
Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset

 , , , , , , , ,
, , , , , , , ,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results
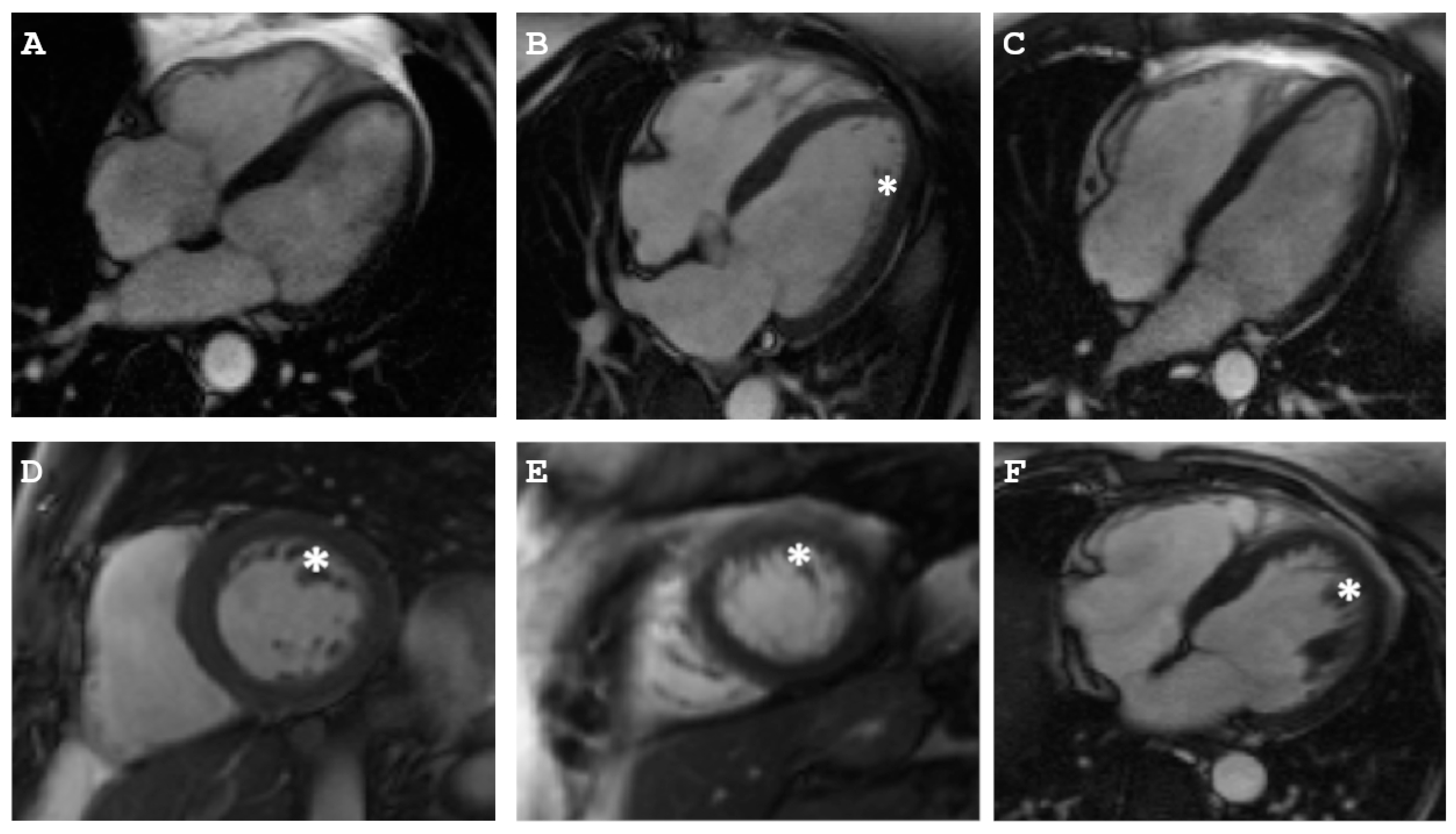
2.1. Clinical Description of the Patients
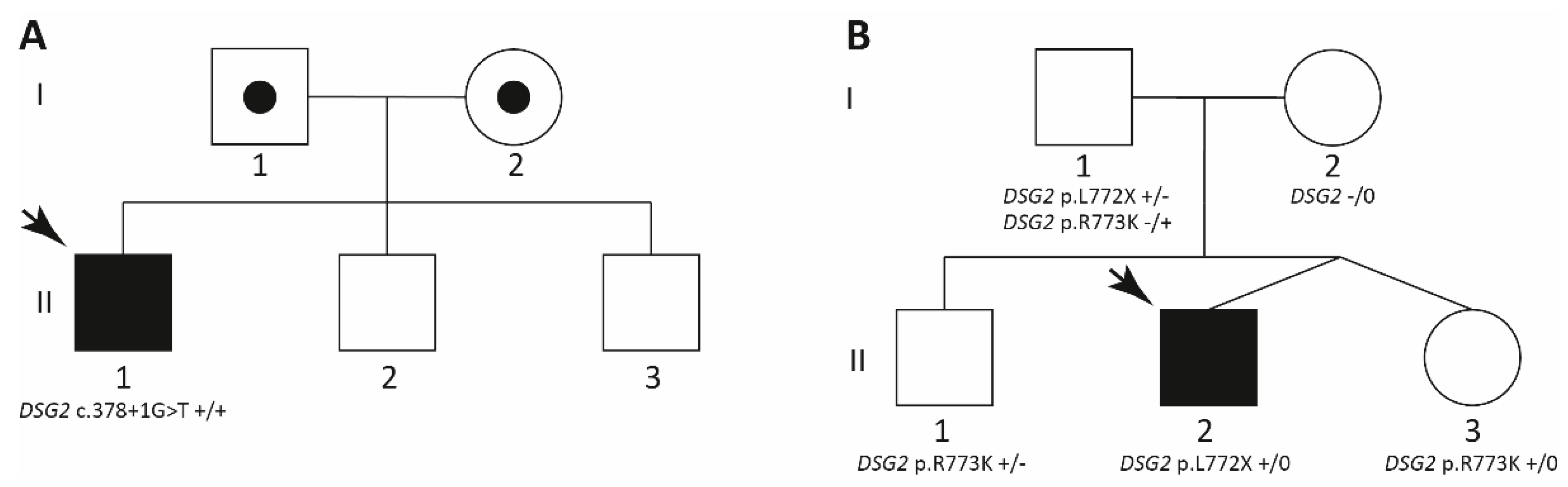
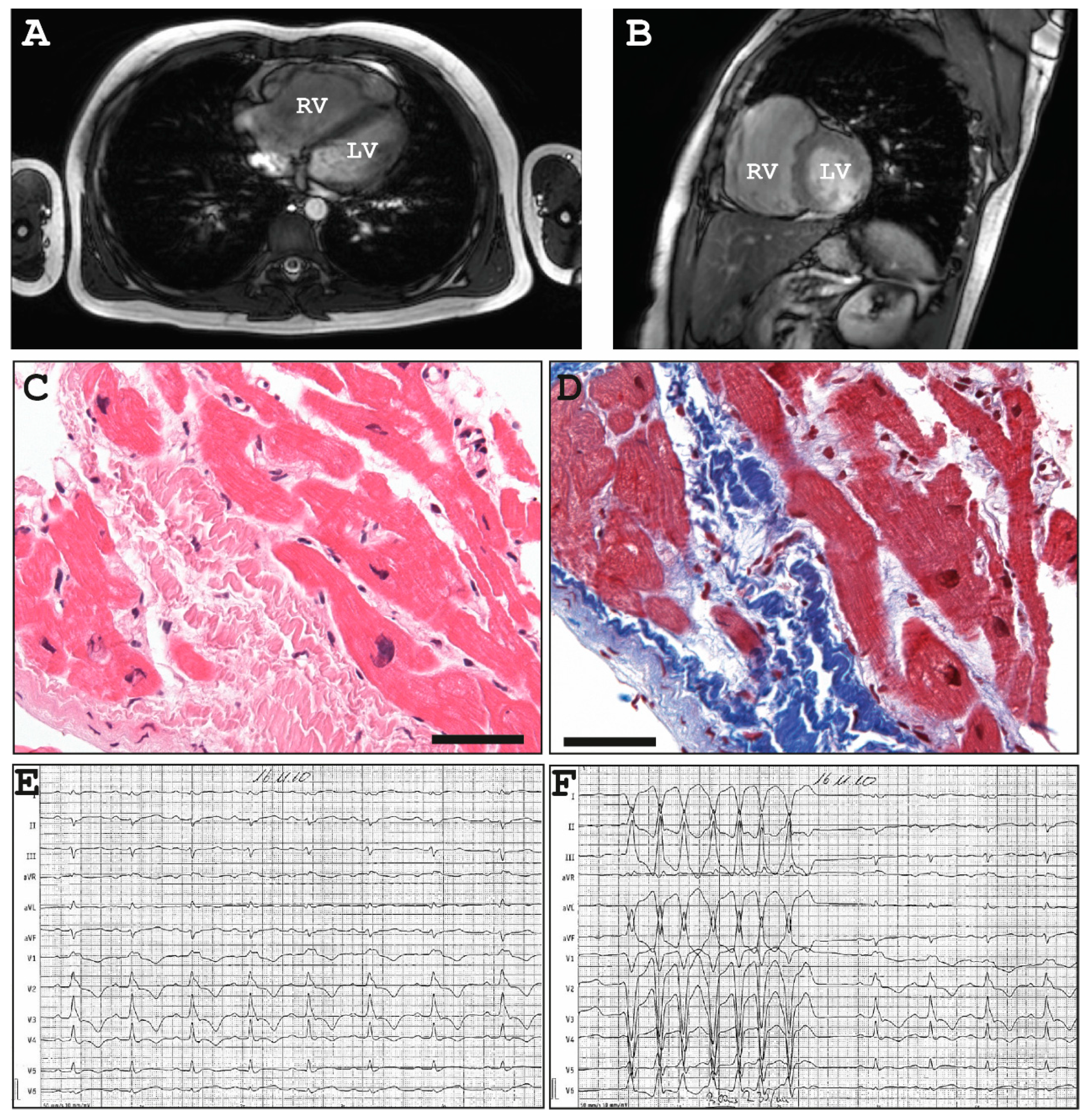
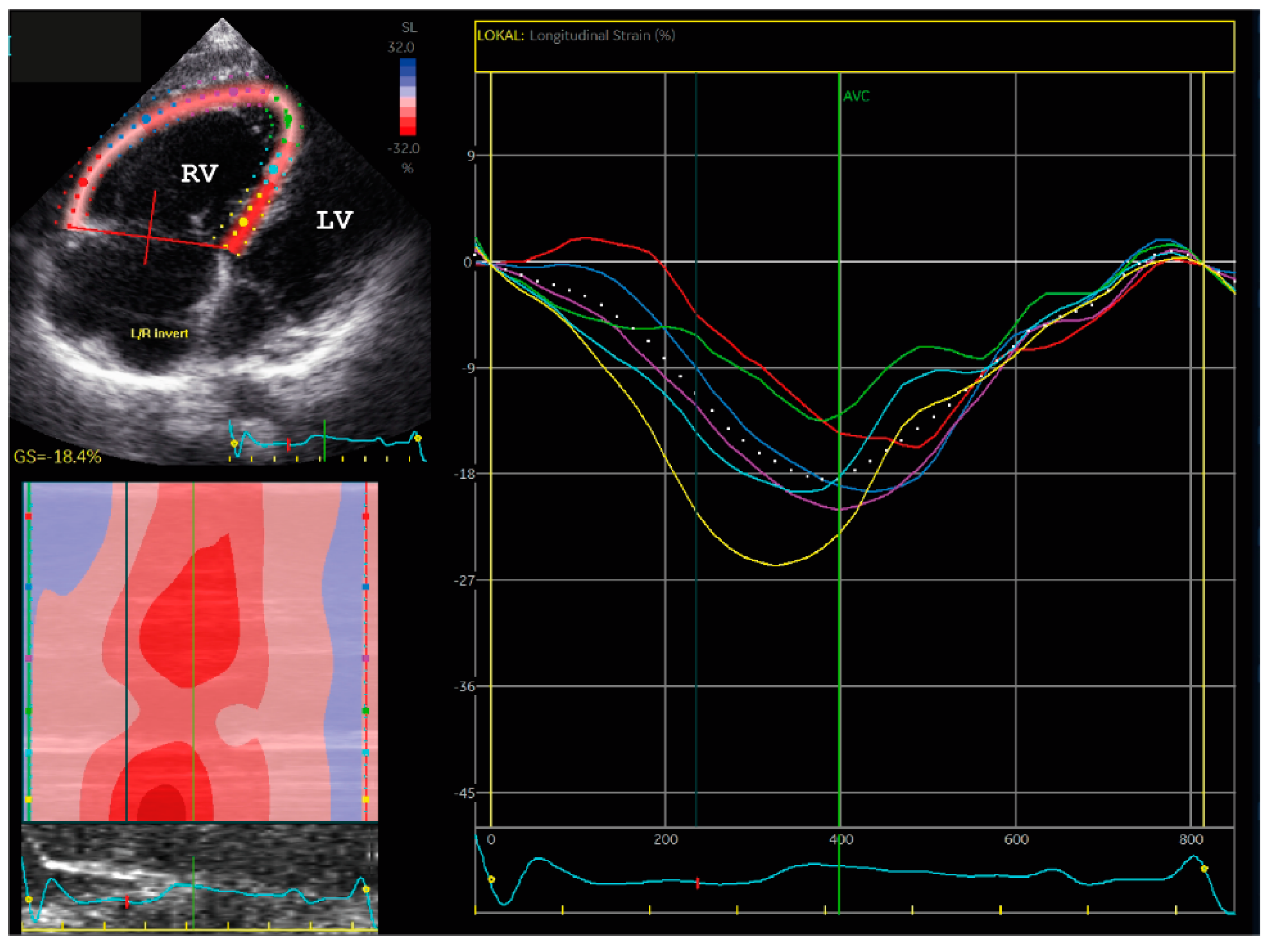
2.1.1. Family A
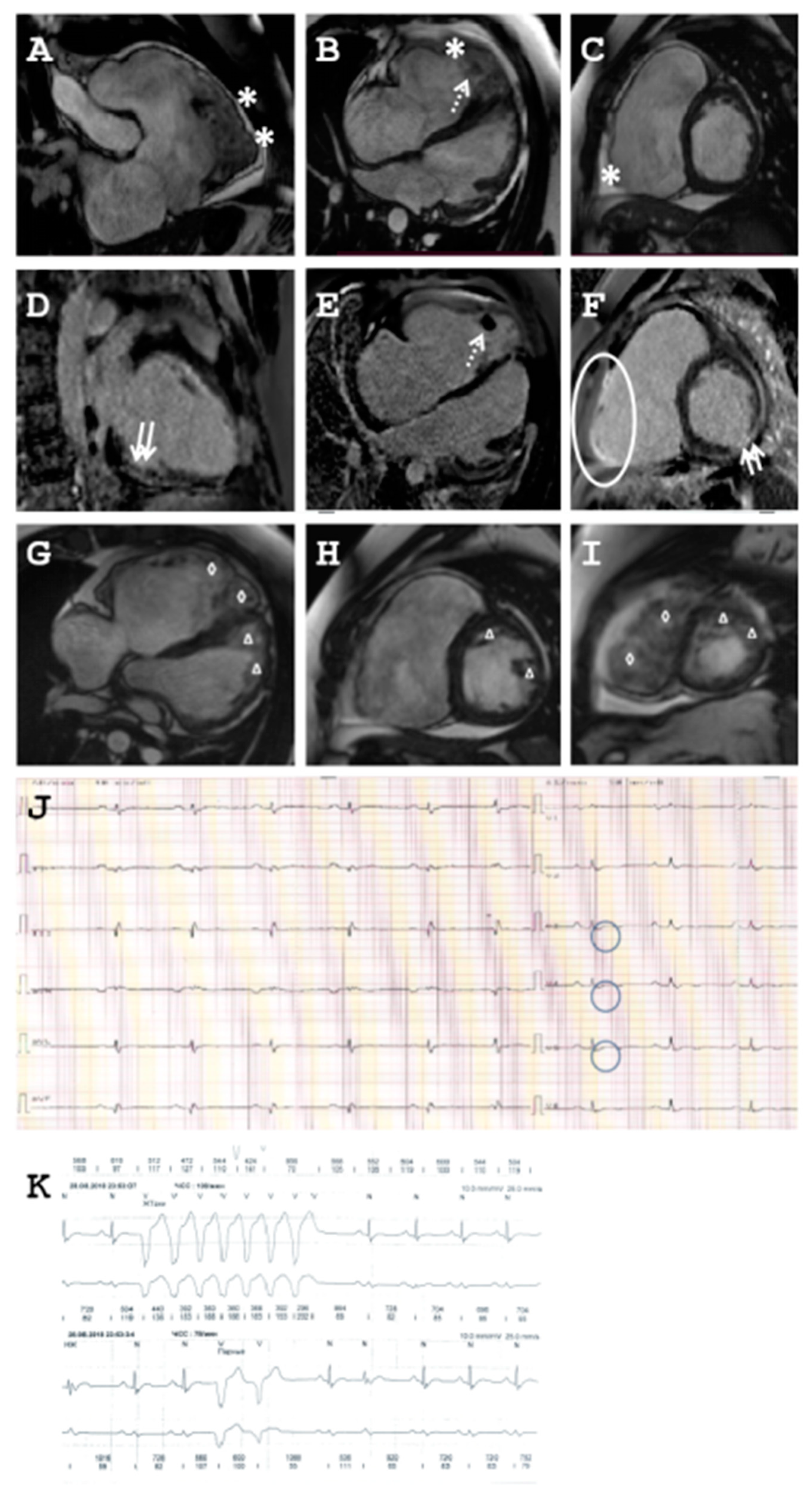
2.1.2. Family B
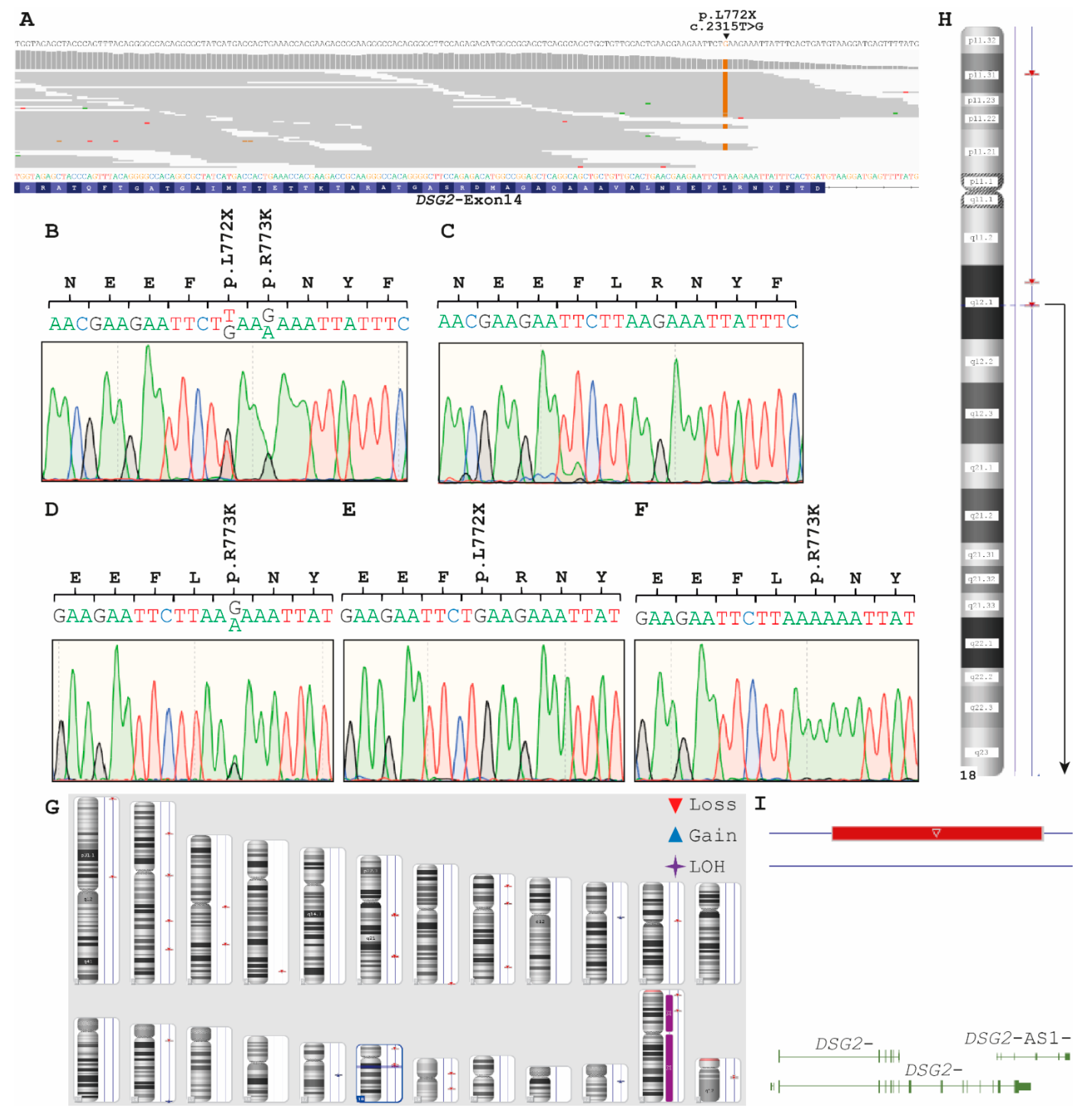
2.2. Genetic Analyses
- Consanguinity of the parents
- An additional large deletion in DSG2 localized on the second chromosome mimicking homozygosity DSG2–c.378+1G>T or
- Uniparental isodisomy (UPD)
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Patel, D.M.; Green, K.J. Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun. Adhes. 2014, 21, 109–128. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Brasch, J.; Lasso, G.; Katsamba, P.S.; Ahlsen, G.; Honig, B.; Shapiro, L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc. Natl. Acad. Sci. USA 2016, 113, 7160–7165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Stanasiuk, C.; Anselmetti, D.; Gummert, J.; Milting, H. Incorporation of desmocollin-2 into the plasma membrane requires N-glycosylation at multiple sites. FEBS Open Bio 2019, 9, 996–1007. [Google Scholar] [CrossRef]
- Kolegraff, K.; Nava, P.; Laur, O.; Parkos, C.A.; Nusrat, A. Characterization of full-length and proteolytic cleavage fragments of desmoglein-2 in native human colon and colonic epithelial cell lines. Cell Adhes. Migr. 2011, 5, 306–314. [Google Scholar] [CrossRef]
- Chen, X.; Bonne, S.; Hatzfeld, M.; van Roy, F.; Green, K.J. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J. Biol. Chem. 2002, 277, 10512–10522. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Gross, J.C.; Pokutta, S.; Weis, W.I. Interactions of plakoglobin and beta-catenin with desmosomal cadherins: Basis of selective exclusion of alpha-and beta-catenin from desmosomes. J. Biol. Chem. 2009, 284, 31776–31788. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, F.; Schuetz, A.; Boldt, L.H.; Martens, K.; Dittmar, G.; Haverkamp, W.; Thierfelder, L.; Heinemann, U.; Gerull, B. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ. Cardiovasc. Genet. 2012, 5, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Favre, B.; Begre, N.; Bouameur, J.E.; Lingasamy, P.; Conover, G.M.; Fontao, L.; Borradori, L. Desmoplakin interacts with the coil 1 of different types of intermediate filament proteins and displays high affinity for assembled intermediate filaments. PLoS ONE 2018, 13, e0205038. [Google Scholar] [CrossRef]
- Kartenbeck, J.; Franke, W.W.; Moser, J.G.; Stoffels, U. Specific attachment of desmin filaments to desmosomal plaques in cardiac myocytes. EMBO J. 1983, 2, 735–742. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Asimaki, A.; Evans, A.; Sen-Chowdhry, S.; Hughes, S.E.; McKenna, W.J. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: A genotype-phenotype characterization of familial disease. Eur. Heart J. 2007, 28, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The novel desmin variant p.Leu115Ile is associated with a unique form of biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2020. [Google Scholar] [CrossRef]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, H.; Cabet, E.; Chevalier, N.R.; Moosmann, J.; Schultheis, D.; Haas, J.; Schowalter, M.; Berwanger, C.; Weyerer, V.; Agaimy, A.; et al. Dual Functional States of R406W-Desmin Assembly Complexes Cause Cardiomyopathy With Severe Intercalated Disc Derangement in Humans and in Knock-In Mice. Circulation 2020, 142, 2155–2171. [Google Scholar] [CrossRef]
- Kulikova, O.; Brodehl, A.; Kiseleva, A.; Myasnikov, R.; Meshkov, A.; Stanasiuk, C.; Gartner, A.; Divashuk, M.; Sotnikova, E.; Koretskiy, S.; et al. The Desmin (DES) Mutation p.A337P Is Associated with Left-Ventricular Non-Compaction Cardiomyopathy. Genes 2021, 12, 121. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef]
- Abdelfatah, N.; Chen, R.; Duff, H.J.; Seifer, C.M.; Buffo, I.; Huculak, C.; Clarke, S.; Clegg, R.; Jassal, D.S.; Gordon, P.M.K.; et al. Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2. JACC Basic Transl. Sci. 2019, 4, 204–221. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: A review. Pacing Clin. Electrophysiol. 1995, 18, 1298–1314. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Bermudez-Jimenez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.A.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Protonotarios, A.; Elliott, P.M. Arrhythmogenic Cardiomyopathy: A Disease or Merely a Phenotype? Eur. Cardiol. 2020, 15, 1–5. [Google Scholar] [CrossRef]
- Burke, A.P.; Farb, A.; Tashko, G.; Virmani, R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: Are they different diseases? Circulation 1998, 97, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, P.M.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The genetic landscape of cardiomyopathies. In Genetic Causes of Cardiac Disease; Springer: New York, NY, USA, 2019; pp. 45–91. [Google Scholar]
- Wada, Y.; Ohno, S.; Aiba, T.; Horie, M. Unique genetic background and outcome of non-Caucasian Japanese probands with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Mol. Genet. Genom. Med. 2017, 5, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, A.; Klauke, B.; Stork, I.; Niehaus, K.; Niemann, G.; Gummert, J.; Milting, H. In vitro functional analyses of arrhythmogenic right ventricular cardiomyopathy-associated desmoglein-2-missense variations. PLoS ONE 2012, 7, e47097. [Google Scholar] [CrossRef] [Green Version]
- Pereira Fernandes, M.; Azevedo, O.; Pereira, V.; Calvo, L.; Lourenco, A. Arrhythmogenic right ventricular cardiomyopathy with left ventricular involvement: A novel splice site mutation in the DSG2 gene. Cardiology 2015, 130, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Gehmlich, K.; Syrris, P.; Reimann, M.; Asimaki, A.; Ehler, E.; Evans, A.; Quarta, G.; Pantazis, A.; Saffitz, J.E.; McKenna, W.J. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc. Pathol. 2012, 21, 275–282. [Google Scholar] [CrossRef]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ventricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Grothoff, M.; Pachowsky, M.; Hoffmann, J.; Posch, M.; Klaassen, S.; Lehmkuhl, L.; Gutberlet, M. Value of cardiovascular MR in diagnosing left ventricular non-compaction cardiomyopathy and in discriminating between other cardiomyopathies. Eur. Radiol. 2012, 22, 2699–2709. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Perazzolo, M.M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Sund, K.L.; Rehder, C.W. Detection and reporting of homozygosity associated with consanguinity in the clinical laboratory. Hum. Hered. 2014, 77, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Eshkind, L.; Tian, Q.; Schmidt, A.; Franke, W.W.; Windoffer, R.; Leube, R.E. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell. Biol. 2002, 81, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Krusche, C.A.; Holthofer, B.; Hofe, V.; van de Sandt, A.M.; Eshkind, L.; Bockamp, E.; Merx, M.W.; Kant, S.; Windoffer, R.; Leube, R.E. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res. Cardiol. 2011, 106, 617–633. [Google Scholar] [CrossRef] [Green Version]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Krull, P.; Eisner, S.; Leube, R.E.; Krusche, C.A. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012, 348, 249–259. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabatelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signalling and miRNA dysregulation. Cardiovasc. Res. 2019, 115, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11, 624. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.A.; Dietz, H.C.; et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehmlich, K.; Asimaki, A.; Cahill, T.J.; Ehler, E.; Syrris, P.; Zachara, E.; Re, F.; Avella, A.; Monserrat, L.; Saffitz, J.E.; et al. Novel missense mutations in exon 15 of desmoglein-2: Role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm 2010, 7, 1446–1453. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gartner, A.; et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29. [Google Scholar] [CrossRef]
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ. Cardiovasc. Genet. 2013, 6, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sabeq, B.; Krahn, A.D.; Conacher, S.; Klein, G.J.; Laksman, Z. Arrhythmogenic right ventricular cardiomyopathy with recessive inheritance related to a new homozygous desmocollin-2 mutation. Can. J. Cardiol. 2014, 30, e691–e693. [Google Scholar] [CrossRef]
- Simpson, M.A.; Mansour, S.; Ahnood, D.; Kalidas, K.; Patton, M.A.; McKenna, W.J.; Behr, E.R.; Crosby, A.H. Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology 2009, 113, 28–34. [Google Scholar] [CrossRef]
- Lorenzon, A.; Pilichou, K.; Rigato, I.; Vazza, G.; De Bortoli, M.; Calore, M.; Occhi, G.; Carturan, E.; Lazzarini, E.; Cason, M.; et al. Homozygous Desmocollin-2 Mutations and Arrhythmogenic Cardiomyopathy. Am. J. Cardiol. 2015, 116, 1245–1251. [Google Scholar] [CrossRef]
- van Lint, F.H.M.; Murray, B.; Tichnell, C.; Zwart, R.; Amat, N.; Lekanne Deprez, R.H.; Dittmann, S.; Stallmeyer, B.; Calkins, H.; van der Smagt, J.J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ. Genom. Precis. Med. 2019, 12, e002467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Mura, I.E.; Bauce, B.; Nava, A.; Fanciulli, M.; Vazza, G.; Mazzotti, E.; Rigato, I.; De Bortoli, M.; Beffagna, G.; Lorenzon, A.; et al. Identification of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Lazzarini, E.; Rigato, I.; Celeghin, R.; De Bortoli, M.; Perazzolo Marra, M.; Cason, M.; Jongbloed, J.; Calore, M.; Rizzo, S.; et al. Large Genomic Rearrangements of Desmosomal Genes in Italian Arrhythmogenic Cardiomyopathy Patients. Circ. Arrhythm. Electrophysiol. 2017, 10. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Genomic Coordinates | Transcript | Kind of Mutation | Protein Change | MAF 1 | ACMG Classification |
|---|---|---|---|---|---|---|
| DSG2 | 18:29100928 | NM_001943.3 | Splice Site Mutation | Unknown | novel | Likely pathogenic |
| TBX3 | 12:115120963 | NM_016569.3 | Missense | p.M15V | novel | VUS |
| TRIM63 | 1:26380423 | NM_032588.3 | Missense | p.D338Y | 0.0002671 | VUS |
| PKP2 | 12:32949101 | NM_004572.3 | Missense | p.R811S | 0.00002828 | VUS |
| SDHA | 5:236628 | NM_004168.2 | Missense | p.A449V | 0.000003543 | VUS |
| TTN | 2:179650454 | NM_001267550.1 | Missense | p.D796N | 0.00007782 | VUS |
| LTBP2 | 14:74975348 | NM_000428.2 | Missense | p.A1204V | 0.0008008 | VUS |
| Gene | Genomic Coordinates | Transcript | Kind of Mutation | Protein or cDNA Change | MAF 1 | ACMG Classification |
|---|---|---|---|---|---|---|
| DSG2 | 18:29122796 | NM_001943.5 | nonsense | p.L772X | Novel | Likely pathogenic |
| TTN | 2:179497960 | NM_001256850.1 | missense | p.V12706A | 0.000008059 | VUS |
| PRDM16 | 1:3331216 | NM_022114.3 | unknown | c.2691+5G>A | 0.0002009 | Likely benign |
| VCL | 10:75849080 | NM_014000.3 | missense | p.S383R | Novel | VUS |
| SYNE2 | 14:64545208 | NM_182914.2 | missense | p.S3683T | 0.0002230 | VUS |
| BAG3 | 10:121431767 | NM_004281.4 | missense | p.R170W | 0.000004054 | VUS |
| PRDM16 | 1:3328948 | NM_022114.4 | missense | p.F729L | 0.0003899 | Likely benign |
| RYR1 | 19:39014565 | NM_000540.3 | missense | p.I3484T | 0.00005576 | VUS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodehl, A.; Meshkov, A.; Myasnikov, R.; Kiseleva, A.; Kulikova, O.; Klauke, B.; Sotnikova, E.; Stanasiuk, C.; Divashuk, M.; Pohl, G.M.; et al. Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset. Int. J. Mol. Sci. 2021, 22, 3786. https://doi.org/10.3390/ijms22073786
Brodehl A, Meshkov A, Myasnikov R, Kiseleva A, Kulikova O, Klauke B, Sotnikova E, Stanasiuk C, Divashuk M, Pohl GM, et al. Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset. International Journal of Molecular Sciences. 2021; 22(7):3786. https://doi.org/10.3390/ijms22073786
Chicago/Turabian StyleBrodehl, Andreas, Alexey Meshkov, Roman Myasnikov, Anna Kiseleva, Olga Kulikova, Bärbel Klauke, Evgeniia Sotnikova, Caroline Stanasiuk, Mikhail Divashuk, Greta Marie Pohl, and et al. 2021. "Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset" International Journal of Molecular Sciences 22, no. 7: 3786. https://doi.org/10.3390/ijms22073786