Foam Cell Induction Activates AMPK But Uncouples Its Regulation of Autophagy and Lysosomal Homeostasis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
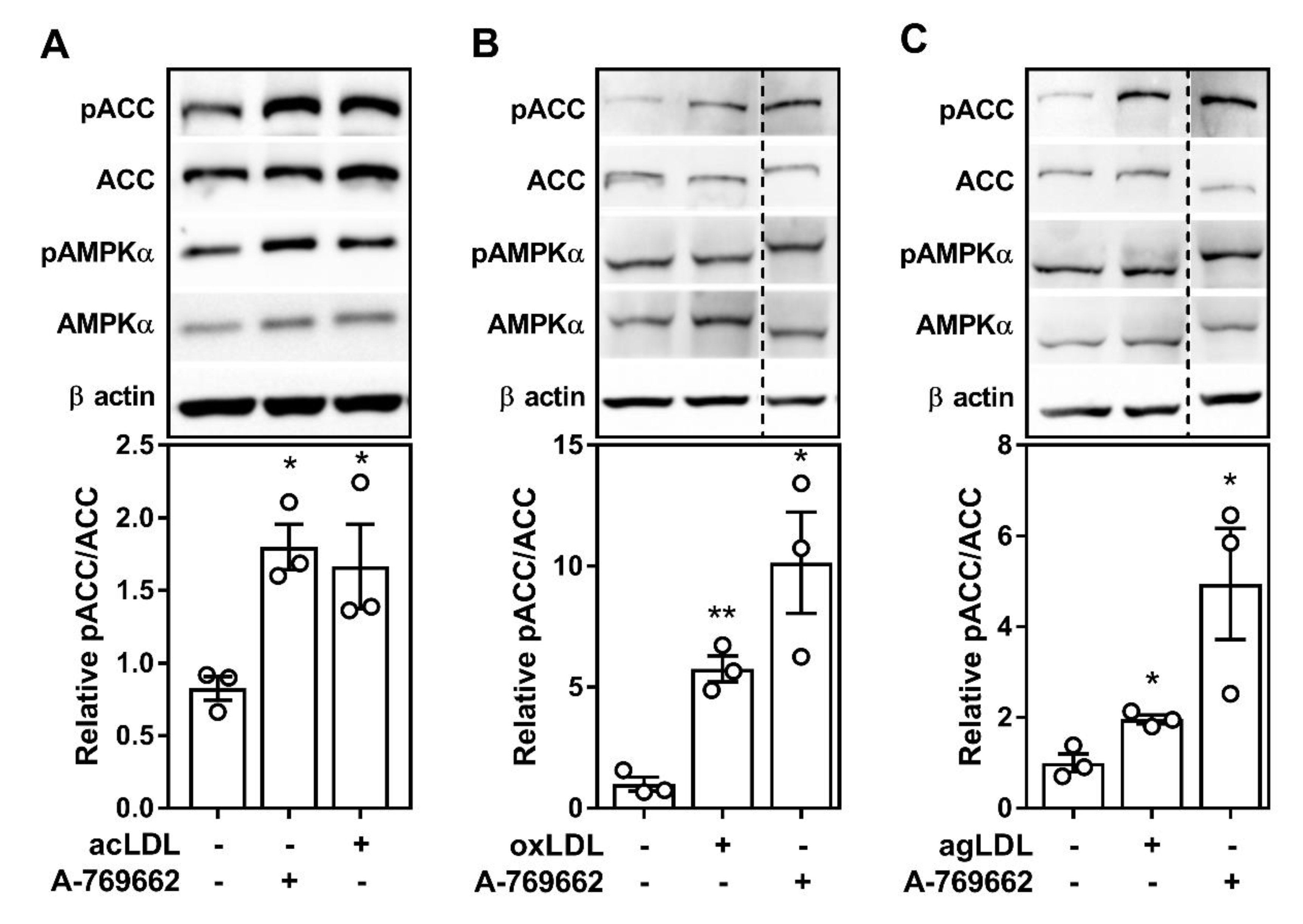
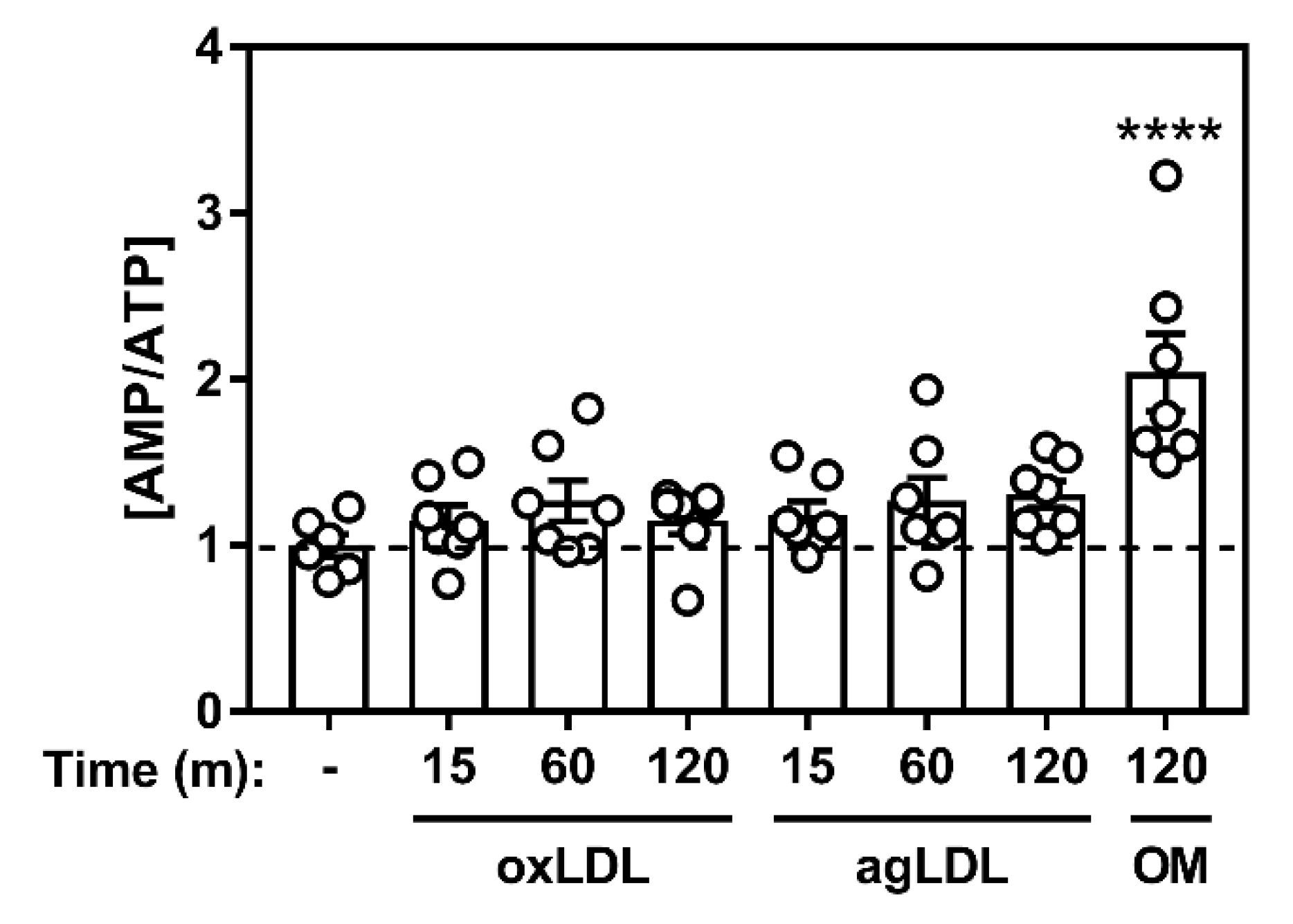
2.1. Atherogenic Lipids Activate AMPK in Bone Marrow-Derived Macrophages
2.2. Atherogenic Lipoproteins Activate Macrophage AMPK Partially via CaMKK2
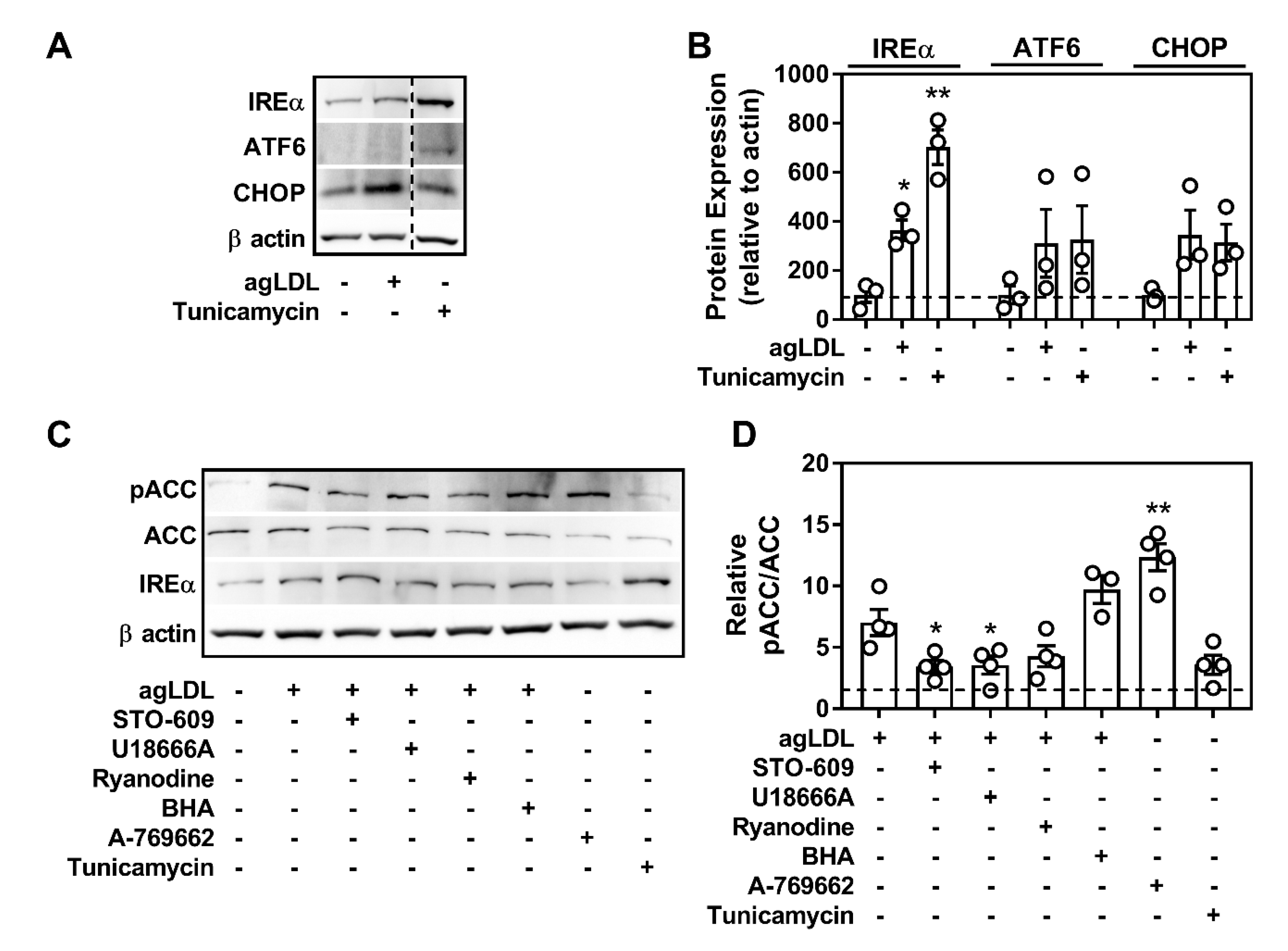
2.3. CD36 Links Atherogenic Lipids, AMPK Signaling, and Autophagy
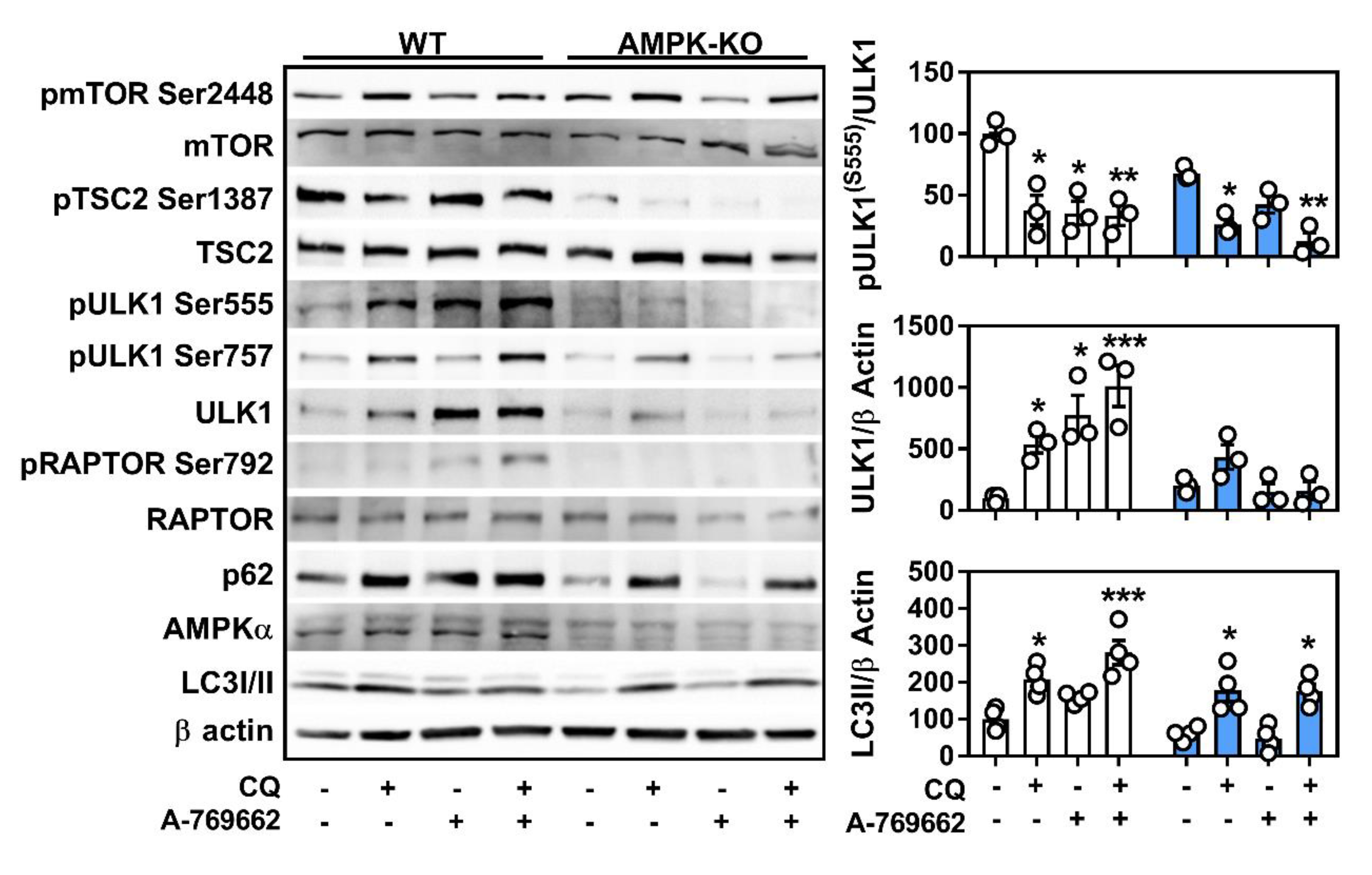
2.4. Macrophage AMPK Regulates Autophagy Signaling
2.5. Transcriptional Control of Lysosomal Programs by Macrophage AMPK Is TFEB-Dependent
2.6. Lysosomal Programs Are Partly Regulated by Macrophage AMPK
2.7. Atherogenic Lipids Overwhelm AMPK Regulation of Lysosomal Programs
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| acLDL | acetylated-LDL |
| ACC | acetyl-CoA carboxylase |
| agLDL | aggregated-LDL |
| AMPK | AMP-activated protein kinase |
| BMDM | bone marrow-derived macrophages |
| CaMKK2 | calmodulin-dependent protein kinase kinase-2 |
| LAL | lysosomal acid lipase |
| LDL | low-density lipoprotein |
| LKB1 | liver kinase B1 |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| oxLDL | oxidized-LDL |
| TFEB | transcription factor EB |
References
- Van de Vijver, L.P.; Kardinaal, A.F.; van Duyvenvoorde, W.; Kruijssen, D.A.; Grobbee, D.E.; van Poppel, G.; Princen, H.M. LDL Oxidation and Extent of Coronary Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinecke, J.W. Lipoprotein oxidation in cardiovascular disease: Chief culprit or innocent bystander? J. Exp. Med. 2006, 203, 813–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanson, M.; Augé, N.; Vindis, C.; Muller, C.; Bando, Y.; Thiers, J.-C.; Marachet, M.-A.; Zarkovic, K.; Sawa, Y.; Salvayre, R.; et al. Oxidized Low-Density Lipoproteins Trigger Endoplasmic Reticulum Stress in Vascular Cells. Circ. Res. 2009, 104, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Gursky, O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol. Concepts 2013, 4, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [Green Version]
- Moore Kathryn, J.; Freeman Mason, W. Scavenger Receptors in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP Activates the AMP-activated Protein Kinase Cascade, and Ca2+/Calmodulin Activates the Calmodulin-dependent Protein Kinase I Cascade, via Three Independent Mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/Calmodulin-dependent Protein Kinase Kinases Are AMP-activated Protein Kinase Kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [Green Version]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. AMP-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. Baltim. Md 1950 2008, 181, 8633–8641. [Google Scholar]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B. Macrophage α1 AMP-activated Protein Kinase (α1AMPK) Antagonizes Fatty Acid-induced Inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.-P.; van Denderen, B.J.; et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin–sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, D.; Wang, Y.; Ling, W.; Feng, X.; Xia, M. Adenosine Monophosphate-activated Protein Kinase Induces Cholesterol Efflux from Macrophage-derived Foam Cells and Alleviates Atherosclerosis in Apolipoprotein E-deficient Mice. J. Biol. Chem. 2010, 285, 33499–33509. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, M.D.; Ford, R.J.; McGregor, C.P.; LeBlond, N.D.; Snider, S.A.; Stypa, S.A.; Day, E.A.; Lhoták, Š.; Schertzer, J.D.; Austin, R.C.; et al. Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. J. Lipid Res. 2015, 56, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhu, H.; Ding, Y.; Liu, Z.; Cai, Z.; Zou, M.-H. AMP-activated Protein Kinase α1 Promotes Atherogenesis by Increasing Monocyte-to-macrophage Differentiation. J. Biol. Chem. 2017, 292, 7888–7903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm. 2019, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Guan, X. Autophagy: A new target for the treatment of atherosclerosis. Front. Lab. Med. 2018, 2, 68–71. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Young, N.P.; Kamireddy, A.; Van Nostrand, J.L.; Eichner, L.J.; Shokhirev, M.N.; Dayn, Y.; Shaw, R.J. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 2016, 30, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Yu, L.; Xue, H.; Yang, Z.; Yin, Y.; Zhang, B.; Chen, M.; Ma, H. Nuclear AMPK regulated CARM1 stabilization impacts autophagy in aged heart. Biochem. Biophys. Res. Commun. 2017, 486, 398–405. [Google Scholar] [CrossRef]
- Shin, H.-J.R.; Kim, H.; Oh, S.; Lee, J.-G.; Kee, M.; Ko, H.-J.; Kweon, M.-N.; Won, K.-J.; Baek, S.H. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.-H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collodet, C.; Foretz, M.; Deak, M.; Bultot, L.; Metairon, S.; Viollet, B.; Lefebvre, G.; Raymond, F.; Parisi, A.; Civiletto, G.; et al. AMPK promotes induction of the tumor suppressor FLCN through activation of TFEB independently of mTOR. FASEB J. 2019, 33, 12374–12391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.N.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of Lysosomal Biogenesis in Atherosclerotic Macrophages Can Rescue Lipid-Induced Lysosomal Dysfunction and Downstream Sequelae. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic Reticulum Stress Promotes Macrophage-derived Foam Cell Formation by Up-regulating Cluster of Differentiation 36 (CD36) Expression. J. Biol. Chem. 2014, 289, 4032–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Samovski, D.; Sun, J.; Pietka, T.; Gross, R.W.; Eckel, R.H.; Su, X.; Stahl, P.D.; Abumrad, N.A. Regulation of AMPK Activation by CD36 Links Fatty Acid Uptake to β-Oxidation. Diabetes 2015, 64, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Samovski, D.; Su, X.; Xu, Y.; Abumrad, N.A.; Stahl, P.D. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J. Lipid Res. 2012, 53, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primer 2019, 5, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pinkosky, S.L.; Scott, J.W.; Desjardins, E.M.; Smith, B.K.; Day, E.A.; Ford, R.J.; Langendorf, C.G.; Ling, N.X.Y.; Nero, T.L.; Loh, K.; et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK β1 isoforms. Nat. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Corbalán-Campos, J.; Gurung, R.; Natarelli, L.; Zhu, M.; Exner, N.; Erhard, F.; Greulich, F.; Geißler, C.; Uhlenhaut, N.H.; et al. Dicer in Macrophages Prevents Atherosclerosis by Promoting Mitochondrial Oxidative Metabolism. Circulation 2018, 138, 2007–2020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Guo, H.; Zhang, C.-S.; Lin, S.-Y.; Yin, Z.; Peng, Y.; Luo, H.; Shi, Y.; Lian, G.; Zhang, C.; et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Bayry, J.; Tschan, M.P.; Muller, S. Progress and Challenges in The Use of MAP1LC3 as a Legitimate Marker for Measuring Dynamic Autophagy In Vivo. Cells 2020, 9, 1321. [Google Scholar] [CrossRef]
- Tian, W.; Alsaadi, R.; Guo, Z.; Kalinina, A.; Carrier, M.; Tremblay, M.-E.; Lacoste, B.; Lagace, D.; Russell, R.C. An antibody for analysis of autophagy induction. Nat. Methods 2019. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, T.; Yang, W.; Zhang, L.; Wu, S.; Yan, C.; Li, Q. Astragalus membranaceus Injection Suppresses Production of Interleukin-6 by Activating Autophagy through the AMPK-mTOR Pathway in Lipopolysaccharide-Stimulated Macrophages. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, P.; Han, F.; Zheng, J.; Xia, X.; Xue, W.; Ding, X.; Ding, C. Comparison of the characteristics of macrophages derived from murine spleen, peritoneal cavity, and bone marrow. J. Zhejiang Univ. Sci. B 2017, 18, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.; van Denderen, B.J.W.; Hevener, A.L.; Jørgensen, S.B.; Honeyman, J.; Galic, S.; Chen, Z.-P.; Watt, M.J.; Campbell, D.J.; Steinberg, G.R.; et al. AMPK β1 Deletion Reduces Appetite, Preventing Obesity and Hepatic Insulin Resistance. J. Biol. Chem. 2010, 285, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauffer, W.; Sheng, H.; Lim, H.N. EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep. 2018, 8, 15764. [Google Scholar] [CrossRef] [PubMed]
- García-Tardón, N.; Guigas, B. Determination of Adenine Nucleotide Concentrations in Cells and Tissues by High-Performance Liquid Chromatography. Methods Mol. Biol. Clifton NJ 2018, 1732, 229–237. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeBlond, N.D.; Nunes, J.R.C.; Smith, T.K.T.; O’Dwyer, C.; Robichaud, S.; Gadde, S.; Côté, M.; Kemp, B.E.; Ouimet, M.; Fullerton, M.D. Foam Cell Induction Activates AMPK But Uncouples Its Regulation of Autophagy and Lysosomal Homeostasis. Int. J. Mol. Sci. 2020, 21, 9033. https://doi.org/10.3390/ijms21239033
LeBlond ND, Nunes JRC, Smith TKT, O’Dwyer C, Robichaud S, Gadde S, Côté M, Kemp BE, Ouimet M, Fullerton MD. Foam Cell Induction Activates AMPK But Uncouples Its Regulation of Autophagy and Lysosomal Homeostasis. International Journal of Molecular Sciences. 2020; 21(23):9033. https://doi.org/10.3390/ijms21239033
Chicago/Turabian StyleLeBlond, Nicholas D., Julia R. C. Nunes, Tyler K. T. Smith, Conor O’Dwyer, Sabrina Robichaud, Suresh Gadde, Marceline Côté, Bruce E. Kemp, Mireille Ouimet, and Morgan D. Fullerton. 2020. "Foam Cell Induction Activates AMPK But Uncouples Its Regulation of Autophagy and Lysosomal Homeostasis" International Journal of Molecular Sciences 21, no. 23: 9033. https://doi.org/10.3390/ijms21239033