Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
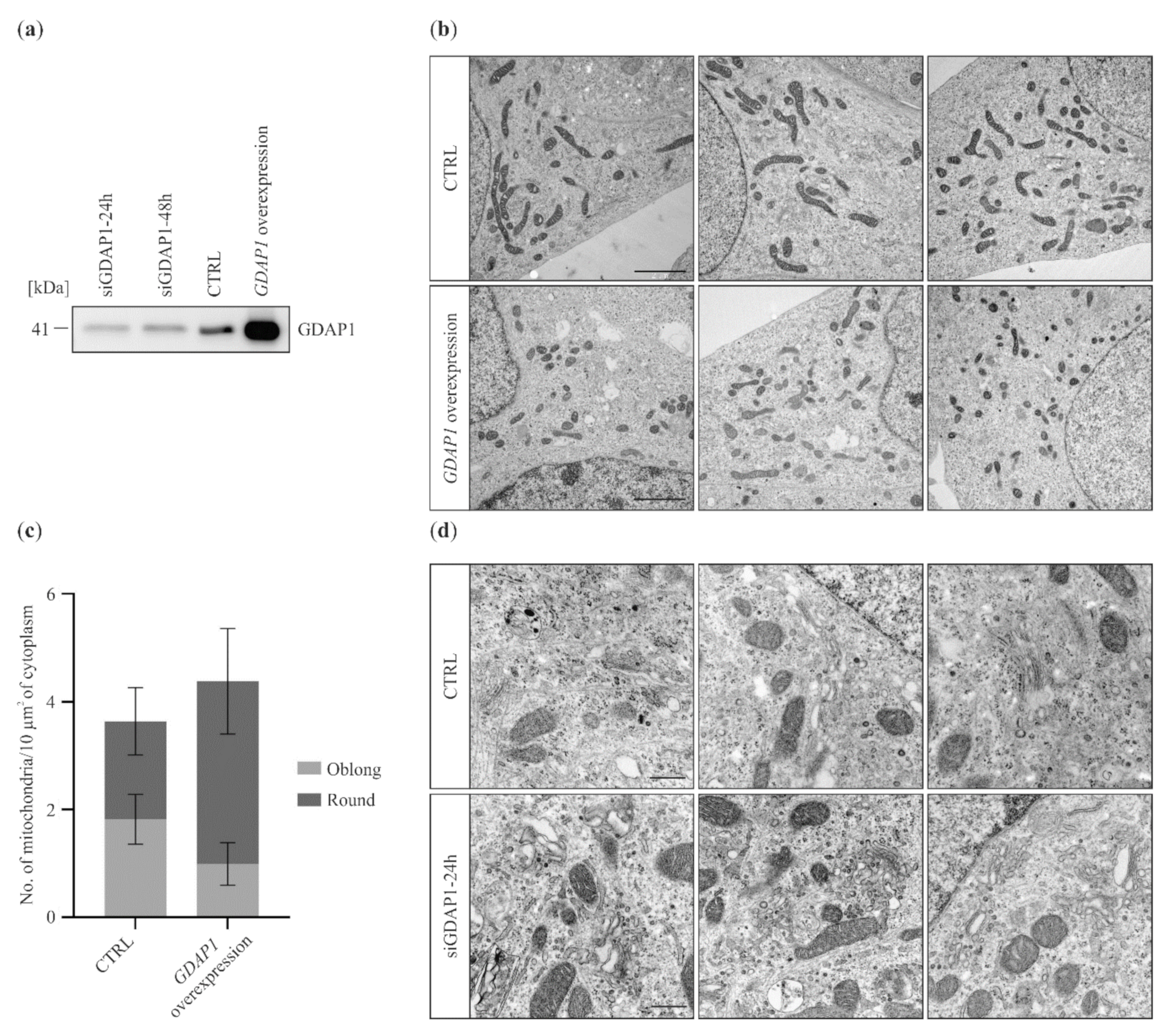
2.1. GDAP1 Protein Levels Influence Mitochondria and Golgi Morphology in SH-SY5Y Cells
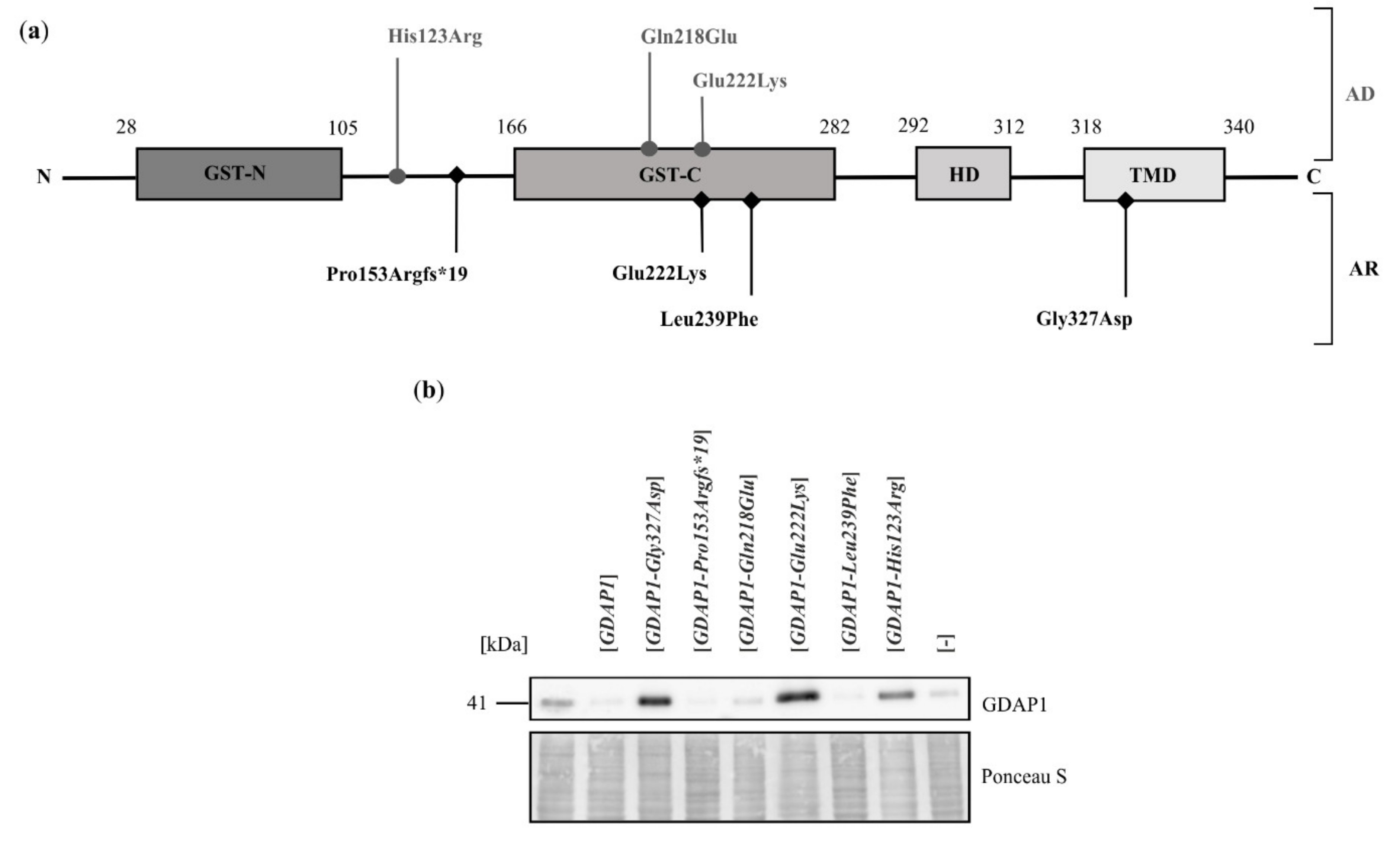
2.2. Exogenous Expression of Particular GDAP1 Variants Affects the Endogenous GDAP1 Protein Levels in HeLa Cells
2.3. A Reduction of GDAP1 Expression Alters Golgi Morphology in HeLa Cells
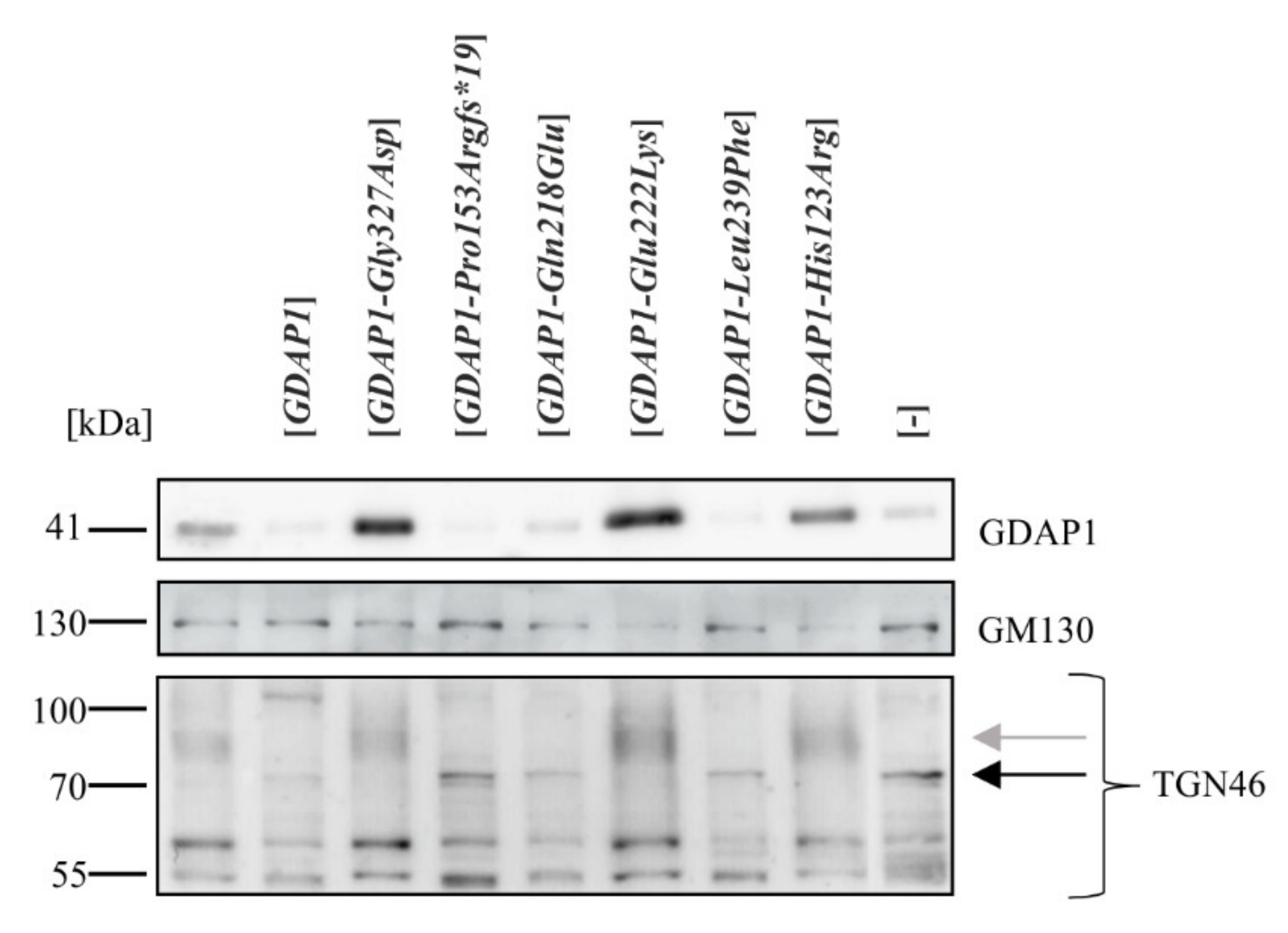
2.4. A Reduction of GDAP1 Expression Results in Changes to Post-Translational Modifications of TGN46
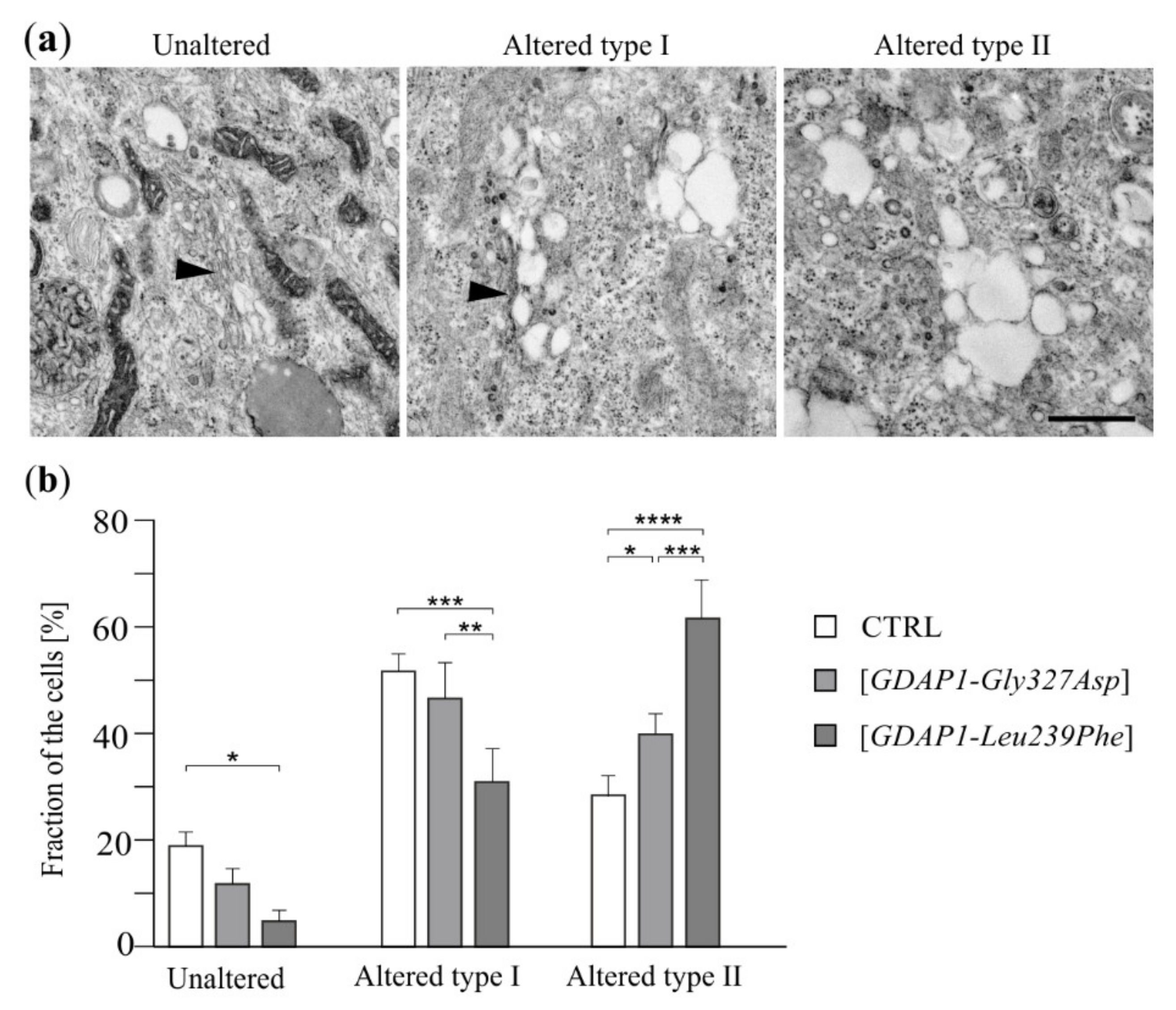
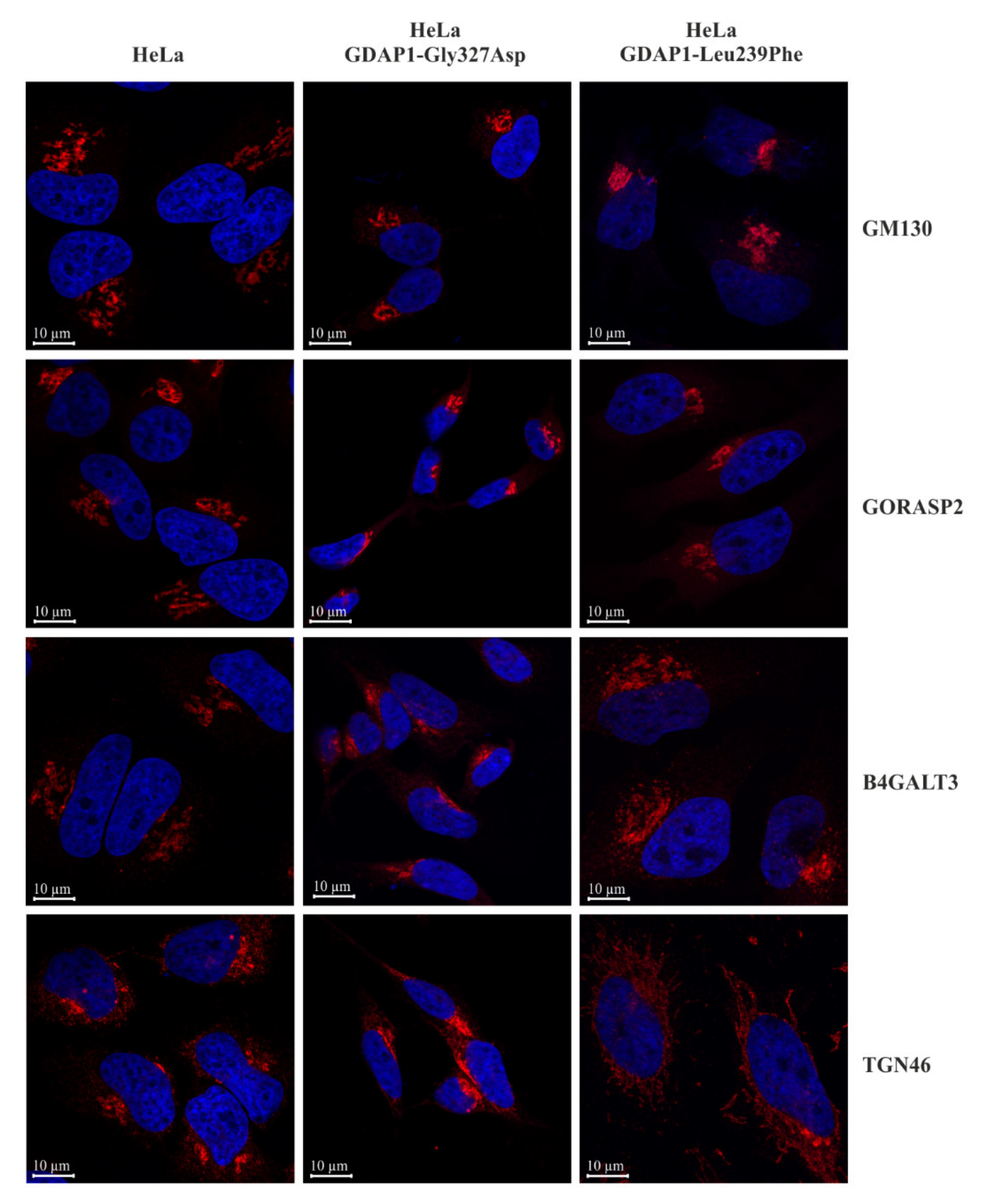
2.5. Localization of TGN46 Is Altered in HeLa Cells Expressing the GDAP1-Leu239Phe Variant
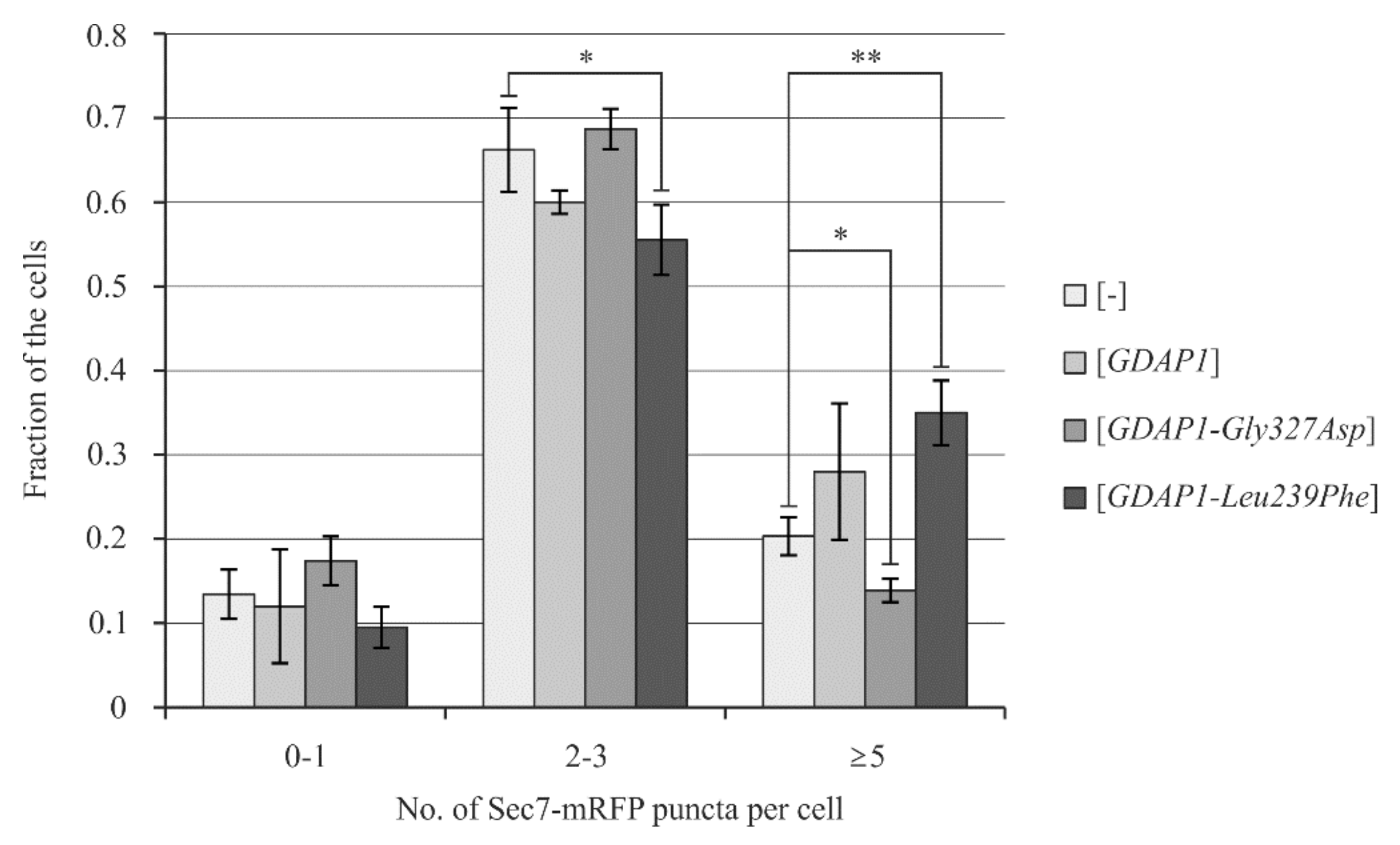
2.6. Yeast Cells Transformed with the GDAP1-Leu239Phe Gene Allele Exhibit Changes in the Localization of Sec7
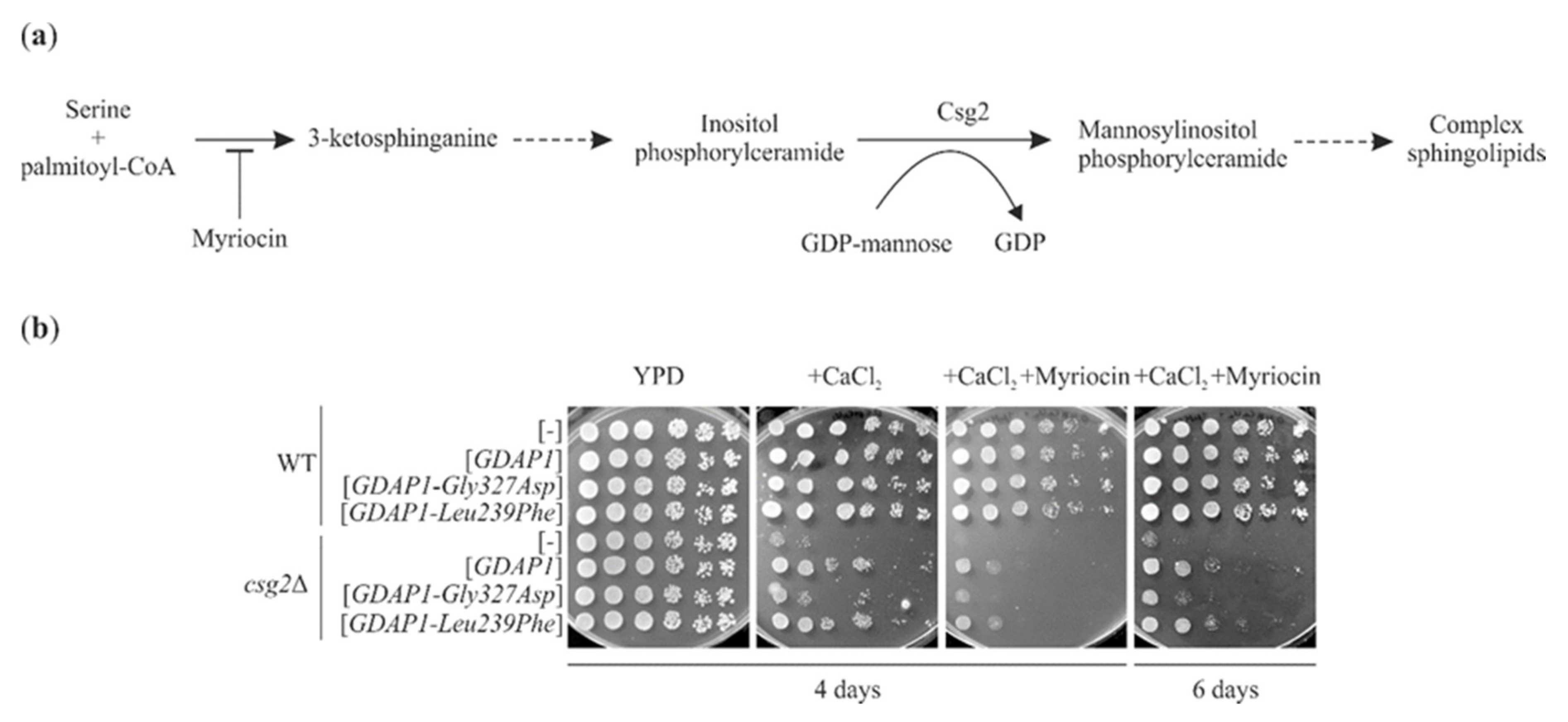
2.7. GDAP1-Mediated Suppression of the Calcium Sensitivity of csg2Δ Does Not Require Ongoing Sphingolipid Synthesis
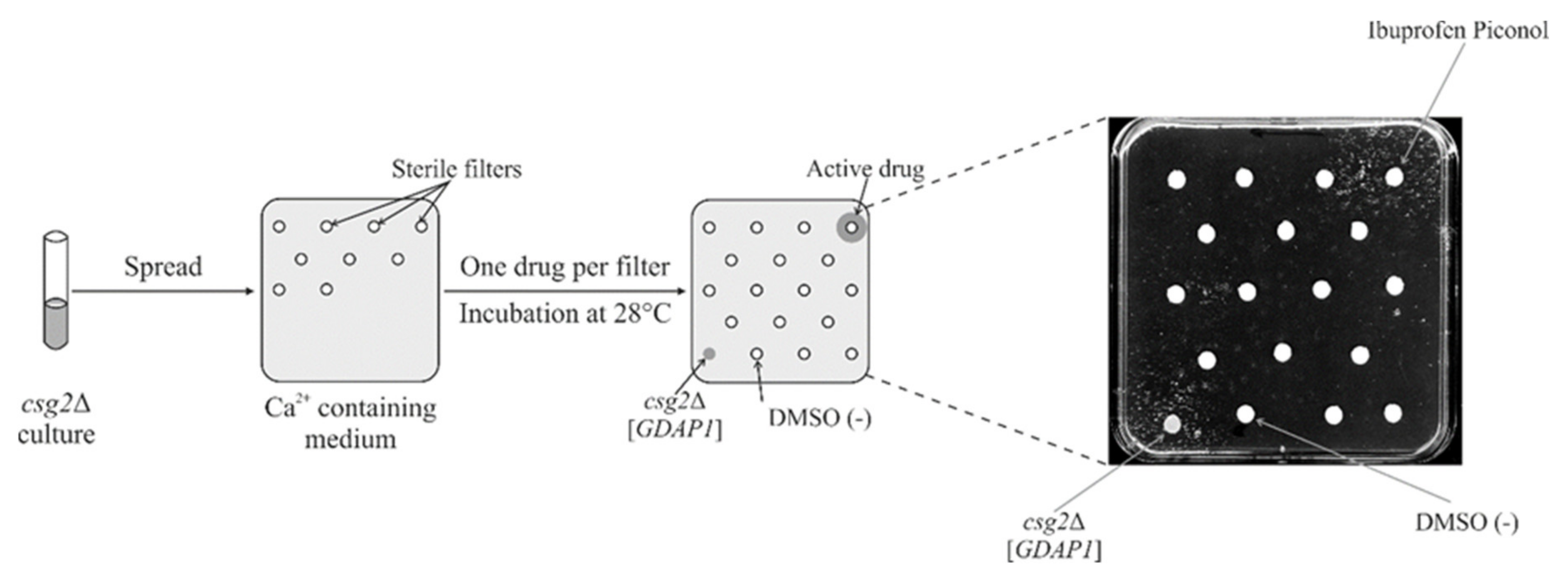
2.8. Screening a Drug Library Using the Calcium Hypersensitivity of the csg2ΔStrain Revealed Ibuprofen Piconol as an Active Compound
3. Discussion
4. Materials and Methods
4.1. Yeast Strains, Media and Growth Conditions
4.2. Cell Transfection and GDAP1 Silencing
4.3. Plasmids and DNA Manipulations
4.4. Protein Extracts and Western Blot Analysis
4.5. Drug Screening Assay
4.6. Confocal Microscopy
4.7. Transmission Electron Microscopy
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sevilla, T.; Jaijo, T.; Nauffal, D.; Collado, D.; Chumillas, M.J.; Vilchez, J.J.; Muelas, N.; Bataller, L.; Domenech, R.; Espinós, C.; et al. Vocal cord paresis and diaphragmatic dysfunction are severe and frequent symptoms of GDAP1-associated neuropathy. Brain 2008, 131, 3051–3061. [Google Scholar] [CrossRef] [Green Version]
- Kabzińska, D.; Niemann, A.; Drac, H.; Huber, N.; Potulska-Chromik, A.; Hausmanowa-Petrusewicz, I.; Suter, U.; Kochański, A. A new missense GDAP1 mutation disturbing targeting to the mitochondrial membrane causes a severe form of AR-CMT2C disease. Neurogenetics 2011, 12, 145–153. [Google Scholar] [CrossRef]
- Rzepnikowska, W.; Kochański, A. A role for the GDAP1 gene in the molecular pathogenesis of Charcot-Marie-Tooth disease. Acta Neurobiol. Exp. 2018, 78, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Nakagawa, T.; Kanematsu, T.; Uchida, T.; Tsuji, S. Isolation of 10 differentially expressed cDNAs in differentiated Neuro2a cells induced through controlled expression of the GD3 synthase gene. J. Neurochem. 1999, 72, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.V.; Othmane, K.B.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Hamida, M.B.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2002, 30, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; García-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marín, I.; Vílchez, J.J.; Palau, F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 2002, 30, 22–25. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, P.; Satrústegui, J.; Palau, F.; Del Arco, A. Calcium deregulation and mitochondrial bioenergetics in GDAP1-related CMT disease. Int. J. Mol. Sci. 2019, 20, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef]
- Marco, A.; Cuesta, A.; Pedrola, L.; Palau, F.; Marín, I. Evolutionary and Structural Analyses of GDAP1, Involved in Charcot-Marie-Tooth Disease, Characterize a Novel Class of Glutathione Transferase-Related Genes. Mol. Biol. Evol. 2004, 21, 176–187. [Google Scholar] [CrossRef]
- Shield, A.J.; Murray, T.P.; Board, P.G. Functional characterisation of ganglioside-induced differentiation-associated protein 1 as a glutathione transferase. Biochem. Biophys. Res. Commun. 2006, 347, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Googins, M.R.; Woghiren-Afegbua, A.O.; Calderon, M.; St. Croix, C.M.; Kiselyov, K.I.; VanDemark, A.P. Structural and functional divergence of GDAP1 from the glutathione S-transferase superfamily. FASEB J. 2020, 34, 7192–7207. [Google Scholar] [CrossRef] [Green Version]
- Pla-Martín, D.; Calpena, E.; Lupo, V.; Márquez, C.; Rivas, E.; Sivera, R.; Sevilla, T.; Palau, F.; Espinós, C. Junctophilin-1 is a modifier gene of GDAP1-related Charcot-Marie-Tooth disease. Hum. Mol. Genet. 2015, 24, 213–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pla-Martín, D.; Rueda, C.B.; Estela, A.; Sánchez-Piris, M.; González-Sánchez, P.; Traba, J.; de la Fuente, S.; Scorrano, L.; Renau-Piqueras, J.; Alvarez, J.; et al. Silencing of the Charcot-Marie-Tooth disease-associated gene GDAP1 induces abnormal mitochondrial distribution and affects Ca2+ homeostasis by reducing store-operated Ca2+ entry. Neurobiol. Dis. 2013, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Barneo-Muñoz, M.; Juárez, P.; Civera-Tregón, A.; Yndriago, L.; Pla-Martin, D.; Zenker, J.; Cuevas-Martín, C.; Estela, A.; Sánchez-Aragó, M.; Forteza-Vila, J.; et al. Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy. PLoS Genet. 2015, 11, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Kochański, A. Pathogenic effect of GDAP1 gene mutations in a yeast model. Genes 2020, 11, 310. [Google Scholar] [CrossRef] [Green Version]
- Huber, N.; Guimaraes, S.; Schrader, M.; Suter, U.; Niemann, A. Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission. EMBO Rep. 2013, 14, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Wagner, K.M.; Ruegg, M.; Suter, U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 2009, 36, 509–520. [Google Scholar] [CrossRef]
- Cassereau, J.; Chevrollier, A.; Gueguen, N.; Malinge, M.C.; Letournel, F.; Nicolas, G.; Richard, L.; Ferre, M.; Verny, C.; Dubas, F.; et al. Mitochondrial complex i deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics 2009, 10, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; zu Hörste, G.M.; Stettner, M.; Kieseier, B.C.; et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef] [Green Version]
- García-Sobrino, T.; Blanco-Arias, P.; Palau, F.; Espinós, C.; Ramirez, L.; Estela, A.; Millán, B.S.; Arias, M.; Sobrido, M.J.; Pardo, J. Phenotypical features of a new dominant GDAP1 pathogenic variant (p.R226del) in axonal Charcot-Marie-Tooth disease. Neuromuscul. Disord. 2017, 27, 667–672. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, P.; Pla-Martín, D.; Martínez-Valero, P.; Rueda, C.B.; Calpena, E.; Del Arco, A.; Palau, F.; Satrústegui, J. CMT-linked loss-of-function mutations in GDAP1 impair store-operated Ca 2+ entry-stimulated respiration. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassereau, J.; Chevrollier, A.; Codron, P.; Goizet, C.; Gueguen, N.; Verny, C.; Reynier, P.; Bonneau, D.; Lenaers, G.; Procaccio, V. Oxidative stress contributes differentially to the pathophysiology of Charcot-Marie-Tooth disease type 2K. Exp. Neurol. 2020, 323, 113069. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Civera-Tregón, A.; Cantarero, L.; Herrer, I.; Juarez, P.; Hoenicka, J.; Palau, F. Neuroinflammation in the pathogenesis of axonal Charcot-Marie-Tooth disease caused by lack of GDAP1. Exp. Neurol. 2019, 320, 113004. [Google Scholar] [CrossRef]
- Wei, J.H.; Seemann, J. Unraveling the Golgi ribbon. Traffic 2010, 11, 1391–1400. [Google Scholar] [CrossRef]
- Li, J.; Ahat, E.; Wang, Y. Golgi Structure and Function in Health, Stress, and Diseases. In Results and Problems in Cell Differentiation; Springer: Berlin/Heidelberg, Germany, 2019; Volume 67, pp. 441–485. [Google Scholar]
- Martínez-Menárguez, J.A. Intra-Golgi Transport: Roles for Vesicles, Tubules, and Cisternae. ISRN Cell Biol. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pedrola, L.; Espert, A.; Valdés-Sánchez, T.; Sánchez-Piris, M.; Sirkowski, E.E.; Scherer, S.S.; Fariñas, I.; Palau, F. Cell expression of GDAP1 in the nervous system and pathogenesis of Charcot-Marie-Tooth type 4A disease. J. Cell. Mol. Med. 2008, 12, 679–689. [Google Scholar] [CrossRef]
- Zimoń, M.; Baets, J.; Fabrizi, G.M.; Jaakkola, E.; Kabzińska, D.; Pilch, J.; Schindler, A.B.; Cornblath, D.R.; Fischbeck, K.H.; Auer-Grumbach, M.; et al. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 2011, 77, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Kabzińska, D.; Strugalska-Cynowska, H.; Kostera-Pruszczyk, A.; Ryniewicz, B.; Posmyk, R.; Midro, A.; Seeman, P.; Baránková, L.; Zimoń, M.; Baets, J.; et al. L239F founder mutation in GDAP1 is associated with a mild Charcot-Marie-Tooth type 4C4 (CMT4C4) phenotype. Neurogenetics 2010, 11, 357–366. [Google Scholar] [CrossRef]
- Matsuura-Tokita, K.; Takeuchi, M.; Ichihara, A.; Mikuriya, K.; Nakano, A. Live imaging of yeast Golgi cisternal maturation. Nature 2006, 441, 1007–1010. [Google Scholar] [CrossRef]
- Wood, C.S.; Hung, C.S.; Huoh, Y.S.; Mousley, C.J.; Stefan, C.J.; Bankaitis, V.; Ferguson, K.M.; Burd, C.G. Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol. Biol. Cell 2012, 23, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Bach, S.; Talarek, N.; Andrieu, T.; Vierfond, J.M.; Mettey, Y.; Galons, H.; Dormont, D.; Meijer, L.; Cullin, C.; Blondel, M. Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat. Biotechnol. 2003, 21, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Couplan, E.; Aiyar, R.S.; Kucharczyk, R.; Kabala, A.; Ezkurdia, N.; Gagneur, J.; Onge, R.P.S.; Salin, B.; Soubigou, F.; Le Cann, M.; et al. A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 11989–11994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soczewka, P.; Flis, K.; Tribouillard-Tanvier, D.; Di Rago, J.P.; Santos, C.N.; Menezes, R.; Kaminska, J.; Żołądek, T. Flavonoids as potential drugs for VPS13-dependent rare neurodegenerative diseases. Genes 2020, 11, 828. [Google Scholar] [CrossRef]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontés, M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat. Med. 2004, 10, 396–401. [Google Scholar] [CrossRef]
- Zu Horste, G.M.; Prukop, T.; Liebetanz, D.; Mobius, W.; Nave, K.A.; Sereda, M.W. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann. Neurol. 2007, 61, 61–72. [Google Scholar] [CrossRef]
- Cottenie, E.; Kochanski, A.; Jordanova, A.; Bansagi, B.; Zimon, M.; Horga, A.; Jaunmuktane, Z.; Saveri, P.; Rasic, V.M.; Baets, J.; et al. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am. J. Hum. Genet. 2014, 95, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Fourriere, L.; Jimenez, A.J.; Perez, F.; Boncompain, G. The role of microtubules in secretory protein transport. J. Cell Sci. 2020, 133, jcs237016. [Google Scholar] [CrossRef]
- Brunden, K.R.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q.; Ballatore, C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol. Dis. 2017, 105, 328–335. [Google Scholar] [CrossRef]
- Sancho, P.; Bartesaghi, L.; Miossec, O.; García-García, F.; Ramírez-Jiménez, L.; Siddell, A.; Åkesson, E.; Hedlund, E.; Laššuthová, P.; Pascual-Pascual, S.I.; et al. Characterization of molecular mechanisms underlying the axonal Charcot-Marie-Tooth neuropathy caused by MORC2 mutations. Hum. Mol. Genet. 2019, 28, 1629–1644. [Google Scholar] [CrossRef]
- Martínez-Menárguez, J.Á.; Tomás, M.; Martínez-Martínez, N.; Martínez-Alonso, E. Golgi Fragmentation in Neurodegenerative Diseases: Is There a Common Cause? Cells 2019, 8, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, T.; Kuwahara, T.; Eguchi, T.; Sakurai, M.; Komori, T.; Iwatsubo, T. Parkinson’s disease-associated mutant LRRK2 phosphorylates Rab7L1 and modifies trans-Golgi morphology. Biochem. Biophys. Res. Commun. 2018, 495, 1708–1715. [Google Scholar] [CrossRef] [PubMed]
- Aistleitner, K.; Clark, T.; Dooley, C.; Hackstadt, T. Selective fragmentation of the trans-Golgi apparatus by Rickettsia rickettsii. PLoS Pathog. 2020, 16, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, Z.; Xu, X.; Wang, Z.; Sun, L.; Zhou, Y.; Lin, X.; Hong, W.; Wang, T. A Role of Rab29 in the Integrity of the Trans-Golgi Network and Retrograde Trafficking of Mannose-6-Phosphate Receptor. PLoS ONE 2014, 9, e96242. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Yang, Y.; Zhou, R.; Gong, T. Golgi Apparatus: An Emerging Platform for Innate Immunity. Trends Cell Biol. 2020, 30, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Holloway, Z.G.; Grabski, R.; Szul, T.; Styers, M.L.; Coventry, J.A.; Monaco, A.P.; Sztul, E. Activation of ADP-ribosylation factor regulates biogenesis of the ATP7A-containing trans-Golgi network compartment and its Cu-induced trafficking. Am. J. Physiol. Cell Physiol. 2007, 293, C1753–C1767. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, P.A.; Lock, J.G.; Luke, M.R.; Stow, J.L. Domains of the TGN: Coats, tethers and G proteins. Traffic 2004, 5, 315–326. [Google Scholar] [CrossRef]
- Bitoun, M.; Durieux, A.; Prudhon, B.; Bevilacqua, J.A.; Herledan, A.; Sakanyan, V.; Urtizberea, A.; Cartier, L.; Romero, N.B.; Guicheney, P. Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum. Mutat. 2009, 30, 1419–1427. [Google Scholar] [CrossRef]
- Gouttenoire, E.A.; Lupo, V.; Calpena, E.; Bartesaghi, L.; Schüpfer, F.; Médard, J.J.; Maurer, F.; Beckmann, J.S.; Senderek, J.; Palau, F.; et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia 2013, 61, 1041–1051. [Google Scholar] [CrossRef]
- Uemura, S.; Kihara, A.; Iwaki, S.; Inokuchi, J.-I.; Igarashi, Y. Regulation of the transport and protein levels of the inositol phosphorylceramide mannosyltransferases Csg1 and Csh1 by the Ca2+−binding protein Csg2. J. Biol. Chem. 2007, 282, 8613–8621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Source |
|---|---|---|
| Yeast Plasmids: | ||
| p425-PTDH3 | 2µ; LEU2 | [53] |
| p425-PTDH3-GDAP1 | 2µ; LEU2; GDAP1 | [16] |
| p425-PTDH3-GDAP1m2 | 2µ; LEU2; GDAP1 c.980G > A; p.Gly327Asp | [16] |
| p425-PTDH3-GDAP1m5 | 2µ; LEU2; GDAP1 c.715C > T; p.Leu239Phe | [16] |
| Sec7-mRFP | URA3; SEC7-mRFP | [31] |
| Sed5-mRFP | URA3; SED5-mRFP | [31] |
| Mammalian Expression Plasmids: | ||
| pCMV6-XL5-GDAP1 | Human Untagged Clone GDAP1 cDNA | OriGene |
| pCMV6-XL5-GDAP1m1 | GDAP1 c.456delC; p.Pro153Argfs*19 | This study |
| pCMV6-XL5-GDAP1m2 | GDAP1 c.980G > A; p.Gly327Asp | This study |
| pCMV6-XL5-GDAP1m3 | GDAP1 c.652C > G; p.Gln218Glu | This study |
| pCMV6-XL5-GDAP1m4 | GDAP1 c.664G > A; p.Glu222Lys | This study |
| pCMV6-XL5-GDAP1m5 | GDAP1 c.715C > T; p.Leu239Phe | This study |
| pCMV6-XL5-GDAP1m6 | GDAP1 c.368A > G; p.His123Arg | This study |
| pIRES2-AcGFP1 | Bicistronic vector with GFP | TAKARA Bio |
| pIRES2-AcGFP1-GDAP1 | GDAP1 WT | This study |
| pIRES2-AcGFP1-GDAP1m1 | GDAP1 c.456delC; p.Pro153Argfs*19 | This study |
| pIRES2-AcGFP1-GDAP1m2 | GDAP1 c.980G > A; p.Gly327Asp | This study |
| pIRES2-AcGFP1-GDAP1m3 | GDAP1 c.652C > G; p.Gln218Glu | This study |
| pIRES2-AcGFP1-GDAP1m4 | GDAP1 c.664G > A; p.Glu222Lys | This study |
| pIRES2-AcGFP1-GDAP1m5 | GDAP1 c.715C > T; p.Leu239Phe | This study |
| pIRES2-AcGFP1-GDAP1m6 | GDAP1 c.368A > G; p.His123Arg | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binięda, K.; Rzepnikowska, W.; Kolakowski, D.; Kaminska, J.; Szczepankiewicz, A.A.; Nieznańska, H.; Kochański, A.; Kabzińska, D. Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network. Int. J. Mol. Sci. 2021, 22, 914. https://doi.org/10.3390/ijms22020914
Binięda K, Rzepnikowska W, Kolakowski D, Kaminska J, Szczepankiewicz AA, Nieznańska H, Kochański A, Kabzińska D. Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network. International Journal of Molecular Sciences. 2021; 22(2):914. https://doi.org/10.3390/ijms22020914
Chicago/Turabian StyleBinięda, Katarzyna, Weronika Rzepnikowska, Damian Kolakowski, Joanna Kaminska, Andrzej Antoni Szczepankiewicz, Hanna Nieznańska, Andrzej Kochański, and Dagmara Kabzińska. 2021. "Mutations in GDAP1 Influence Structure and Function of the Trans-Golgi Network" International Journal of Molecular Sciences 22, no. 2: 914. https://doi.org/10.3390/ijms22020914