
Carfilzomib Promotes the Unfolded Protein Response and Apoptosis in Cetuximab-Resistant Colorectal Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
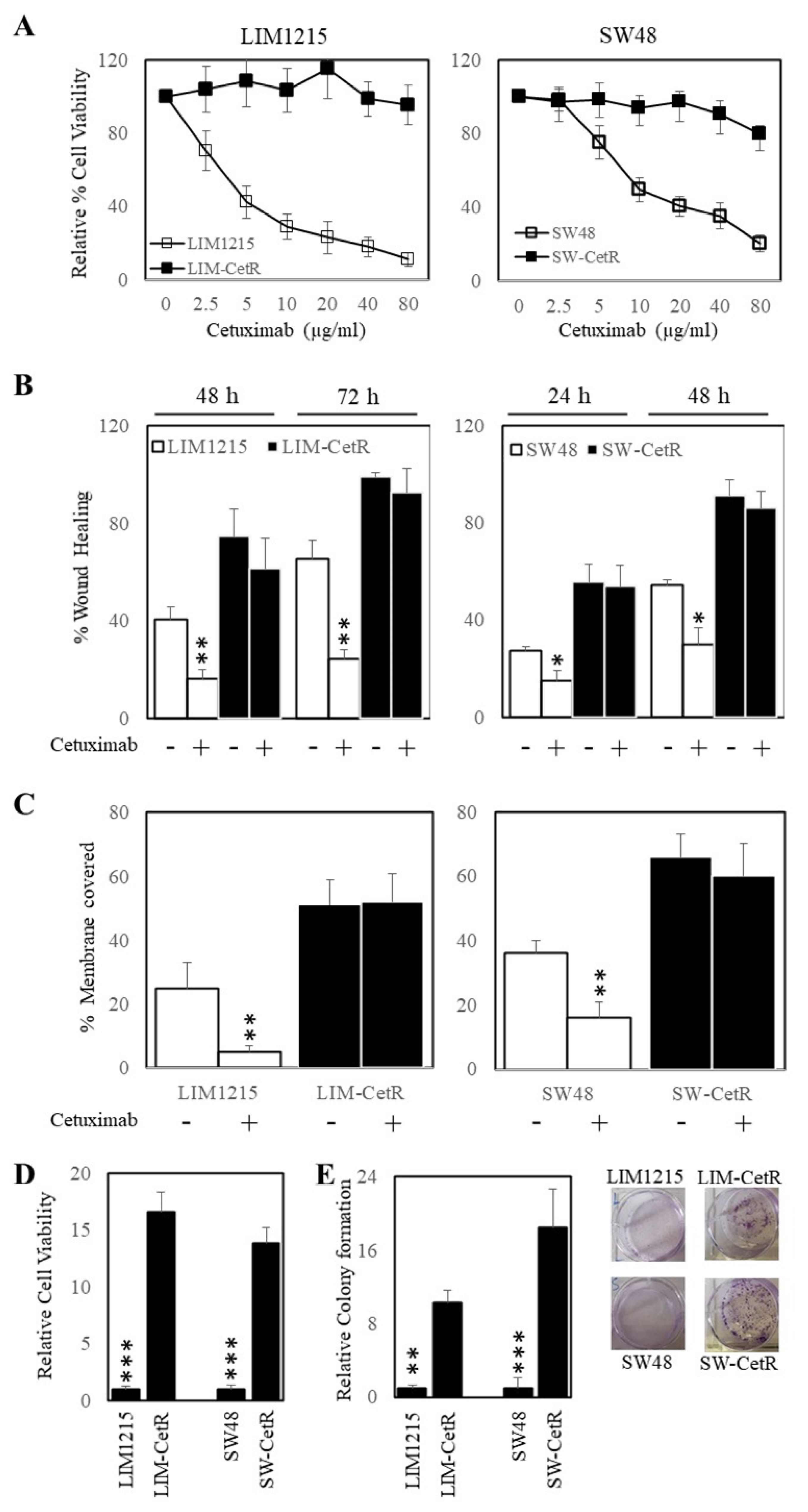
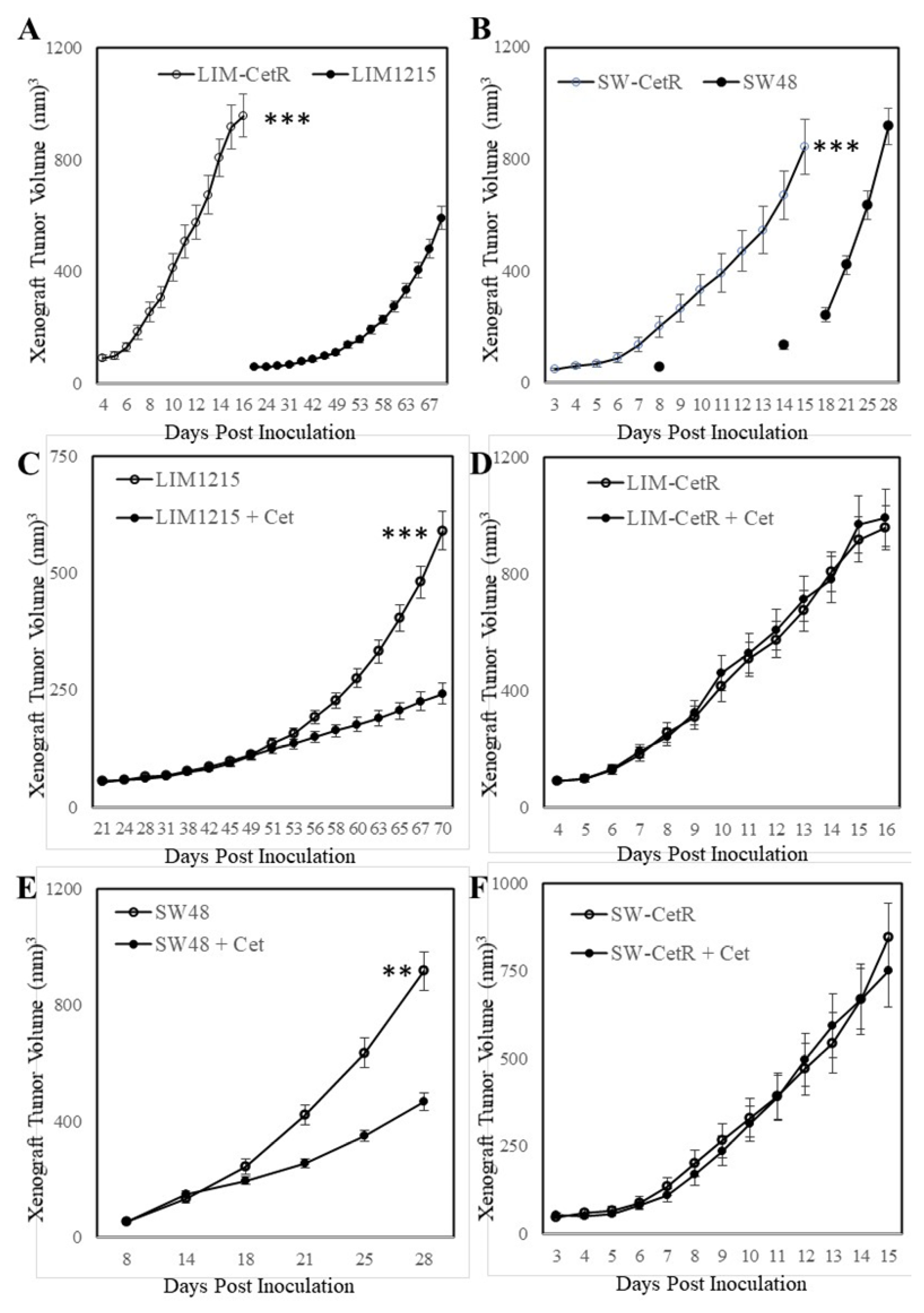
3.1. Generation and Characterisation of Acquired Cetuximab-Resistant Cell Lines
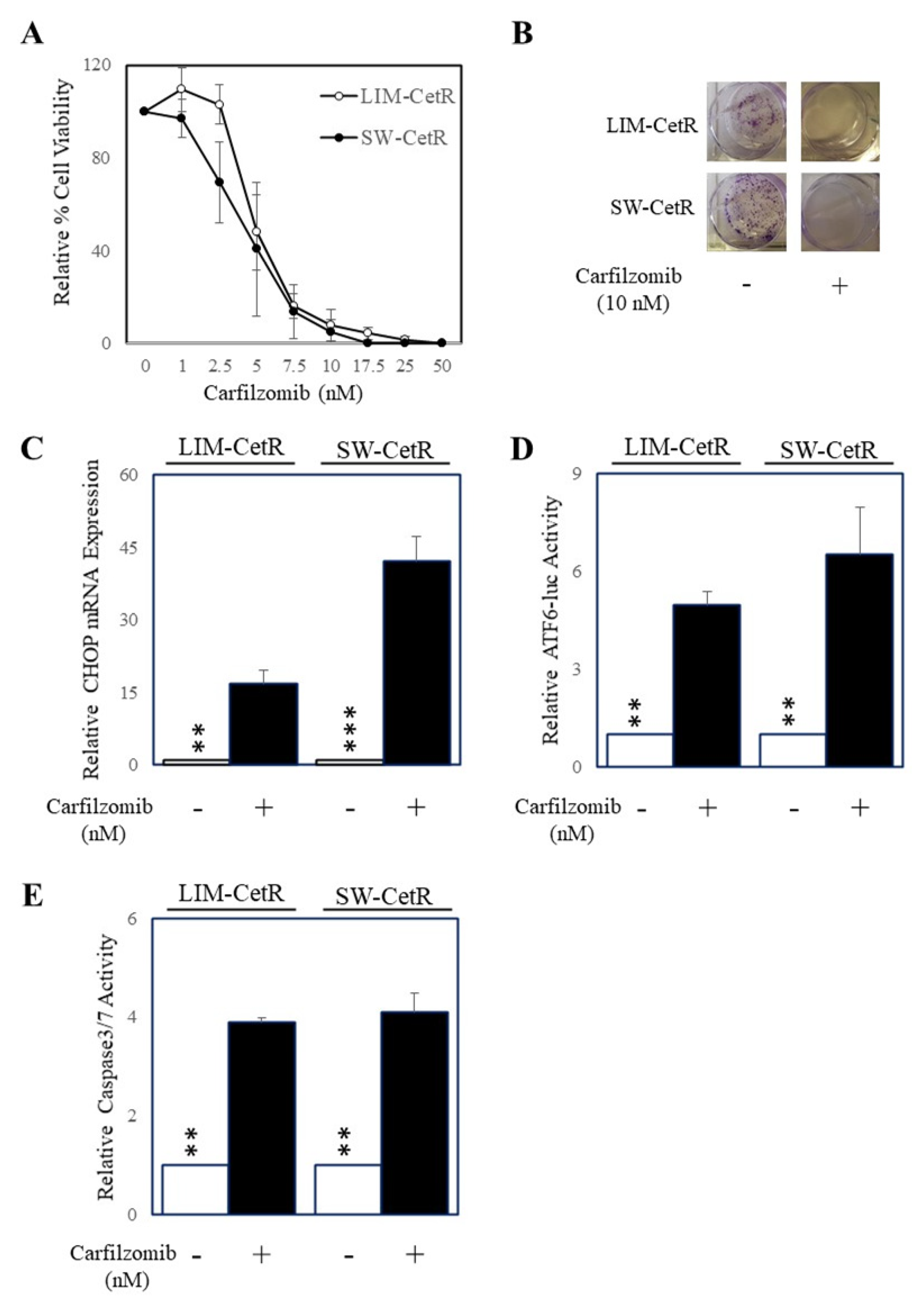
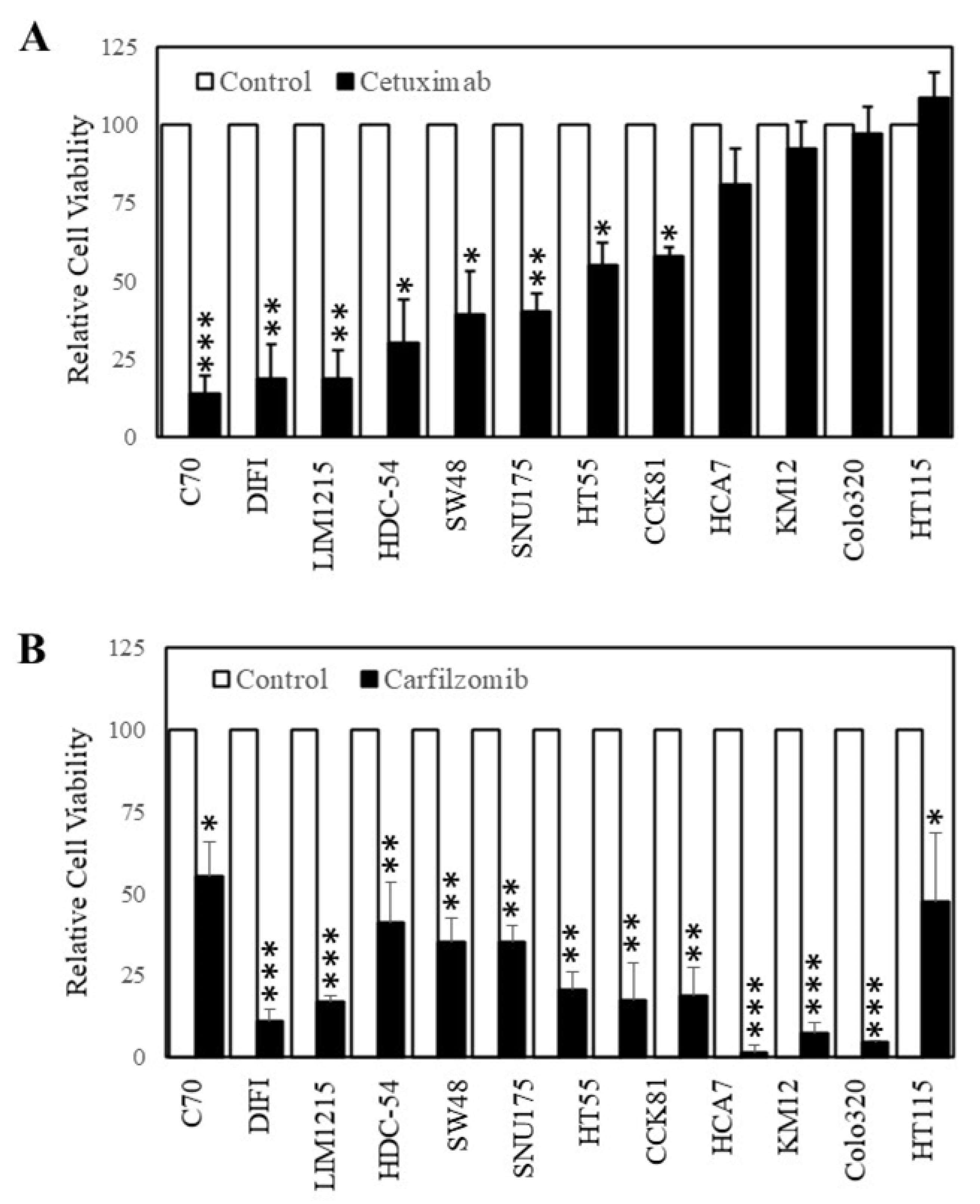
3.2. Carfilzomib Inhibits Colorectal Cancer Cells with Acquired Resistance to Cetuximab
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Andre, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Hoff, P.M.; Harper, P.; Bukowski, R.M.; Cunningham, D.; Dufour, P.; Graeven, U.; Lokich, J.; Madajewicz, S.; Maroun, J.A.; et al. Oral capecitabine vs intravenous 5-fluorouracil and leucovorin: Integrated efficacy data and novel analyses from two large, randomised, phase III trials. Br. J. Cancer 2004, 90, 1190–1197. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Alberts, S.R.; Sargent, D.J.; Nair, S.; Mahoney, M.R.; Mooney, M.; Thibodeau, S.N.; Smyrk, T.C.; Sinicrope, F.A.; Chan, E.; Gill, S.; et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: A randomized trial. JAMA 2012, 307, 1383–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gramont, A.; Van Cutsem, E.; Schmoll, H.J.; Tabernero, J.; Clarke, S.; Moore, M.J.; Cunningham, D.; Cartwright, T.H.; Hecht, J.R.; Rivera, F.; et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): A phase 3 randomised controlled trial. Lancet Oncol. 2012, 13, 1225–1233. [Google Scholar] [CrossRef]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.J.; Oliveira, J.; Group, E.G.W. Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann. Oncol. 2008, 19, ii33–ii34. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Sartore-Bianchi, A.; Di Nicolantonio, F.; Zanon, C.; Moroni, M.; Veronese, S.; Siena, S.; Bardelli, A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007, 67, 2643–2648. [Google Scholar] [CrossRef] [Green Version]
- Datta, K.; Suman, S.; Kumar, S.; Fornace, A.J., Jr. Colorectal Carcinogenesis, Radiation Quality, and the Ubiquitin-Proteasome Pathway. J. Cancer 2016, 7, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yu, Y.; Wang, Z.; Wang, H.; Bieerkehazhi, S.; Zhao, Y.; Suzuk, L.; Zhang, H. Second-generation proteasome inhibitor carfilzomib enhances doxorubicin-induced cytotoxicity and apoptosis in breast cancer cells. Oncotarget 2016, 7, 73697–73710. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Garcia, T.A.; Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Mitsiades, C.; Anderson, K.C.; Richardson, P.G. The power of proteasome inhibition in multiple myeloma. Expert Rev. Proteomics 2018, 15, 1033–1052. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.; Klaus, J.O.; Stockerl-Goldstein, K. Carfilzomib: A second-generation proteasome inhibitor for the treatment of multiple myeloma. Am. J. Health Syst. Pharm. 2015, 72, 353–360. [Google Scholar] [CrossRef]
- Jakubowiak, A.J. Evolution of carfilzomib dose and schedule in patients with multiple myeloma: A historical overview. Cancer Treat. Rev. 2014, 40, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Arastu-Kapur, S.; Anderl, J.L.; Kraus, M.; Parlati, F.; Shenk, K.D.; Lee, S.J.; Muchamuel, T.; Bennett, M.K.; Driessen, C.; Ball, A.J.; et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: A link to clinical adverse events. Clin. Cancer Res. 2011, 17, 2734–2743. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Su, G.; Li, J.; Liao, J.; Chen, S.; Huang, C.; Liu, F.; Chen, Q.; Ye, Y. Enhanced anti-colorectal cancer effects of carfilzomib combined with CPT-11 via downregulation of nuclear factor-kappaB in vitro and in vivo. Int J. Oncol. 2014, 45, 995–1010. [Google Scholar] [CrossRef] [Green Version]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef]
- Forsythe, N.; Refaat, A.; Javadi, A.; Khawaja, H.; Weir, J.A.; Emam, H.; Allen, W.L.; Burkamp, F.; Popovici, V.; Jithesh, P.V.; et al. The Unfolded Protein Response: A Novel Therapeutic Target for Poor Prognostic BRAF Mutant Colorectal Cancer. Mol. Cancer Ther. 2018, 17, 1280–1290. [Google Scholar] [CrossRef] [Green Version]
- Zecchin, D.; Boscaro, V.; Medico, E.; Barault, L.; Martini, M.; Arena, S.; Cancelliere, C.; Bartolini, A.; Crowley, E.H.; Bardelli, A.; et al. BRAF V600E is a determinant of sensitivity to proteasome inhibitors. Mol. Cancer Ther. 2013, 12, 2950–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ri, M. Endoplasmic-reticulum stress pathway-associated mechanisms of action of proteasome inhibitors in multiple myeloma. Int. J. Hematol. 2016, 104, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Penaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef]
- Sansalone, L.; Veliz, E.A.; Myrthil, N.G.; Stathias, V.; Walters, W.; Torrens, I.I.; Schurer, S.C.; Vanni, S.; Leblanc, R.M.; Graham, R.M. Novel Curcumin Inspired Bis-Chalcone Promotes Endoplasmic Reticulum Stress and Glioblastoma Neurosphere Cell Death. Cancers 2019, 11, 357. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Truscott, K.N.; Bezawork-Geleta, A.; Dougan, D.A. Unfolded protein responses in bacteria and mitochondria: A central role for the ClpXP machine. IUBMB Life 2011, 63, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masud, A.; Mohapatra, A.; Lakhani, S.A.; Ferrandino, A.; Hakem, R.; Flavell, R.A. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J. Biol. Chem. 2007, 282, 14132–14139. [Google Scholar] [CrossRef] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Zmijewska, A.A.; Jope, R.S. HSP105 interacts with GRP78 and GSK3 and promotes ER stress-induced caspase-3 activation. Cell Signal. 2008, 20, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Lamothe, B.; Wierda, W.G.; Keating, M.J.; Gandhi, V. Carfilzomib Triggers Cell Death in Chronic Lymphocytic Leukemia by Inducing Proapoptotic and Endoplasmic Reticulum Stress Responses. Clin. Cancer Res. 2016, 22, 4712–4726. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Lim, S.H.; Yu, A.R.; Hwang, C.Y.; Kang, I.; Yeo, E.J. Carfilzomib in Combination with Bortezomib Enhances Apoptotic Cell Death in B16-F1 Melanoma Cells. Biology 2021, 10, 153. [Google Scholar] [CrossRef]
- Qian, G.; Yao, W.; Zhang, S.; Bajpai, R.; Hall, W.D.; Shanmugam, M.; Lonial, S.; Sun, S.Y. Co-inhibition of BET and proteasome enhances ER stress and Bim-dependent apoptosis with augmented cancer therapeutic efficacy. Cancer Lett. 2018, 435, 44–54. [Google Scholar] [CrossRef]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.H.; Putoczki, T.L.; Lou, J.; Hinde, E.; Hollande, F.; Giraud, J.; Stylli, S.S.; Paradiso, L.; Zhu, H.J.; Sieber, O.M.; et al. Ponatinib Inhibits Multiple Signaling Pathways Involved in STAT3 Signaling and Attenuates Colorectal Tumor Growth. Cancers 2018, 10, 526. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Luwor, R.B.; Johns, T.G.; Murone, C.; Huang, H.J.; Cavenee, W.K.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001, 61, 5355–5361. [Google Scholar] [PubMed]
- Benavente, S.; Huang, S.; Armstrong, E.A.; Chi, A.; Hsu, K.T.; Wheeler, D.L.; Harari, P.M. Establishment and characterization of a model of acquired resistance to epidermal growth factor receptor targeting agents in human cancer cells. Clin. Cancer Res. 2009, 15, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Boeckx, C.; Blockx, L.; de Beeck, K.O.; Limame, R.; Camp, G.V.; Peeters, M.; Vermorken, J.B.; Specenier, P.; Wouters, A.; Baay, M.; et al. Establishment and characterization of cetuximab resistant head and neck squamous cell carcinoma cell lines: Focus on the contribution of the AP-1 transcription factor. Am. J. Cancer Res. 2015, 5, 1921–1938. [Google Scholar] [PubMed]
- Ciardiello, F.; Bianco, R.; Caputo, R.; Caputo, R.; Damiano, V.; Troiani, T.; Melisi, D.; De Vita, F.; De Placido, S.; Bianco, A.R.; et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin. Cancer Res. 2004, 10, 784–793. [Google Scholar] [CrossRef] [Green Version]
- De Pauw, I.; Lardon, F.; Van den Bossche, J.; Baysal, H.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Wouters, A. Overcoming Intrinsic and Acquired Cetuximab Resistance in RAS Wild-Type Colorectal Cancer: An In Vitro Study on the Expression of HER Receptors and the Potential of Afatinib. Cancers 2019, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Iida, M.; Brand, T.M.; Starr, M.M.; Huppert, E.J.; Luthar, N.; Bahrar, H.; Coan, J.P.; Pearson, H.E.; Salgia, R.; Wheeler, D.L. Overcoming acquired resistance to cetuximab by dual targeting HER family receptors with antibody-based therapy. Mol. Cancer 2014, 13, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Li, X.; Liang, K.; Luwor, R.; Siddik, Z.H.; Mills, G.B.; Mendelsohn, J.; Fan, Z. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007, 67, 8240–8247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, S.; Martini, G.; Rinaldi, B.; Martinelli, E.; Donniacuo, M.; Berrino, L.; Vitagliano, D.; Morgillo, F.; Barra, G.; De Palma, R.; et al. Primary and Acquired Resistance of Colorectal Cancer to Anti-EGFR Monoclonal Antibody Can Be Overcome by Combined Treatment of Regorafenib with Cetuximab. Clin. Cancer Res. 2015, 21, 2975–2983. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | LIM1215 | LIM-CetR | SW48 | SW-CetR |
|---|---|---|---|---|
| DMSO control | 100.00 | 100.00 | 100.00 | 100.00 |
| Acadesine | 119.77 | 95.23 | 93.28 | 100.77 |
| Afatinib | 18.78 | 93.71 | 55.16 | 97.41 |
| Aminosalicylate sodium | 132.93 | 97.98 | 123.89 | 101.23 |
| Apatinib | 180.49 | 98.62 | 141.19 | 105.17 |
| Axitinib | 136.60 | 97.67 | 130.47 | 104.04 |
| Azelnidipine | 148.98 | 95.80 | 127.15 | 104.25 |
| Bisoprolol | 148.70 | 97.21 | 128.14 | 107.63 |
| Bosutinib | 150.12 | 94.05 | 89.78 | 114.99 |
| Cabozantinib | 168.01 | 96.34 | 132.71 | 101.71 |
| Carfilzomib | 0.17 | 0.34 | 0.36 | 0.02 |
| Crizotinib | 121.20 | 99.78 | 89.24 | 93.82 |
| Dasatinib | 66.63 | 72.63 | 38.00 | 55.49 |
| Dexamethasone | 116.76 | 100.73 | 103.43 | 97.36 |
| Diclofenac | 123.72 | 99.24 | 119.04 | 98.46 |
| Enzalutamide | 80.98 | 98.58 | 59.50 | 99.57 |
| Erlotinib HCl | 126.71 | 98.35 | 107.96 | 99.11 |
| Everolimus | 81.51 | 74.39 | 73.61 | 48.96 |
| Evista | 133.94 | 96.29 | 105.28 | 95.96 |
| Gefitinib | 61.67 | 86.68 | 55.08 | 88.92 |
| Ibrutinib | 23.89 | 95.84 | 52.37 | 96.63 |
| Imatinib mesylate | 117.71 | 100.22 | 117.19 | 96.93 |
| Irinotecan | 83.54 | 62.15 | 34.01 | 53.35 |
| Lapatinib | 32.16 | 96.55 | 66.56 | 97.60 |
| Lenalidomide | 130.12 | 97.45 | 123.61 | 91.34 |
| Masitinib | 131.84 | 95.78 | 93.71 | 89.78 |
| Niclosamide | 133.66 | 94.45 | 130.87 | 92.32 |
| Nilotinib | 183.86 | 82.47 | 191.23 | 88.46 |
| Nystatin (Mycostatin) | 126.32 | 93.01 | 154.25 | 92.84 |
| Pazopanib HCl | 91.72 | 80.10 | 80.85 | 88.57 |
| Ponatinib | 119.96 | 56.59 | 108.29 | 69.50 |
| Regorafenib (BAY 73-4506) | 107.05 | 92.17 | 98.12 | 88.42 |
| Resveratrol | 91.12 | 97.65 | 109.41 | 90.56 |
| Ruxolitinib (INCB018424) | 100.87 | 93.08 | 104.89 | 85.55 |
| Temsirolimus (Torisel) | 76.89 | 66.05 | 74.76 | 52.96 |
| Tofacitinib citrate (CP-690550) | 101.68 | 94.48 | 105.54 | 86.10 |
| Vemurafenib | 106.57 | 59.66 | 121.68 | 44.70 |
| Vismodegib | 92.23 | 85.23 | 112.89 | 73.74 |
| Vorinostat (SAHA) | 87.65 | 80.84 | 116.45 | 74.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zulkifli, A.; Tan, F.H.; Areeb, Z.; Stuart, S.F.; Gomez, J.; Paradiso, L.; Luwor, R.B. Carfilzomib Promotes the Unfolded Protein Response and Apoptosis in Cetuximab-Resistant Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 7114. https://doi.org/10.3390/ijms22137114
Zulkifli A, Tan FH, Areeb Z, Stuart SF, Gomez J, Paradiso L, Luwor RB. Carfilzomib Promotes the Unfolded Protein Response and Apoptosis in Cetuximab-Resistant Colorectal Cancer. International Journal of Molecular Sciences. 2021; 22(13):7114. https://doi.org/10.3390/ijms22137114
Chicago/Turabian StyleZulkifli, Ahmad, Fiona H. Tan, Zammam Areeb, Sarah F. Stuart, Juliana Gomez, Lucia Paradiso, and Rodney B. Luwor. 2021. "Carfilzomib Promotes the Unfolded Protein Response and Apoptosis in Cetuximab-Resistant Colorectal Cancer" International Journal of Molecular Sciences 22, no. 13: 7114. https://doi.org/10.3390/ijms22137114