
Both Specific Endothelial and Proximal Tubular Adam17 Deletion Protect against Diabetic Nephropathy

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Confirmation of Adam17 Deletion on Endothelial and Renal Proximal Tubular Cells
2.2. Tubular Adam17 Knockout Protects from DN Hyperfiltration
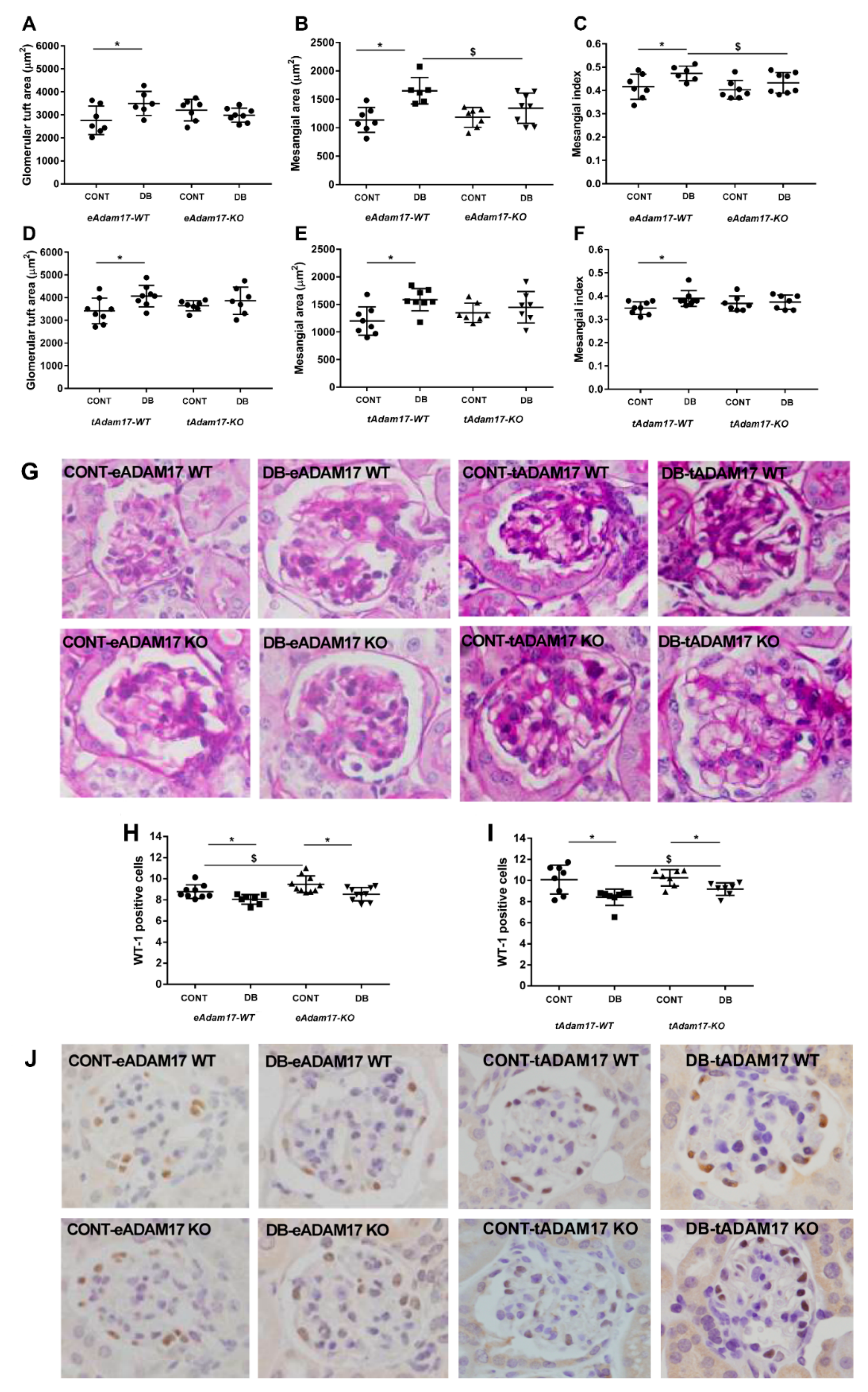
2.3. Diabetic Kidneys of Adam17-Deficient Mice at Either Proximal Tubular Cells or Endothelial Cells Are Protected from Matrix Mesangial Expansion
2.4. Adam17 Deletion in Proximal Tubular Cells Protects against Podocyte Loss
2.5. Adam17 Deletion in Proximal Tubular Cells Attenuates the Renin Angiotensin System
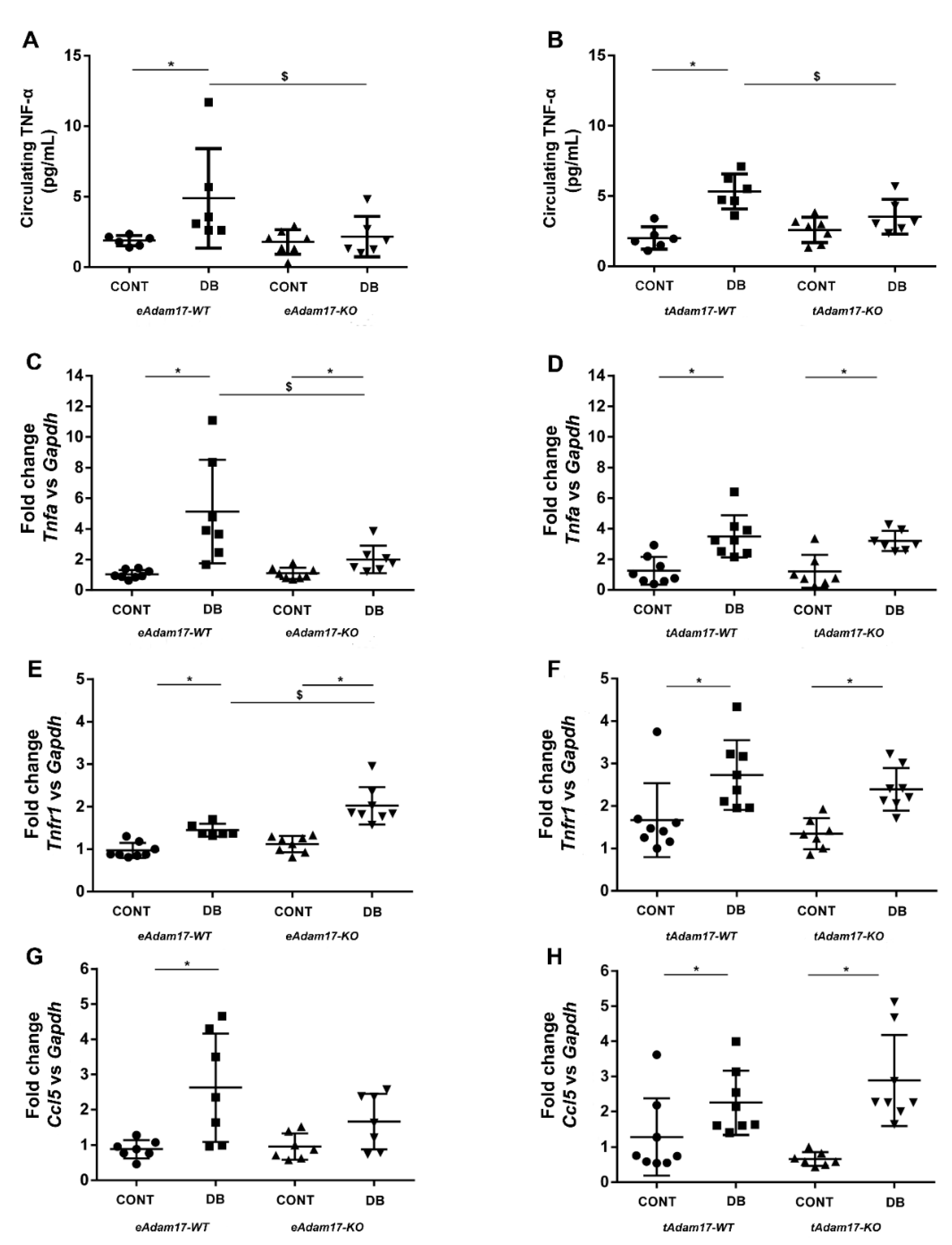
2.6. Adam17 Deletion Decreased Inflammation in DN
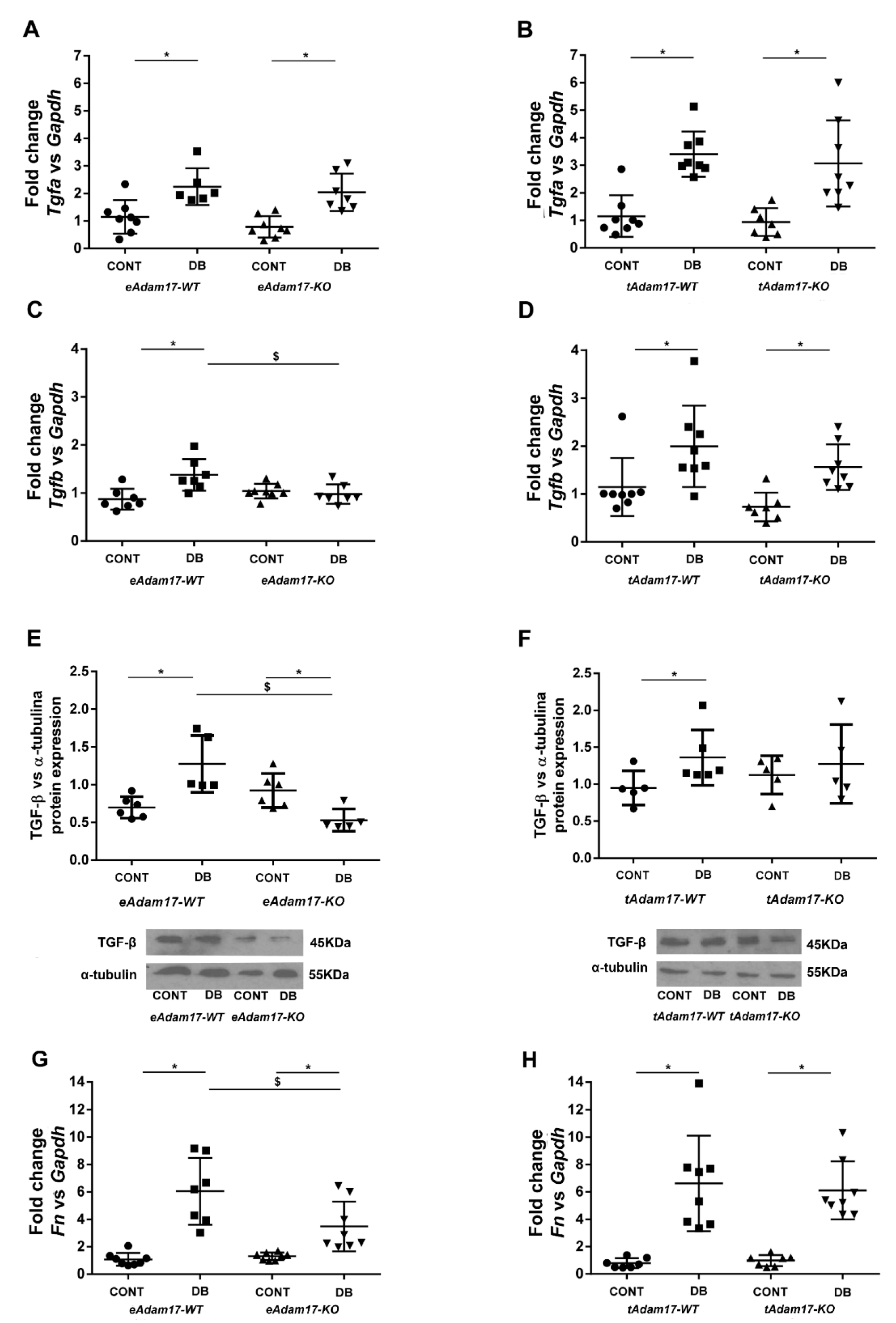
2.7. Diabetic Kidneys of Adam17 Deficient Mice in Either Proximal Tubular Cells or Endothelial Cells Are Protected from Renal Fibrosis
2.8. Fibrotic Markers Expression in Adam17-Deleted HKC-8 Spheroids
3. Discussion
4. Materials and Methods
4.1. Animal Models and Diabetes Induction
4.2. Generation of the Specific Endothelial or Tubular Adam17KO Mice
4.3. Urine Albumin Creatinine Ratio
4.4. Immunohistochemistry on OCT-Embedded Tissue
4.5. Immunohistochemistry on Paraffined-Embedded Tissue
4.6. Western Blot
4.7. Soluble TNF-α ELISA
4.8. 3D Cell Culture Set up and CRISPR/Cas9 Adam17 Silencing
4.9. Immunofluorescence in Tubular Spheroids
4.10. Gene Expression
4.11. ACE2 Enzymatic Activity
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riera, M.; Márquez, E.; Clotet, S.; Gimeno, J.; Roca-Ho, H.; Lloreta, J.; Juanpere, N.; Batlle, D.; Pascual, J.; Soler, M.J. Effect of insulin on ACE2 activity and kidney function in the non-obese diabetic mouse. PLoS ONE 2014, 9, e84683. [Google Scholar] [CrossRef]
- Diez-Sampedro, A.; Lenz, O.; Fornoni, A. Podocytopathy in diabetes: A metabolic and endocrine disorder. Am. J. Kidney Dis. 2011, 58, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Gross, J.L.; De Azevedo, M.J.; Silveiro, S.P.; Canani, L.H.; Caramori, M.L.; Zelmanovitz, T. Diabetic nephropathy: Diagnosis, prevention, and treatment. Diabetes Care 2005, 28, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Feldman, E.; Pennathur, S.; Kretzler, M.; Brosius, F.C. From fibrosis to sclerosis: Mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes 2008, 57, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Tesch, G.H. Macrophages and diabetic nephropathy. Semin. Nephrol. 2010, 30, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.P.; Sharma, K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev. Mol. Med. 2009, 11, e13. [Google Scholar] [CrossRef]
- Zhu, Y.; Casado, M.; Vaulont, S.; Sharma, K. Role of upstream stimulatory factors in regulation of renal transforming growth factor-β1. Diabetes 2005, 54, 1976–1984. [Google Scholar] [CrossRef] [Green Version]
- Salem, E.S.B.; Grobe, N.; Elased, K.M. Insulin treatment attenuates renal ADAM17 and ACE2 shedding in diabetic Akita mice. AJP Ren. Physiol. 2014, 306, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Chodavarapu, H.; Grobe, N.; Somineni, H.K.; Salem, E.S.B.; Madhu, M.; Elased, K.M. Rosiglitazone Treatment of Type 2 Diabetic db/db Mice Attenuates Urinary Albumin and Angiotensin Converting Enzyme 2 Excretion. PLoS ONE 2013, 8, e62833. [Google Scholar] [CrossRef]
- Riera, M.; Anguiano, L.; Clotet, S.; Roca-Ho, H.; Rebull, M.; Pascual, J.; Soler, M.J. Paricalcitol modulates ACE2 shedding and renal ADAM17 in NOD mice beyond proteinuria. Am. J. Physiol. Physiol. 2016, 310, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Xia, L.; Goldberg, H.J.; Lee, K.W.K.; Shah, A.; Stavar, L.; Masson, E.A.Y.; Momen, A.; Shikatani, E.A.; John, R.; et al. Inhibition of src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes 2013, 62, 3874–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, B.M.; Eid, A.A.; Gooz, M.; Barnes, J.L.; Gorin, Y.C.; Abboud, H.E. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. AJP Ren. Physiol. 2013, 305, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattenist, L.; Ochodnický, P.; Ahdi, M.; Claessen, N.; Leemans, J.C.; Satchell, S.C.; Florquin, S.; Gerdes, V.E.; Roelofs, J.J.T.H. Renal endothelial protein C receptor expression and shedding during diabetic nephropathy. J. Thromb. Haemost. 2016, 14, 1171–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kefaloyianni, E.; Muthu, M.L.; Kaeppler, J.; Sun, X.; Sabbisetti, V.; Chalaris, A.; Rose-John, S.; Wong, E.; Sagi, I.; Waikar, S.S.; et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight 2016, 1, e87023. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Morton, J.; Davidge, S.T.; Kassiri, Z. Loss of smooth muscle cell disintegrin and metalloproteinase 17 transiently suppresses angiotensin II-induced hypertension and end-organ damage. J. Mol. Cell. Cardiol. 2017, 103, 11–21. [Google Scholar] [CrossRef]
- Melenhorst, W.B.; Visser, L.; Timmer, A.; van den Heuvel, M.C.; Stegeman, C.A.; van Goor, H. ADAM17 upregulation in human renal disease: A role in modulating TGF-α availability? Am. J. Physiol. Physiol. 2009, 297, 781–790. [Google Scholar] [CrossRef]
- Kanasaki, K.; Taduri, G.; Koya, D. Diabetic nephropathy: The role of inflammation in fibroblast activation and kidney fibrosis. Front. Endocrinol. 2013, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Giricz, O.; Calvo, V.; Peterson, E.A.; Abouzeid, C.M.; Kenny, P.A. TACE-dependent TGFα shedding drives triple-negative breast cancer cell invasion. Int. J. Cancer 2013, 133, 2587–2595. [Google Scholar] [CrossRef]
- Menghini, R.; Fiorentino, L.; Casagrande, V.; Lauro, R.; Federici, M. The role of ADAM17 in metabolic inflammation. Atherosclerosis 2013, 228, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, Y.; Ning, G.; Deb, D.K.; Kong, J.; Yan, C.L. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proc. Natl. Acad. Sci. USA 2008, 105, 15896–15901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clotet-Freixas, S.; Soler, M.J.; Palau, V.; Anguiano, L.; Gimeno, J.; Konvalinka, A.; Pascual, J.; Riera, M. Sex dimorphism in ANGII-mediated crosstalk between ACE2 and ACE in diabetic nephropathy. Lab. Investig. 2018, 98, 1237–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.W.; Oudit, G.Y.; Reich, H.; Kassiri, Z.; Zhou, J.; Liu, Q.C.; Backx, P.H.; Penninger, J.M.; Herzenberg, A.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am. J. Pathol. 2007, 171, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudit, G.Y.; Liu, G.C.; Zhong, J.C.; Basu, R.; Chow, F.L.; Zhou, J.; Loibner, H.; Janzek, E.; Schuster, M.; Penninger, J.M.; et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 2010, 59, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassy-Prigent, C.; Heudes, D.; Mandet, C.; Bélair, M.F.; Michel, O.; Perdereau, B.; Bariéty, J.; Bruneval, P. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes 2000, 49, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Song, Z.; Zhou, M.; Yang, Y.; Zhao, Y.; Liu, B.; Zhang, X. Infiltrating macrophages in diabetic nephropathy promote podocytes apoptosis via TNF-α-ROS-p38MAPK pathway. Oncotarget 2017, 8, 53276–53287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clotet, S.; Soler, M.J.; Rebull, M.; Gimeno, J.; Gurley, S.B.; Pascual, J.; Riera, M. Gonadectomy prevents the increase in blood pressure and glomerular injury in angiotensin-converting enzyme 2 knockout diabetic male mice. Effects on renin-angiotensin system. J. Hypertens. 2016, 34, 1752–1765. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jiang, S.; Tang, X.; Cai, L.; Epstein, P.N.; Cheng, Y.; Sun, W.; Xu, Z.; Tan, Y. Sex differences in progression of diabetic nephropathy in OVE26 type 1 diabetic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165589. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular sirt1 attenuates diabetic albuminuria by epigenetically suppressing claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef] [Green Version]
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activity in diabetic mice. Diabetes 2006, 55, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Yamaleyeva, L.M.; Gilliam-Davis, S.; Almeida, I.; Brosnihan, K.B.; Lindsey, S.H.; Chappell, M.C. Differential regulation of circulating and renal ACE2 and ACE in hypertensive mRen2.Lewis rats with early-onset diabetes. AJP Ren. Physiol. 2012, 302, 1374–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omote, K.; Gohda, T.; Murakoshi, M.; Sasaki, Y.; Kazuno, S.; Fujimura, T.; Ishizaka, M.; Sonoda, Y.; Tomino, Y. Role of the TNF pathway in the progression of diabetic nephropathy in KK-Ay mice. Am. J. Physiol.-Ren. Physiol. 2014, 306, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Kefaloyianni, E.; Raja, M.R.K.; Schumacher, J.; Muthu, M.L.; Krishnadoss, V.; Waikar, S.S.; Herrlich, A. Proximal tubule-derived amphiregulin amplifies and integrates profibrotic EGF receptor signals in kidney fibrosis. J. Am. Soc. Nephrol. 2019, 30, 2370–2383. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Harris, R.C. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- McLennan, S.V.; Fisher, E.; Martell, S.Y.; Death, A.K.; Williams, P.F.; Lyons, J.G.; Yue, D.K. Effects of glucose on matrix metalloproteinase and plasmin activities in mesangial cells: Possible role in diabetic nephropathy. Kidney Int. Suppl. 2000, 77, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Peng, F.; Zhang, B.; Ingram, A.J.; Kelly, D.J.; Gilbert, R.E.; Gao, B.; Krepinsky, J.C. PKC-1 Mediates Glucose-Induced Akt Activation and TGF- 1 Upregulation in Mesangial Cells. J. Am. Soc. Nephrol. 2009, 20, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Overstreet, J.M.; Wang, Y.; Wang, X.; Niu, A.; Gewin, L.S.; Yao, B.; Harris, R.C.; Zhang, M.Z. Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. FASEB J. 2017, 31, 4407–4421. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.L.; Gough, P.J.; Chang, C.A.; Chan, C.K.; Frey, J.M.; Liu, Y.; Braun, K.R.; Chin, M.T.; Wight, T.N.; Raines, E.W. Endothelial deletion of ADAM17 in mice results in defective remodeling of the semilunar valves and cardiac dysfunction in adults. Mech. Dev. 2013, 130, 272–289. [Google Scholar] [CrossRef] [PubMed]
- Claxton, S.; Kostourou, V.; Jadeja, S.; Chambon, P.; Hodivala-Dilke, K.; Fruttiger, M. Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis 2008, 46, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Tomaszewski, J.E.; Haase, V.H. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006, 66, 2576–2583. [Google Scholar] [CrossRef] [Green Version]
- Patel, Y.M.; Yun, J.S.; Liu, J.; McGrane, M.M.; Hanson, R.W. An analysis of regulatory elements in the phosphoenolpyruvate carboxykinase (GTP) gene which are responsible for its tissue-specific expression and metabolic control in transgenic mice. J. Biol. Chem. 1994, 269, 5619–5628. [Google Scholar] [CrossRef]
- Junqueira, L.C.U.; Bignolas, G.; Brentani, R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B.; Mohr, M.A.; Ponti, A.; Emmert, M.Y.; Weibel, F.; Hoerstrup, S.P.; Moll, S.; Certa, U.; Prunotto, M.; Pantazis, P. Monitoring and manipulating cellular crosstalk during kidney fibrosis inside a 3D in vitro co-culture. Sci. Rep. 2017, 7, 14490. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endothelial ADAM17 Model | ||||
|---|---|---|---|---|
| WT-CONT | WT-DB | KO-CONT | KO-DB | |
| Fasting blood glucose (mg/dL) | 175.44 ± 20.26 | 430.29 ± 31.49 * | 185.30 ± 15.68 | 485.9 ± 86.91 * |
| Body weight (g) | 34.61 ± 5.37 | 26.35 ± 3.61 * | 33.09 ± 3.34 | 24.16 ± 2.70 * |
| KW/BW | 0.97 ± 0.19 | 1.45 ± 0.31 * | 1.00 ± 0.11 | 1.41 ± 0.16 * |
| Proximal Tubular ADAM17 Model | ||||
| WT-CONT | WT-DB | KO-CONT | KO-DB | |
| Fasting blood glucose (mg/dL) | 177.13 ± 6.45 | 397.33 ± 17.89 * | 202.67 ± 5.92 | 362.58 ± 27.54 * |
| Body weight (g) | 34.38 ± 4.45 | 28.35 ± 3.27 * | 34.94 ± 4.40 | 27.87 ± 2.15 * |
| KW/BW | 1.07 ± 0.17 | 1.35 ± 0.15 * | 1.00 ± 0.10 | 1.25 ± 0.20 *$ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palau, V.; Nugraha, B.; Benito, D.; Pascual, J.; Emmert, M.Y.; Hoerstrup, S.P.; Riera, M.; Soler, M.J. Both Specific Endothelial and Proximal Tubular Adam17 Deletion Protect against Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 5520. https://doi.org/10.3390/ijms22115520
Palau V, Nugraha B, Benito D, Pascual J, Emmert MY, Hoerstrup SP, Riera M, Soler MJ. Both Specific Endothelial and Proximal Tubular Adam17 Deletion Protect against Diabetic Nephropathy. International Journal of Molecular Sciences. 2021; 22(11):5520. https://doi.org/10.3390/ijms22115520
Chicago/Turabian StylePalau, Vanesa, Bramasta Nugraha, David Benito, Julio Pascual, Maximilian Y. Emmert, Simon P. Hoerstrup, Marta Riera, and Maria José Soler. 2021. "Both Specific Endothelial and Proximal Tubular Adam17 Deletion Protect against Diabetic Nephropathy" International Journal of Molecular Sciences 22, no. 11: 5520. https://doi.org/10.3390/ijms22115520