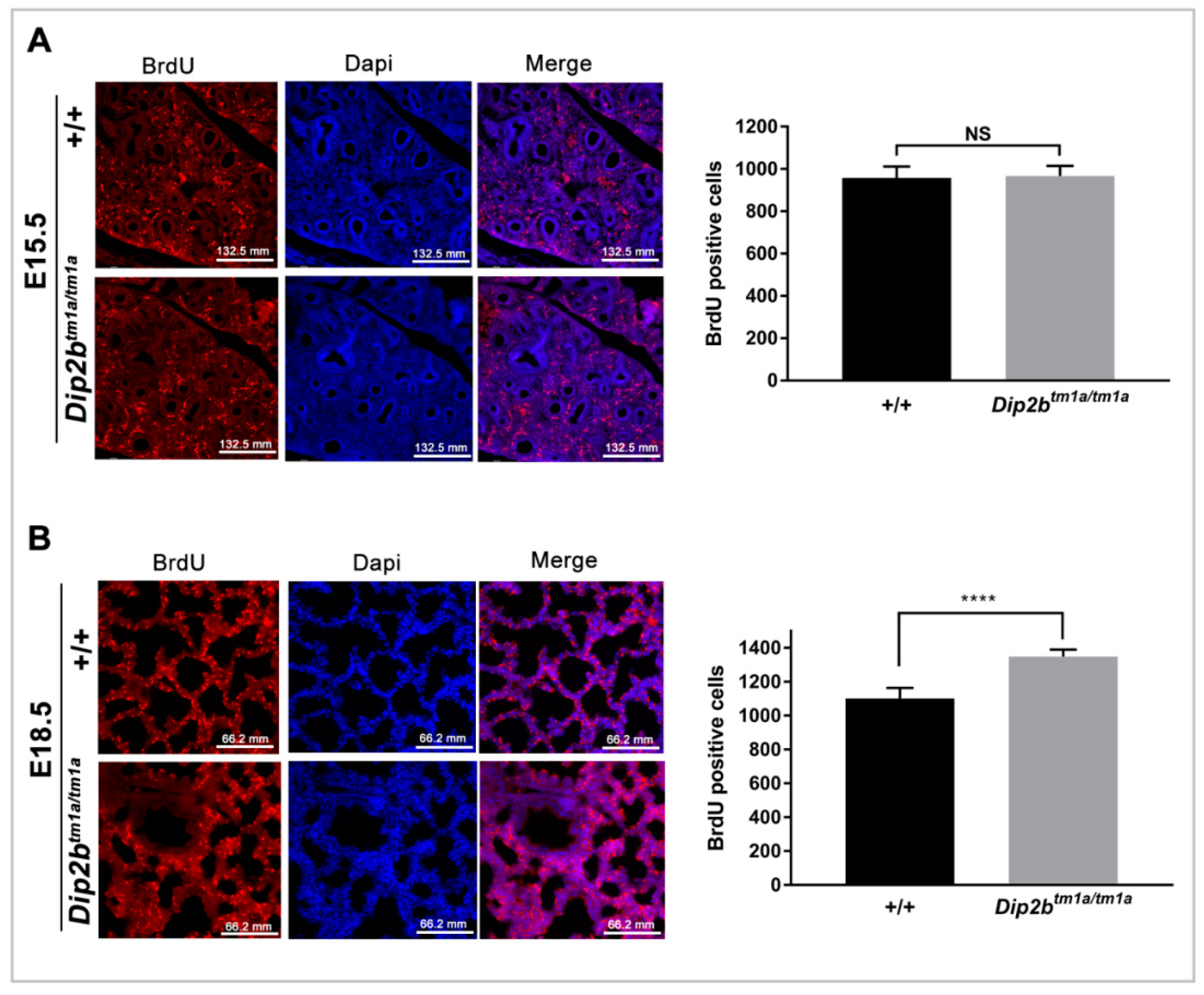
2.4. Dip2b Regulates Cell Proliferation but Not Apoptosis in Lungs
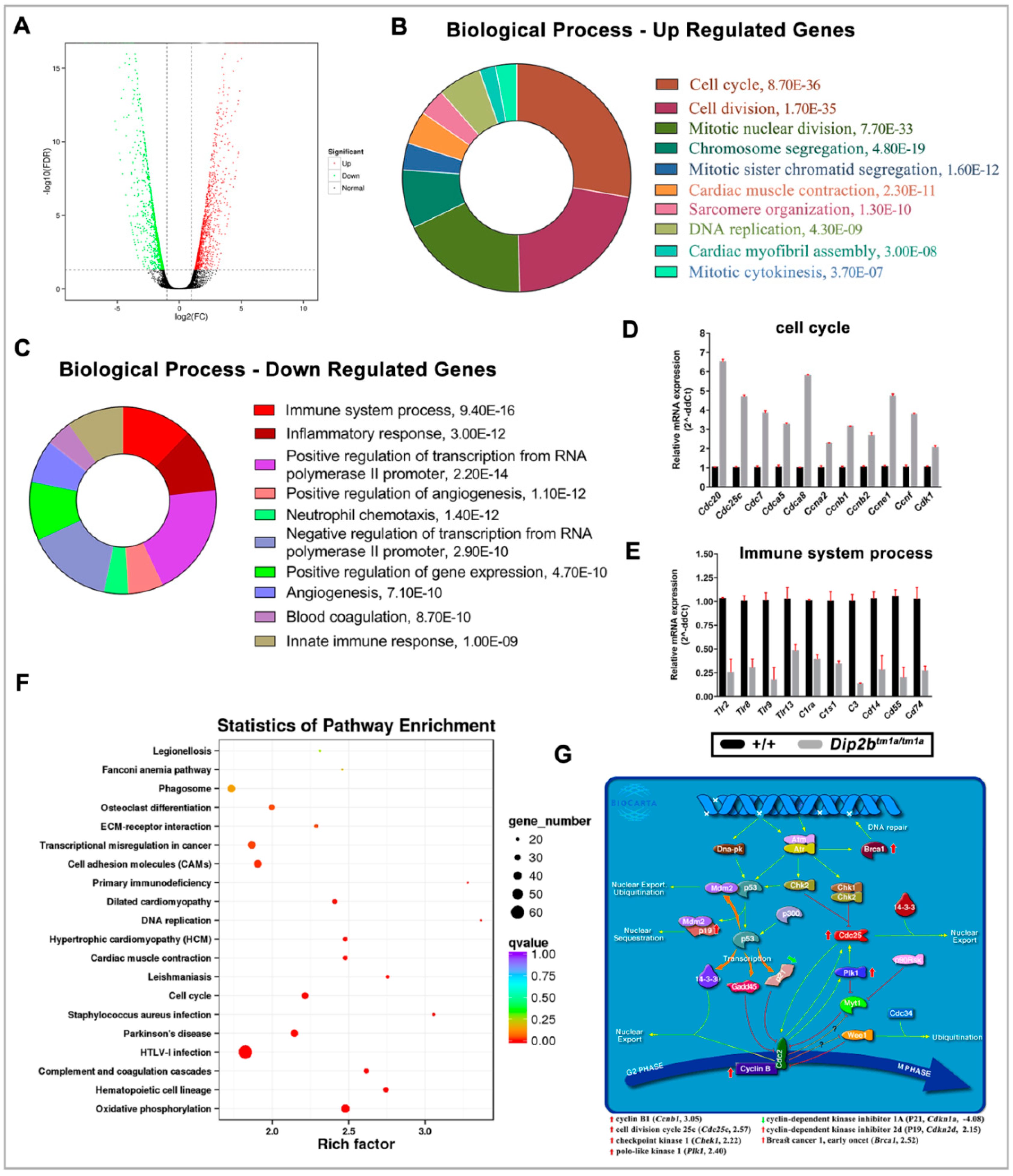
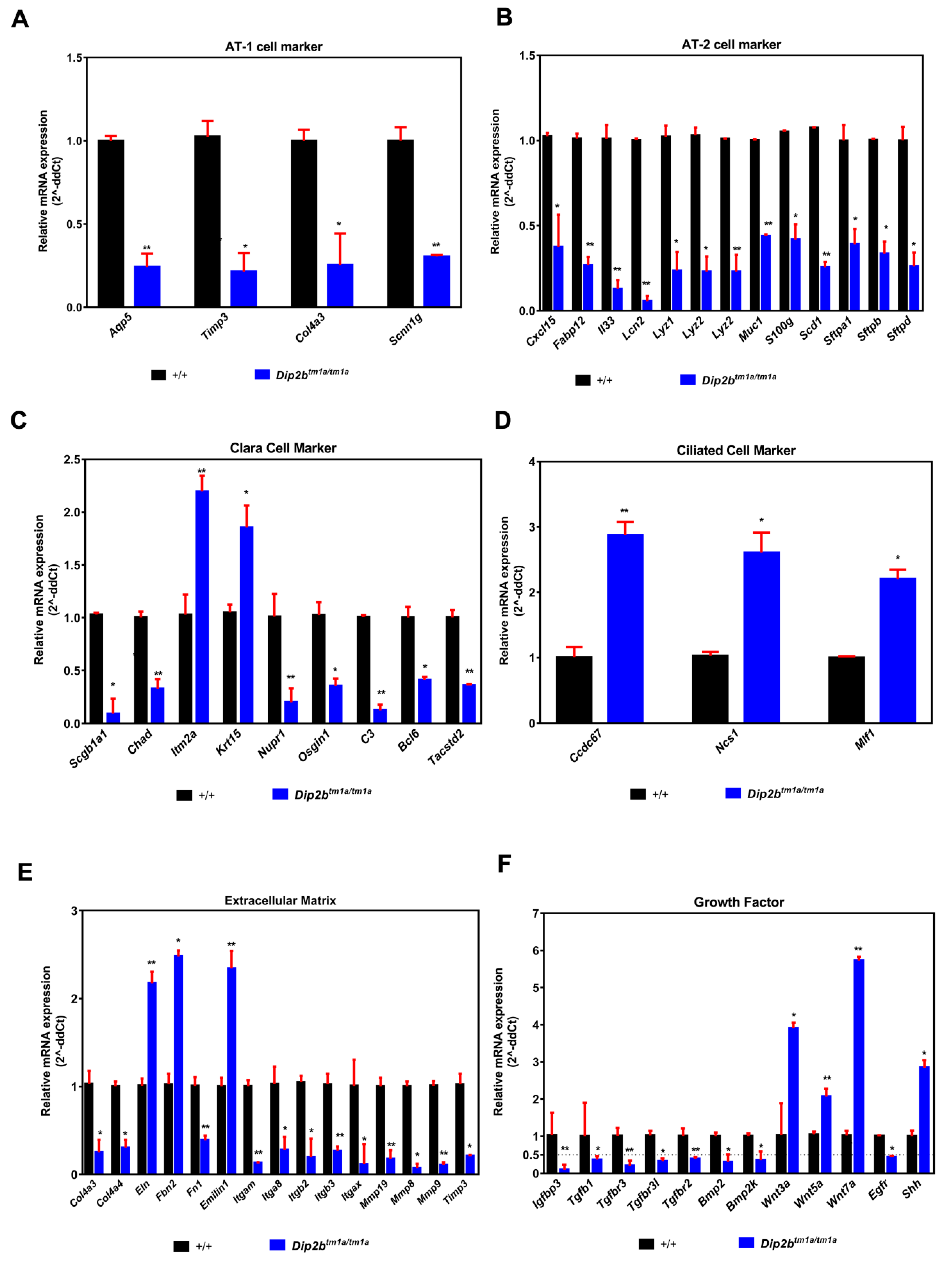
2.5. Dip2b Regulates Expression of Cell Cycle Genes, Epithelial Cell Differentiation Markers and Lung Mediators
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. LacZ Staining
4.3. Cesarean Delivery and Weight Measurements
4.4. Hydrostatic Lung Test
4.5. Weight Measurement of Lungs
4.6. Lung Histology/Staining
4.7. Analysis of Lung Branching Morphogenesis
4.8. Quantification of Lung Alveolar Sac Formation
4.9. 5-Bromo-2-Deoxy-Uridine (BrdU) Labeling
4.10. TUNEL Assay
4.11. RNA-Sequencing (RNA-Seq)
4.12. Quantitative Real Time PCR (qPCR) Validation of RNA-Seq Results
4.13. Statistical Analysis
4.14. Database
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Dip2a | Disco Interacting protein 2 Homolog a |
| Dip2b | Disco Interacting protein 2 Homolog b |
| Dip2c | Disco Interacting protein 2 Homolog c |
| LacZ | β-Galactosidase |
| BrdU | 5-Bromo-2-Deoxy-Uridine |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Tanaka, M.; Murakami, K.; Ozaki, S.; Imura, Y.; Tong, X.-P.; Watanabe, T.; Sawaki, T.; Kawanami, T.; Kawabata, D.; Fujii, T.; et al. DIP2 disco-interacting protein 2 homolog A (Drosophila) is a candidate receptor for follistatin-related protein/follistatin-like 1—Analysis of their binding with TGF-β superfamily proteins. FEBS J. 2010, 277, 4278–4289. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, M.; Pelka, P.; DeSousa, D.; Kablar, B.; Schindler, A.; Rudnicki, M.A.; Campos, A.R. Cloning, genomic organization and expression pattern of a novel Drosophila gene, the disco-interacting protein 2 (dip2), and its murine homolog. Gene 2002, 293, 59–65. [Google Scholar] [CrossRef]
- Nitta, Y.; Yamazaki, D.; Sugie, A.; Hiroi, M.; Tabata, T. DISCO Interacting Protein 2 regulates axonal bifurcation and guidance of Drosophila mushroom body neurons. Dev. Biol. 2017, 421, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Nitta, Y.; Sugie, A. DISCO interacting protein 2 determines direction of axon projection under the regulation of c-Jun N-terminal kinase in the Drosophila mushroom body. Biochem. Biophys. Res. Commun. 2017, 487, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Noblett, N.; Wu, Z.; Ding, Z.H.; Park, S.; Roenspies, T.; Flibotte, S.; Chisholm, A.D.; Jin, Y.; Colavita, A. DIP-2 suppresses ectopic neurite sprouting and axonal regeneration in mature neurons. J. Cell Biol. 2019, 218, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Asaumi, Y.; Ohashi, K.; Higuchi, A.; Sono-Romanelli, S.; Oshima, Y.; Walsh, K. DIP2A functions as a FSTL1 receptor. J. Biol. Chem. 2010, 285, 7127–7134. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Hu, Q.; Li, B.; McBride, D.; Bian, H.; Spagnoli, P.; Chen, D.; Tang, J.; Zhang, J.H. Follistatin-like 1 attenuates apoptosis via disco-interacting protein 2 homolog A/Akt pathway after middle cerebral artery occlusion in rats. Stroke 2014, 45, 3048–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, E.; Miao, F.; Jin, X.; Wu, W.; Zhou, X.; Zeng, A.; Yu, T.; Zhi, T.; Shi, Z.; Wang, Y.; et al. Fstl1/DIP2A/MGMT signaling pathway plays important roles in temozolomide resistance in glioblastoma. Oncogene 2019, 38, 2706–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo-Saito, C.; Ishida, A.; Shouya, Y.; Teramoto, K.; Igarashi, T.; Kon, R.; Saito, K.; Awada, C.; Ogiwara, Y.; Toyoura, M. Blocking the FSTL1-DIP2A Axis Improves Anti-tumor Immunity. Cell Rep. 2018, 24, 1790–1801. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhang, L.-Q.; He, Z.-X.; He, X.-X.; Wang, Y.-J.; Jian, Y.-L.; Wang, X.; Zhang, B.-B.; Su, C.; Lu, J.; et al. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 2019, 17, e3000461. [Google Scholar] [CrossRef]
- Egger, G.; Roetzer, K.M.; Noor, A.; Lionel, A.C.; Mahmood, H.; Schwarzbraun, T.; Boright, O.; Mikhailov, A.; Marshall, C.R.; Windpassinger, C.; et al. Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 2014, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Poelmans, G.; Engelen, J.J.M.; Van Lent-Albrechts, J.; Smeets, H.J.; Schoenmakers, E.; Franke, B.; Buitelaar, J.K.; Wuisman-Frerker, M.; Erens, W.; Steyaert, J.; et al. Identification of novel dyslexia candidate genes through the analysis of a chromosomal deletion. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2009, 150B, 140–147. [Google Scholar] [CrossRef] [PubMed]
- DeScipio, C.; Conlin, L.; Rosenfeld, J.; Tepperberg, J.; Pasion, R.; Patel, A.; McDonald, M.T.; Aradhya, S.; Ho, D.; Goldstein, J.; et al. Subtelomeric deletion of chromosome 10p15.3: Clinical findings and molecular cytogenetic characterization. Am. J. Med. Genet. A 2012, 158A, 2152–2161. [Google Scholar] [CrossRef] [Green Version]
- Larsson, C.; Ali, M.A.; Pandzic, T.; Lindroth, A.M.; He, L.; Sjöblom, T. Loss of DIP2C in RKO cells stimulates changes in DNA methylation and epithelial-mesenchymal transition. BMC Cancer 2017, 17, 487. [Google Scholar] [CrossRef]
- Winnepenninckx, B.; Debacker, K.; Ramsay, J.; Smeets, D.; Smits, A.; FitzPatrick, D.R.; Kooy, R.F. CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet. 2007, 80, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Lombaert, I.M.A.; Hauser, B.R.; Patel, V.N.; Hoffman, M.P. Exosomal MicroRNA Transport from Salivary Mesenchyme Regulates Epithelial Progenitor Expansion during Organogenesis. Dev. Cell 2017, 40, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; He, A.; Zhu, F.; Ding, M.; Hao, J.; Fan, Q.; Li, P.; Liu, L.; Du, Y.; Liang, X.; et al. Integrating genome-wide association study and expression quantitative trait locus study identifies multiple genes and gene sets associated with schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 81, 50–54. [Google Scholar] [CrossRef]
- Gong, J.; Qiu, C.; Huang, D.; Zhang, Y.; Yu, S.; Zeng, C. Integrative functional analysis of super enhancer SNPs for coronary artery disease. J. Hum. Genet. 2018, 63, 627–638. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, X.; Wang, C.; Li, C. The correlation analysis of miRNAs and target genes in metastasis of cervical squamous cell carcinoma. Epigenomics 2018, 10, 259–275. [Google Scholar] [CrossRef]
- Closa, A.; Cordero, D.; Sanz-Pamplona, R.; Solé, X.; Crous-Bou, M.; Paré-Brunet, L.; Berenguer, A.; Guino, E.; Lopez-Doriga, A.; Guardiola, J.; et al. Identification of candidate susceptibility genes for colorectal cancer through eQTL analysis. Carcinogenesis 2014, 35, 2039–2046. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jia, R.; Palange, N.J.; Satheka, A.C.; Togo, J.; An, Y.; Humphrey, M.; Ban, L.; Ji, Y.; Jin, H.; et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS ONE 2015, 10, e0120396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Mabwi, H.A.; Palange, N.J.; Jia, R.; Ma, J.; Bah, F.B.; Sah, R.K.; Li, D.; Wang, D.; Bah, F.B.M.; et al. Expression Patterns and Potential Biological Roles of Dip2a. PLoS ONE 2015, 10, e0143284. [Google Scholar] [CrossRef]
- Ma, J.; Chen, L.; He, X.X.; Wang, Y.J.; Yu, H.L.; He, Z.X.; Zhang, L.Q.; Zheng, Y.W.; Zhu, X.J. Functional prediction and characterization of Dip2 gene in mice. Cell Biol. Int. 2019, 43, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.K.; Yang, A.; Bah, F.B.; Adlat, S.; Bohio, A.A.; Oo, Z.M.; Wang, C.; Myint, M.Z.Z.; Bahadar, N.; Zhang, L.; et al. Correction: Transcriptome profiling of mouse brain and lung under Dip2a regulation using RNA-sequencing. PLoS ONE 2019, 14, e0225570. [Google Scholar] [CrossRef]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Stiles, A.D.; Chrysis, D.; Jarvis, H.W.; Brighton, B.; Moats-Staats, B.M. Programmed cell death in normal fetal rat lung development. Exp. Lung Res. 2001, 27, 569–587. [Google Scholar] [CrossRef]
- Warburton, D.; El-Hashash, A.; Carraro, G.; Tiozzo, C.; Sala, F.; Rogers, O.; De Langhe, S.; Kemp, P.J.; Riccardi, D.; Torday, J.; et al. Lung organogenesis. Curr. Top. Dev. Biol. 2010, 90, 73–158. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Horowitz, J.C.; Naba, A.; Ambalavanan, N.; Atabai, K.; Balestrini, J.; Bitterman, P.B.; Corley, R.A.; Ding, B.-S.; Engler, A.J.; et al. Extracellular matrix in lung development, homeostasis and disease. Matrix Biol. 2018, 73, 77–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, T.J.; Cardoso, W.V. Growth factors in lung development and disease: Friends or foe? Respir. Res. 2001, 3, 2. [Google Scholar] [CrossRef]
- Speer, C.P. Neonatal respiratory distress syndrome: An inflammatory disease? Neonatology 2011, 99, 316–319. [Google Scholar] [CrossRef]
- Morrisey, E.E.; Hogan, B.L.M. Preparing for the first breath: Genetic and cellular mechanisms in lung development. Dev. Cell 2010, 18, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Whitsett, J.A.; Weaver, T.E. Alveolar development and disease. Am. J. Respir. Cell Mol. Biol. 2015, 53, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herriges, M.; Morrisey, E.E. Lung development: Orchestrating the generation and regeneration of a complex organ. Development 2014, 141, 502–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, R.H.; Kalinichenko, V.V.; Lim, L. Transcription factors in mouse lung development and function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L823–L838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodhan, P.; Kinane, T.B. Developmental paradigms in terminal lung development. Bioessays 2002, 24, 1052–1059. [Google Scholar] [CrossRef]
- Warburton, D.; Schwarz, M.; Tefft, D.; Flores-Delgado, G.; Anderson, K.D.; Cardoso, W.V. The molecular basis of lung morphogenesis. Mech. Dev. 2000, 92, 55–81. [Google Scholar] [CrossRef]
- Carraro, G.; del Moral, P.-M.; Warburton, D. Mouse embryonic lung culture, a system to evaluate the molecular mechanisms of branching. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [Green Version]
- Effect of corticosteroids for fetal maturation on perinatal outcomes. NIH Consens. Statement 1994, 12, 1–24.
- Muglia, L.J.; Bae, D.S.; Brown, T.T.; Vogt, S.K.; Alvarez, J.G.; Sunday, M.E.; Majzoub, J.A. Proliferation and differentiation defects during lung development in corticotropin-releasing hormone-deficient mice. Am. J. Respir. Cell Mol. Biol. 1999, 20, 181–188. [Google Scholar] [CrossRef]
- Bird, A.D.; Tan, K.H.; Olsson, P.F.; Zieba, M.; Flecknoe, S.J.; Liddicoat, D.R.; Mollard, R.; Hooper, S.B.; Cole, T.J. Identification of glucocorticoid-regulated genes that control cell proliferation during murine respiratory development. J. Physiol. 2007, 585, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Heisterkamp, N.; Groffen, J.; Zhao, J.; Warburton, D.; Kaartinen, V. TGF-beta3-null mutation does not abrogate fetal lung maturation in vivo by glucocorticoids. Am. J. Physiol. 1999, 277, L1205–L1213. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, J.L.; Watkins-Chow, D.E.; Baxter, L.L.; Yokoyama, T.; Zerfas, P.M.; Starost, M.F.; Gahl, W.A.; Malicdan, M.C.V.; Porter, F.D.; Platt, F.M.; et al. NPC1 Deficiency in Mice is Associated with Fetal Growth Restriction, Neonatal Lethality and Abnormal Lung Pathology. J. Clin. Med. 2019, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Colarossi, C.; Chen, Y.; Obata, H.; Jurukovski, V.; Fontana, L.; Dabovic, B.; Rifkin, D.B. Lung alveolar septation defects in Ltbp-3-null mice. Am. J. Pathol. 2005, 167, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Steele-Perkins, G.; Plachez, C.; Butz, K.G.; Yang, G.; Bachurski, C.J.; Kinsman, S.L.; Litwack, E.D.; Richards, L.J.; Gronostajski, R.M. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol. Cell. Biol. 2005, 25, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Manwani, N.; Gagnon, S.; Post, M.; Joza, S.; Muglia, L.; Cornejo, S.; Kaplan, F.; Sweezey, N.B. Reduced viability of mice with lung epithelial-specific knockout of glucocorticoid receptor. Am. J. Respir. Cell Mol. Biol. 2010, 43, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, H.; Peschon, J.J.; Shi, W.; Zhang, Y.; Frank, S.J.; Warburton, D. Pulmonary hypoplasia in mice lacking tumor necrosis factor-alpha converting enzyme indicates an indispensable role for cell surface protein shedding during embryonic lung branching morphogenesis. Dev. Biol. 2001, 232, 204–218. [Google Scholar] [CrossRef] [Green Version]
- Boase, N.A.; Rychkov, G.Y.; Townley, S.L.; Dinudom, A.; Candi, E.; Voss, A.K.; Tsoutsman, T.; Semsarian, C.; Melino, G.; Koentgen, F.; et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat. Commun. 2011, 2, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Li, Y.; Ren, J.; Man Lam, S.; Zhang, Y.; Hou, Y.; Zhang, X.; Xu, R.; Shui, G.; Ma, R.Z. Neonatal Respiratory Failure with Retarded Perinatal Lung Maturation in Mice Caused by Reticulocalbin 3 Disruption. Am. J. Respir. Cell Mol. Biol. 2016, 54, 410–423. [Google Scholar] [CrossRef]
- Geng, Y.; Dong, Y.; Yu, M.; Zhang, L.; Yan, X.; Sun, J.; Qiao, L.; Geng, H.; Nakajima, M.; Furuichi, T.; et al. Follistatin-like 1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling antagonist in controlling mouse lung development. Proc. Natl. Acad. Sci. USA 2011, 108, 7058–7063. [Google Scholar] [CrossRef] [Green Version]
- Gould, K.L. Cyclin-dependent protein kinases. In Protein Kinases; Woodgett, J.R., Ed.; IRL Press: Oxford, UK, 1994; pp. 149–176. [Google Scholar]
- Lew, D.J.; Kornbluth, S. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell Biol. 1996, 8, 795–804. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Ohi, R.; Gould, K.L. Regulating the onset of mitosis. Curr. Opin. Cell Biol. 1999, 11, 267–273. [Google Scholar] [CrossRef]
- Nigg, E.A. Polo-like kinases: Positive regulators of cell division from start to finish. Curr. Opin. Cell Biol. 1998, 10, 776–783. [Google Scholar] [CrossRef]
- Qian, Y.W.; Erikson, E.; Li, C.; Maller, J.L. Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis. Mol. Cell. Biol. 1998, 18, 4262–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herriges, M.J.; Swarr, D.T.; Morley, M.P.; Rathi, K.S.; Peng, T.; Stewart, K.M.; Morrisey, E.E. Long noncoding RNAs are spatially correlated with transcription factors and regulate lung development. Genes Dev. 2014, 28, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | +/+ | Dip2btm1a/+ | Dip2btm1a/tm1a | χ2 10df | p Value |
|---|---|---|---|---|---|
| E9.5 | 7 (23.3%) | 15 (50.0%) | 8 (26.6%) | 0.81 | 0.26 |
| E11.5 | 12 (23.5) | 25 (49.0%) | 14 (27.5%) | 0.71 | 0.99 |
| E12.5 | 22 (25.5%) | 40 (46.5%) | 24 (27.9%) | 0.56 | 0.14 |
| E15.5 | 25(24.5%) | 48 (47.0%) | 29 (28.5) | 0.37 | 0.26 |
| E18.5 | 46 (26.2%) | 90 (51.4%) | 39 (22.2%) | 0.35 | 0.71 |
| P1 | 102 (32.2) | 214 (67.8%) | 0 (0%) | 0.61 | 0 |
| Expected | 25% | 50% | 25% |
| Genotype | No. of Pups | Average Body wt (g) | Left Lung DNA/Total Body wt (μg/g) |
|---|---|---|---|
| +/+ | 7 | 1.14 ± 0.027 | 43.6 ± 3.93 |
| Dip2btm1a/+ | 13 | 1.06 ± 0.050 | 52.9 ± 5.08 |
| Dip2btm1a/tm1a | 6 | 0.82 ± 0.108 | 63.1 ± 6.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sah, R.K.; Ma, J.; Bah, F.B.; Xing, Z.; Adlat, S.; Oo, Z.M.; Wang, Y.; Bahadar, N.; Bohio, A.A.; Nagi, F.H.; et al. Targeted Disruption of Mouse Dip2B Leads to Abnormal Lung Development and Prenatal Lethality. Int. J. Mol. Sci. 2020, 21, 8223. https://doi.org/10.3390/ijms21218223
Sah RK, Ma J, Bah FB, Xing Z, Adlat S, Oo ZM, Wang Y, Bahadar N, Bohio AA, Nagi FH, et al. Targeted Disruption of Mouse Dip2B Leads to Abnormal Lung Development and Prenatal Lethality. International Journal of Molecular Sciences. 2020; 21(21):8223. https://doi.org/10.3390/ijms21218223
Chicago/Turabian StyleSah, Rajiv Kumar, Jun Ma, Fatoumata Binta Bah, Zhenkai Xing, Salah Adlat, Zin Ma Oo, Yajun Wang, Noor Bahadar, Ameer Ali Bohio, Farooq Hayel Nagi, and et al. 2020. "Targeted Disruption of Mouse Dip2B Leads to Abnormal Lung Development and Prenatal Lethality" International Journal of Molecular Sciences 21, no. 21: 8223. https://doi.org/10.3390/ijms21218223