Conformational Insights into the Control of CNF1 Toxin Activity by Peptidyl-Prolyl Isomerization: A Molecular Dynamics Perspective

 and
and
Abstract
:1. Introduction
2. Results
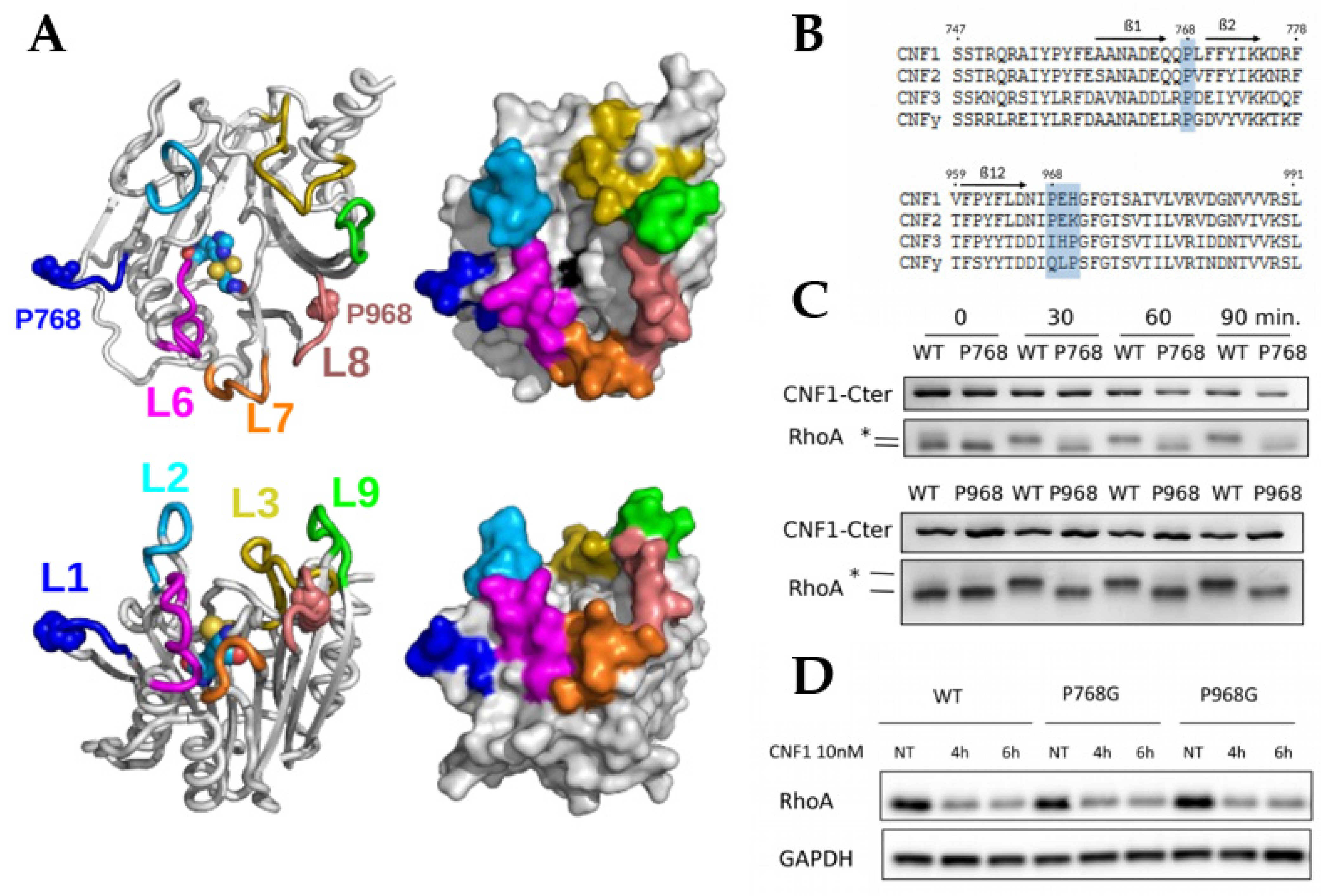
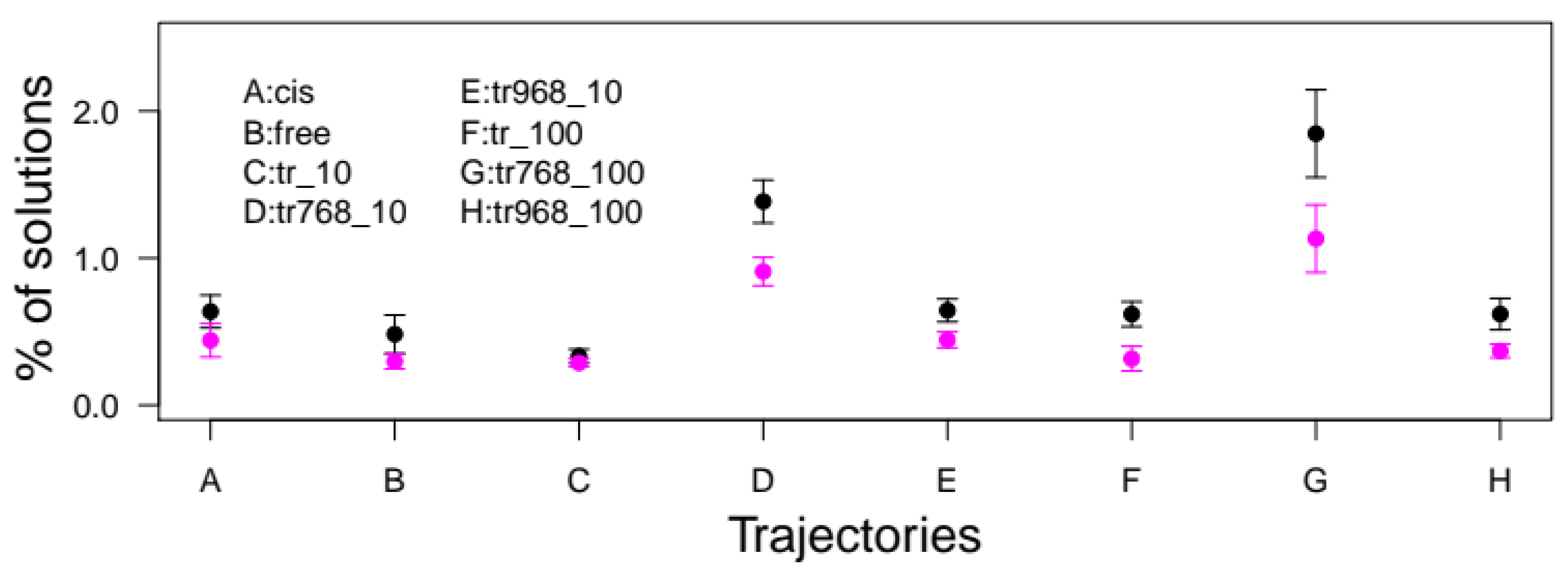
2.1. Essential Roles of P768 and P968 in CNF1-Mediated Deamidation of RhoA In Vitro
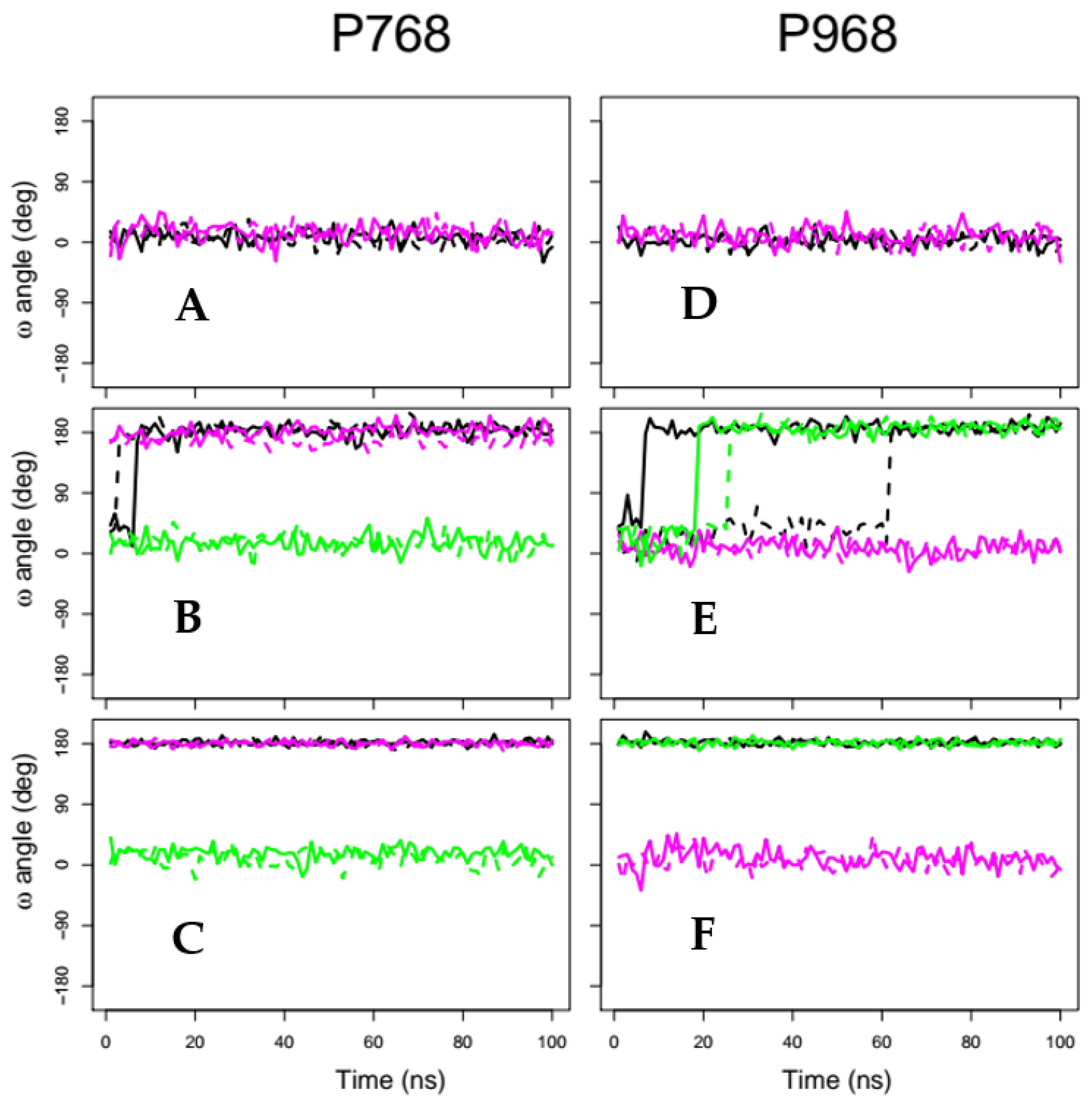
2.2. X-Pro Imide Bond cis-trans Isomerization and Tertiary Structure of CNF1CD
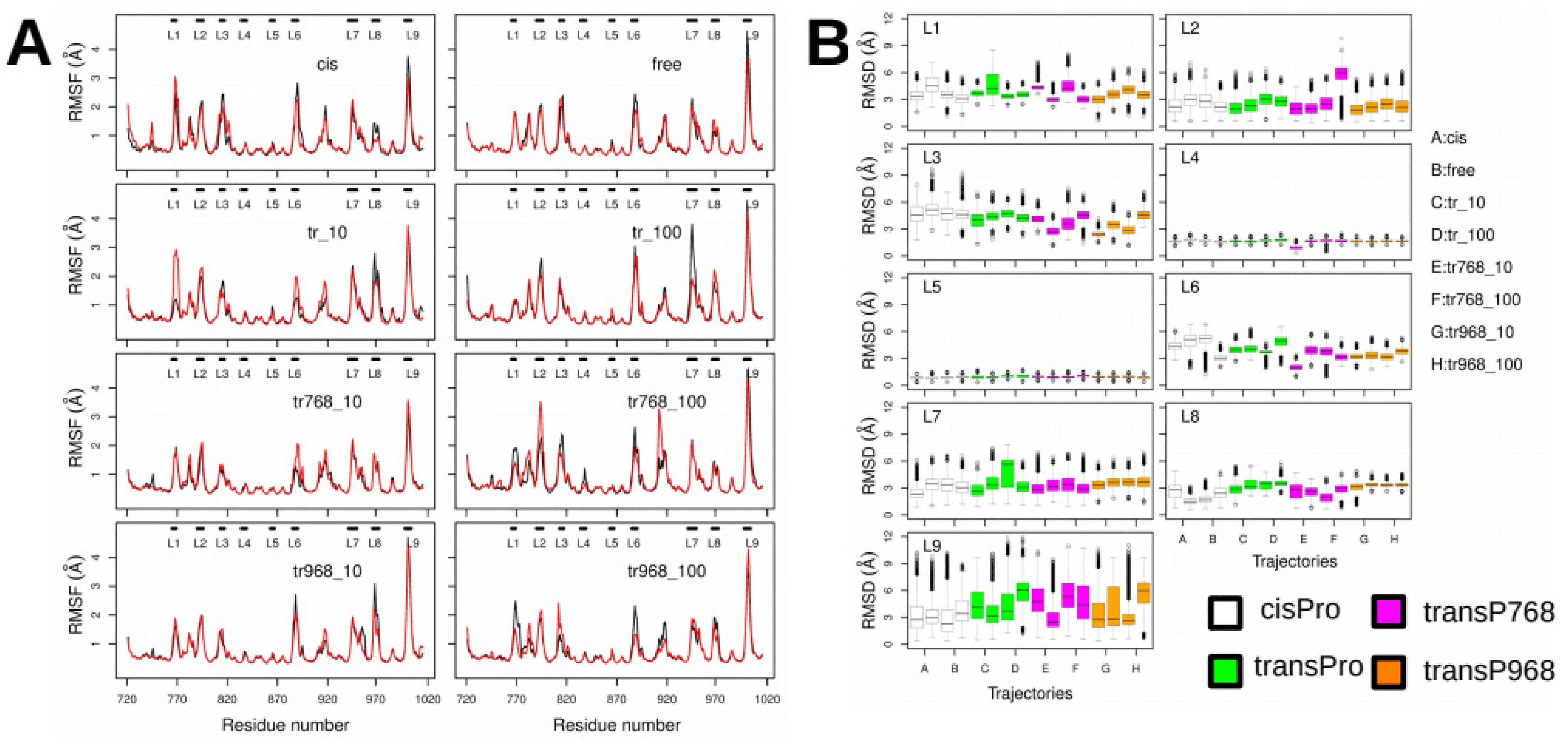
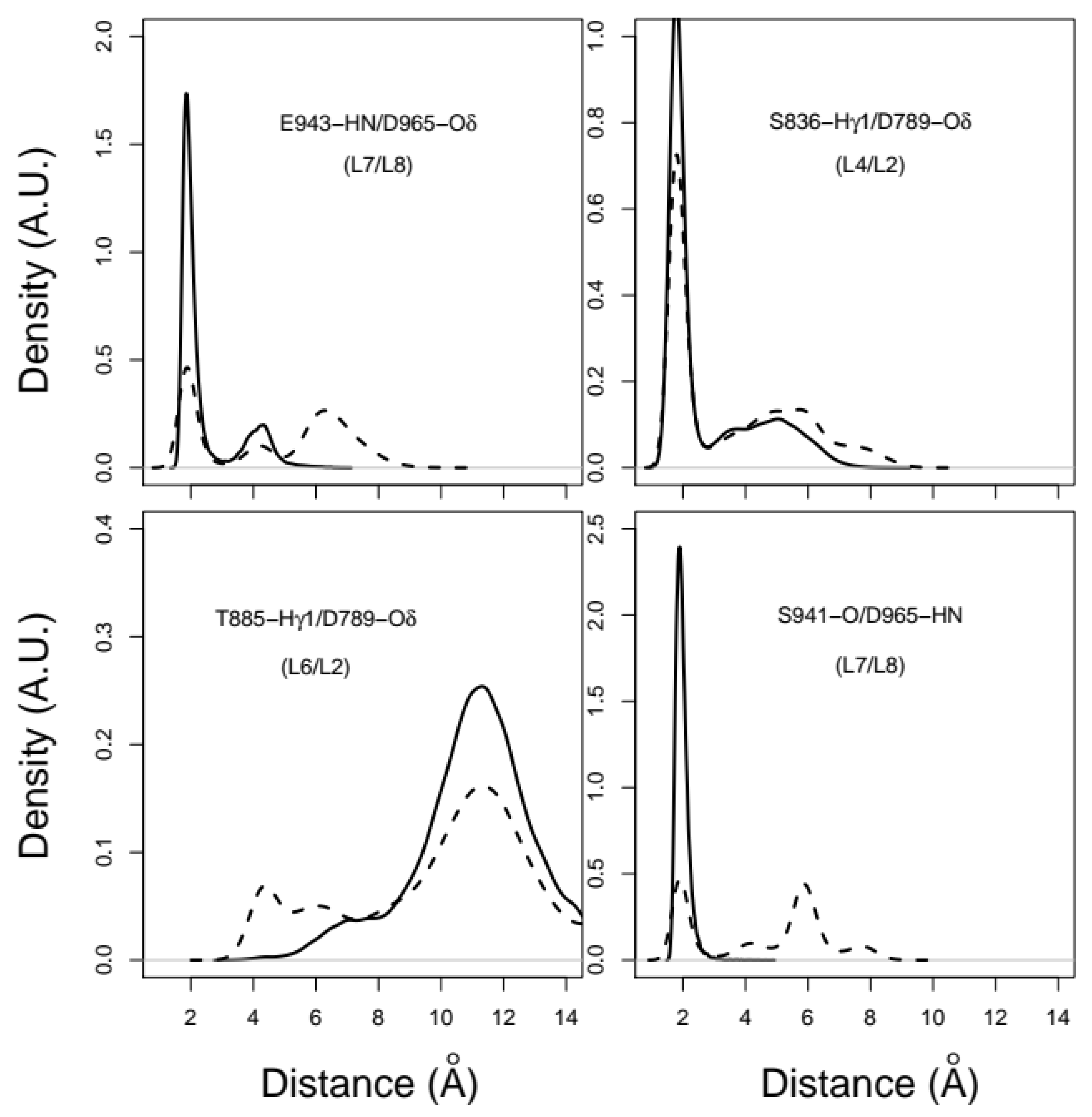
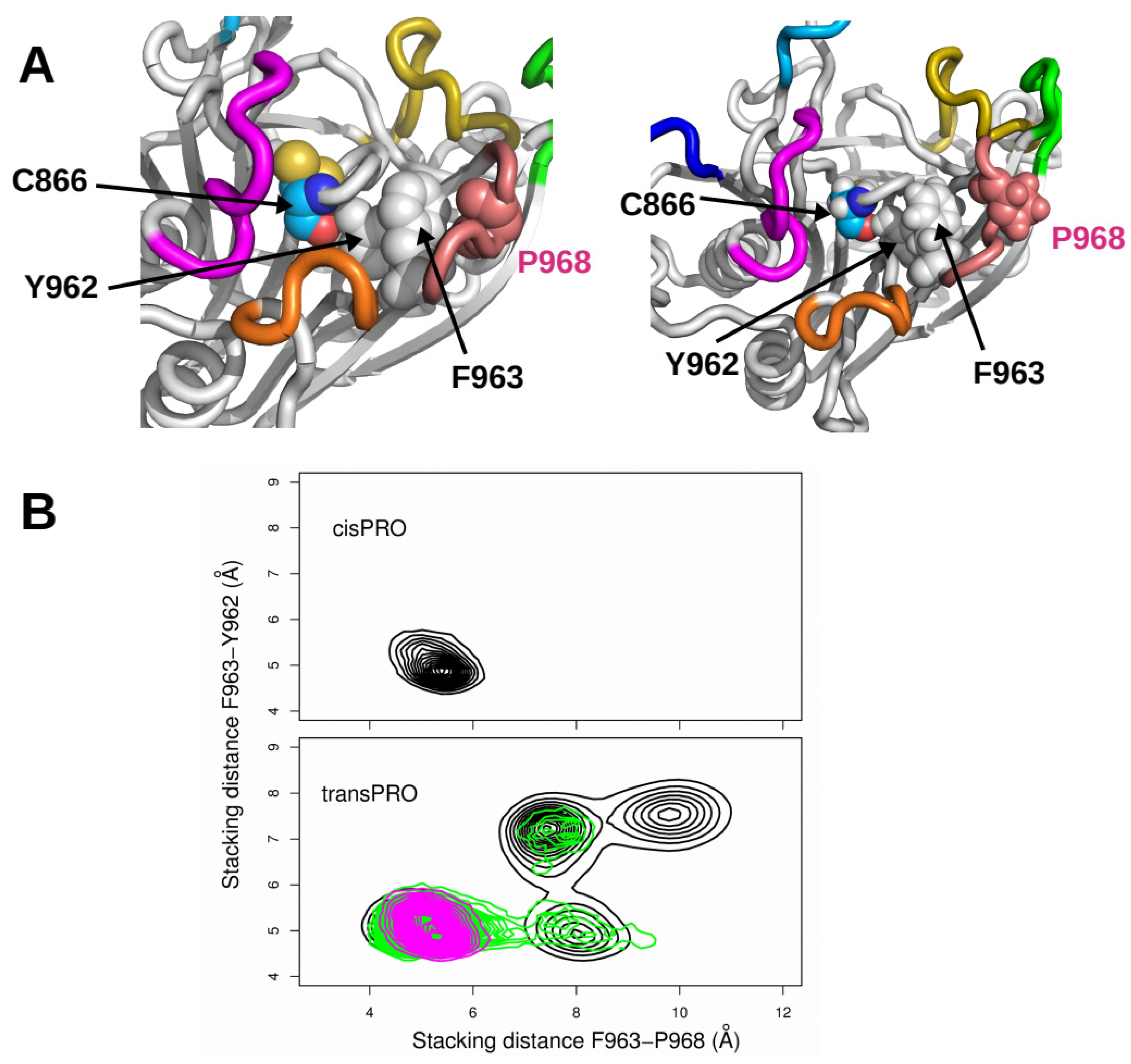
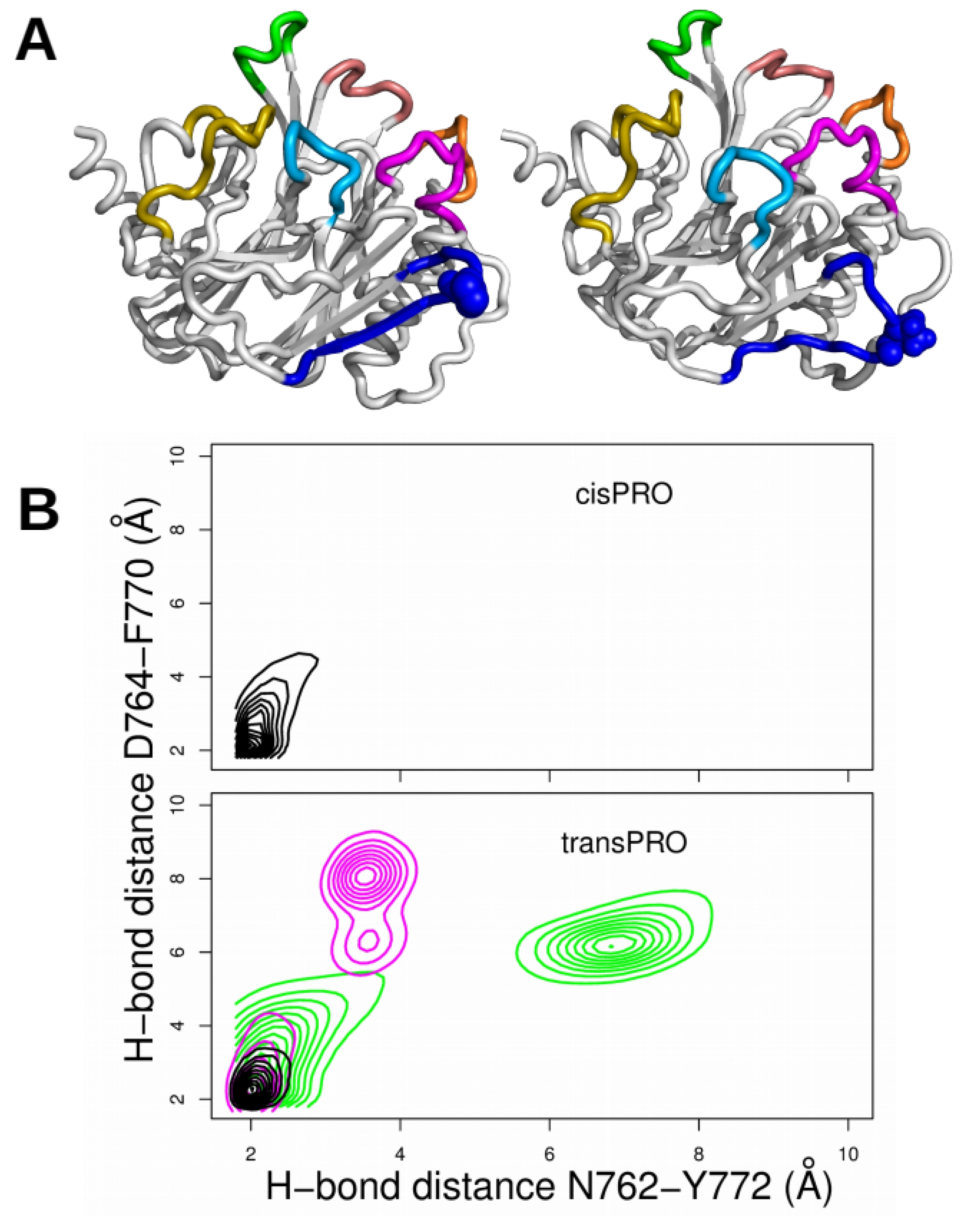
2.3. X-Pro Imide Bond cis-trans Isomerization Affects the Organization of the Network of Loops Restricting Catalytic Cavity Accessibility
2.4. Accessibility to the Catalytic Cavity of CNF1CD
2.5. Docking of the RhoA SWII Target Peptide to CNF1CD cis-trans Isomer Models
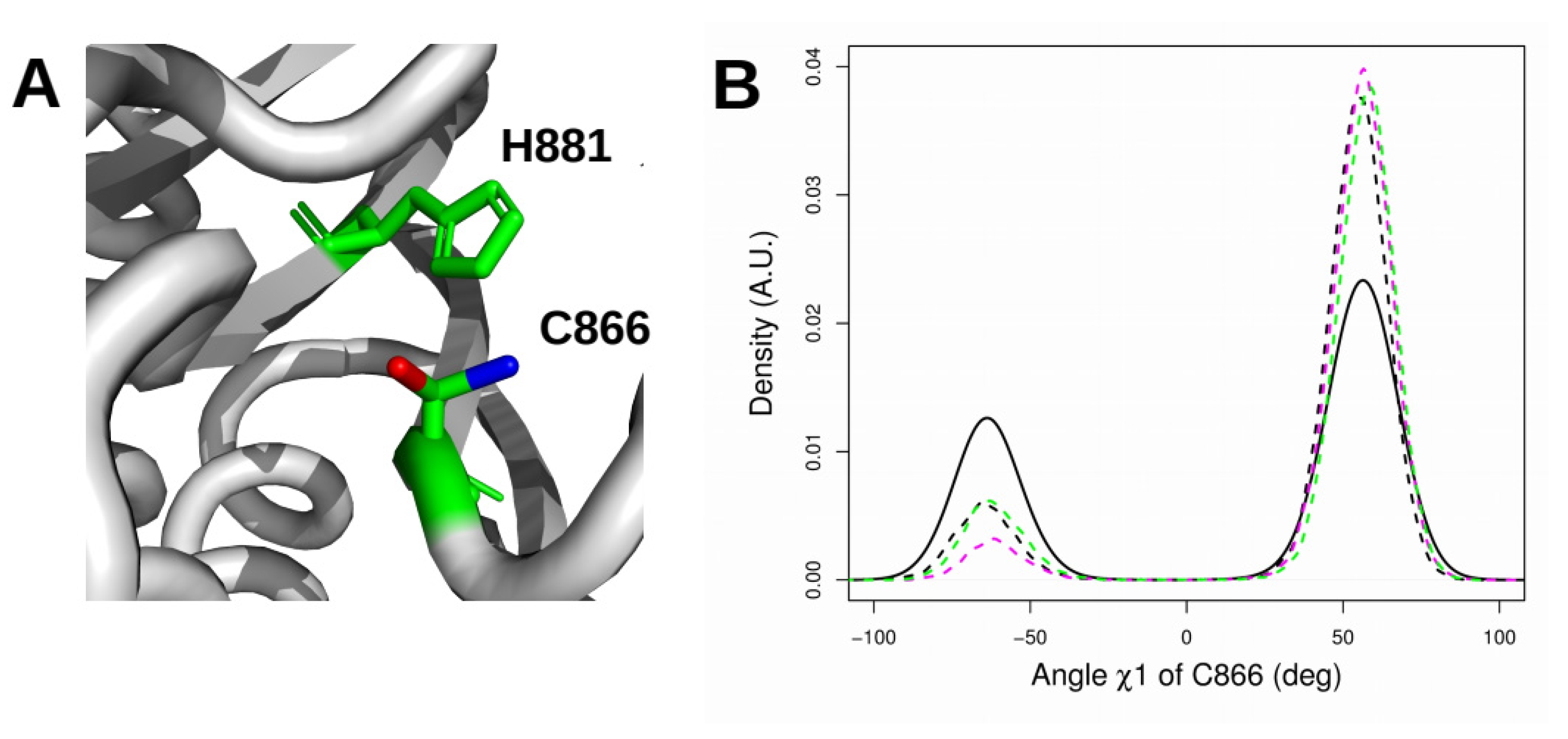
2.6. Orientation of the Thiol Side Chain of the Catalytic Cysteine C866
3. Discussion
4. Materials and Methods
4.1. Preparation of WT in Silico System for MD Simulations
4.2. Restraints on X-Pro Imide Bonds
4.3. Recording MD Trajectories
4.4. Analysis of MD Trajectories
4.5. Clustering of Conformations Sampled Along MD Trajectories
4.6. Peptide Docking
4.7. Selection of Peptide Conformations
4.8. Directed Mutagenesis and Modification of RhoA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: Epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clement, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Munro, P.; Flatau, G.; Doye, A.; Boyer, L.; Oregioni, O.; Mege, J.L.; Landraud, L.; Lemichez, E. Activation and proteasomal degradation of Rho GTPases by cytotoxic necrotizing factor-1 elicit a controlled inflammatory response. J. Biol. Chem. 2004, 279, 35849–35857. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.; Mettouchi, A.; Wilson, B.A.; Lemichez, E. CNF1-like deamidase domains: Common lego bricks among cancer-promoting immunomodulatory bacterial virulence factors. Pathog. Dis. 2018, 76, fty045. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Landraud, L.; Boquet, P.; Bruzzone, M.; Munro, P. Deamidation of RhoA glutamine 63 by the escherichia coli CNF1 toxin requires a short sequence of the GTPase switch 2 domain. Biochem. Biophys. Res. Commun. 2000, 267, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by escherichia coli cytotoxic necrotizing factor 1: Activation of c-Jun N-terminal kinase in hela cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Buetow, L.; Flatau, G.; Chiu, K.; Boquet, P.; Ghosh, P. Structure of the Rho-Activating domain of escherichia coli cytotoxic necrotizing factor 1. Nat. Struct. Biol. 2001, 8, 584–588. [Google Scholar] [CrossRef]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl Cis-Trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef]
- Weiss, M.S.; Jabs, A.; Hilgenfeld, R. Peptide bonds revisited. Nat. Struct. Biol. 1998, 5, 676. [Google Scholar] [CrossRef]
- Jabs, A.; Weiss, M.S.; Hilgenfeld, R. Non-Proline Cis peptide bonds in proteins. J. Mol. Biol. 1999, 286, 291–304. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, A.H. Native state proline isomerization: An intrinsic molecular switch. Biochemistry 2003, 42, 9515–9524. [Google Scholar] [CrossRef]
- Xia, J.; Levy, R.M. Molecular dynamics of the proline switch and its role in crk signaling. J. Phys. Chem. B 2014, 118, 4535–4545. [Google Scholar] [CrossRef]
- Mustafi, S.M.; Brecher, M.; Zhang, J.; Li, H.; Lemaster, D.M.; Hernandez, G. Structural basis of conformational transitions in the active site and 80’s loop in the FK506-binding protein FKBP12. Biochem. J. 2014, 458, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Rasch, J.; Unal, C.M.; Steinert, M. Peptidylprolyl Cis-Trans isomerases of legionella pneumophila: Virulence, moonlighting and novel therapeutic targets. Biochem. Soc. Trans. 2014, 42, 1728–1733. [Google Scholar] [CrossRef]
- Marsolier, J.; Perichon, M.; DeBarry, J.D.; Villoutreix, B.O.; Chluba, J.; Lopez, T.; Garrido, C.; Zhou, X.Z.; Lu, K.P.; Fritsch, L.; et al. Theileria parasites secrete a prolyl isomerase to maintain host leukocyte transformation. Nature 2015, 520, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, K.; Schnell, L.; Barth, H. Host cell chaperones Hsp70/Hsp90 and peptidyl-prolyl Cis-trans isomerases are required for the membrane translocation of bacterial ADP-Ribosylating toxins. Curr. Top. Microbiol. Immunol. 2017, 406, 163–198. [Google Scholar] [PubMed]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-Angstrom Modeling of complexes between flexible peptides and globular proteins. Proteins 2010, 78, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Ghosh, P. Structural elements required for deamidation of RhoA by cytotoxic necrotizing factor 1. Biochemistry 2003, 42, 12784–12791. [Google Scholar] [CrossRef]
- Hoffmann, C.; Aktories, K.; Schmidt, G. Change in substrate specificity of cytotoxic necrotizing factor unmasks proteasome-independent down-regulation of constitutively active RhoA. J. Biol. Chem. 2007, 282, 10826–10832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: Palo Alto, CA, USA, 2002; Available online: http://www.pymol.org (accessed on 1 August 2021).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Zondlo, N.J. Aromatic-Proline interactions: Electronically tunable CH/π interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [Green Version]
- Lerm, M.; Schmidt, G.; Goehring, U.M.; Schirmer, J.; Aktories, K. Identification of the region of rho involved in substrate recognition by escherichia coli cytotoxic necrotizing factor 1 (CNF1). J. Biol. Chem. 1999, 274, 28999–29004. [Google Scholar] [CrossRef] [PubMed]
- Mallis, R.J.; Brazin, K.N.; Fulton, D.B.; Andreotti, A.H. Structural characterization of a proline-driven conformational switch within the Itk SH2 domain. Nat. Struct. Biol. 2002, 9, 900–905. [Google Scholar] [CrossRef]
- Shinoda, K.; Fujitani, H. Initiation of prolyl Cis-Trans isomerisation in the CDR-H3 loop of an antibody in response to antigen binding. Sci. Rep. 2017, 7, 16964. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Kamitani, S.; Miyazawa, M.; Hanajima-Ozawa, M.; Fukui, A.; Miyake, M.; Horiguchi, Y. Crystal structures reveal a thiol protease-like catalytic triad in the C-Terminal region of pasteurella multocida toxin. Proc. Natl. Acad. Sci. USA. 2007, 104, 5139–5144. [Google Scholar] [CrossRef] [Green Version]
- Crow, A.; Hughes, R.K.; Taieb, F.; Oswald, E.; Banfield, M.J. The molecular basis of ubiquitin-like protein NEDD8 deamidation by the bacterial effector protein cif. Proc. Natl. Acad. Sci. USA. 2012, 109, E1830–E1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishide, A.; Kim, M.; Takagi, K.; Himeno, A.; Sanada, T.; Sasakawa, C.; Mizushima, T. Structural basis for the recognition of Ubc13 by the shigella flexneri effector OspI. J. Mol. Biol. 2013, 425, 2623–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.; Zhang, X.; Jin, M.; Xu, L.; Wang, C.; Xia, Z.; Zhu, Y. Complex structure of OspI and Ubc13: The molecular basis of Ubc13 deamidation and convergence of bacterial and host E2 recognition. PLoS Pathog. 2013, 9, e1003322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washington, E.J.; Banfield, M.J.; Dangl, J.L. What a difference a dalton makes: Bacterial virulence factors modulate eukaryotic host cell signaling systems via deamidation. Microbiol. Mol. Biol. Rev. 2013, 77, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Cui, J.; Wang, J.; Li, T.; Wan, X.; Luo, T.; Gong, Y.N.; Xu, Y.; Huang, N.; Shao, F. Structural mechanism of ubiquitin and NEDD8 deamidation catalyzed by bacterial effectors that induce macrophage-specific apoptosis. Proc. Natl. Acad. Sci. USA. 2012, 109, 20395–20400. [Google Scholar] [CrossRef] [Green Version]
- Crow, A.; Race, P.R.; Jubelin, G.; Varela Chavez, C.; Escoubas, J.M.; Oswald, E.; Banfield, M.J. Crystal structures of cif from bacterial pathogens photorhabdus luminescens and burkholderia pseudomallei. PLoS ONE 2009, 4, e5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaoprasid, P.; Lukat, P.; Mühlen, S.; Heidler, T.; Gazdag, E.-M.; Dong, S.; Bi, W.; Rüter, C.; Kirchenwitz, M.; Steffen, A.; et al. Crystal structure of full-length cytotoxic necrotizing factor CNFY reveals molecular building blocks for intoxication. EMBO J. 2020, 40, e105202. [Google Scholar]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields and limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comp. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Martyna, G.; Tobias, D.; Klein, M. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.; Zhang, Y.; Pastor, R.; Brooks, B. Constant pressure molecular dynamics simulation and the langevin piston method. J. Chem. Phys. 1995, 103, 4613–4622. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald and an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints and molecular dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.C. Rattle and a “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comp. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Dotson, D.L.; Domanski, J.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; Volume 32, pp. 102–109. [Google Scholar]
- Mitternacht, S. FreeSASA: An open source c library for solvent accessible surface area calculation. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef]
- Bouvier, G.; Desdouits, N.; Ferber, M.; Blondel, A.; Nilges, M. An automatic tool to analyze and cluster macromolecular conformations based on self-organizing maps. Bioinformatics 2014, 31, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Duclert-Savatier, N.; Bouvier, G.; Nilges, M.; Malliavin, T.E. Building graphs to describe dynamics, kinetics, and energetics in the d-ALa:D-Lac ligase VanA. J. Chem. Inf. Model. 2016, 56, 1762–1775. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Ihara, K.; Muraguchi, S.; Kato, M.; Shimizu, T.; Shirakawa, M.; Kuroda, S.; Kaibuchi, K.; Hakoshima, T. Crystal structure of human rhoa in a dominantly active form complexed with a GTP analogue. J. Biol. Chem. 1998, 273, 9656–9666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longenecker, K.; Read, P.; Derewenda, U.; Dauter, Z.; Liu, X.; Garrard, S.; Walker, L.; Somlyo, A.V.; Nakamoto, R.K.; Somlyo, A.P.; et al. How RhoGDI binds Rho. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1503–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maesaki, R.; Ihara, K.; Shimizu, T.; Kuroda, S.; Kaibuchi, K.; Hakoshima, T. The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol. Cell 1999, 4, 793–803. [Google Scholar] [CrossRef]
- Shimizu, T.; Ihara, K.; Maesaki, R.; Kuroda, S.; Kaibuchi, K.; Hakoshima, T. An Open Conformation of Switch I Revealed by the Crystal Structure of a Mg2+-Free Form of RHOA Complexed with GDP. Implications for the GDP/GTP exchange mechanism. J. Biol. Chem. 2000, 275, 18311–18317. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zhang, Y.; Derewenda, U.; Liu, X.; Minor, W.; Nakamoto, R.K.; Somlyo, A.V.; Somlyo, A.P.; Derewenda, Z.S. Crystal structure of RhoA-GDP and its functional implications. Nat. Struct. Biol. 1997, 4, 699–703. [Google Scholar] [CrossRef]
- Longenecker, K.; Read, P.; Lin, S.K.; Somlyo, A.P.; Nakamoto, R.K.; Derewenda, Z.S. Structure of a constitutively activated RhoA mutant (Q63L) at 1.55 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 876–880. [Google Scholar] [CrossRef]
- Snyder, J.T.; Worthylake, D.K.; Rossman, K.L.; Betts, L.; Pruitt, W.M.; Siderovski, D.P.; Der, C.J.; Sondek, J. Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat. Struct. Biol. 2002, 9, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Dvorsky, R.; Blumenstein, L.; Vetter, I.R.; Ahmadian, M.R. Structural insights into the interaction of ROCKI with the switch regions of RhoA. J. Biol. Chem. 2004, 279, 7098–7104. [Google Scholar] [CrossRef] [Green Version]
- Kristelly, R.; Gao, G.; Tesmer, J.J. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 2004, 279, 47352–47362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derewenda, U.; Oleksy, A.; Stevenson, A.S.; Korczynska, J.; Dauter, Z.; Somlyo, A.P.; Otlewski, J.; Somlyo, A.V.; Derewenda, Z.S. The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca(2+) sensitization pathway in smooth muscle. Structure 2004, 12, 1955–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, S.; Shankaranarayanan, A.; Coco, C.; Ridilla, M.; Nance, M.R.; Vettel, C.; Baltus, D.; Evelyn, C.R.; Neubig, R.R.; Wieland, T.; et al. Structure of Galphaq-P63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 2007, 318, 1923–1927. [Google Scholar] [CrossRef]
- Chen, Z.; Gutowski, S.; Sternweis, P.C. Crystal structures of the PH domains from Lbc family of RhoGEFs bound to activated RhoA GTPase. Data Brief. 2018, 17, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Klink, B.U.; Barden, S.; Heidler, T.V.; Borchers, C.; Ladwein, M.; Stradal, T.E.; Rottner, K.; Heinz, D.W. Structure of Shigella IpgB2 in complex with human RhoA: Implications for the mechanism of bacterial guanine nucleotide exchange factor mimicry. J. Biol. Chem. 2010, 285, 17197–17208. [Google Scholar] [CrossRef] [Green Version]
- Bielnicki, J.A.; Shkumatov, A.V.; Derewenda, U.; Somlyo, A.V.; Svergun, D.I.; Derewenda, Z.S. Insights into the Molecular Activation Mechanism of the RhoA-Specific Guanine Nucleotide Exchange Factor, PDZRhoGEF. J. Biol. Chem. 2011, 286, 35163–35175. [Google Scholar] [CrossRef] [Green Version]
- Jobichen, C.; Pal, K.; Swaminathan, K. Crystal Structure of Mouse RhoA:GTPÎ3S Complex in a Centered Lattice. J. Struct. Funct. Genom. 2012, 13, 241–245. [Google Scholar] [CrossRef]
- Abdul Azeez, K.R.; Knapp, S.; Fernandes, J.M.; Klussmann, E.; Elkins, J.M. The Crystal Structure of the RhoA-AKAP-Lbc DH-PH Domain Complex. Biochem. J. 2014, 464, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Tnimov, Z.; Guo, Z.; Gambin, Y.; Nguyen, U.T.; Wu, Y.W.; Abankwa, D.; Stigter, A.; Collins, B.M.; Waldmann, H.; Goody, R.S.; et al. Quantitative Analysis of Prenylated RhoA Interaction with Its Chaperone, RhoGDI. J. Biol. Chem. 2012, 287, 26549–26562. [Google Scholar] [CrossRef] [Green Version]
- Petit, A.P.; Garcia-Petit, C.; Bueren-Calabuig, J.A.; Vuillard, L.M.; Ferry, G.; Boutin, J.A. A Structural Study of the Complex between Neuroepithelial Cell Transforming Gene 1 (Net1) and RhoA Reveals a Potential Anticancer Drug Hot Spot. J. Biol. Chem. 2018, 293, 9064–9077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Guan, R.; Lee, I.J.; Liu, Y.; Chen, M.; Wang, J.; Wu, J.Q.; Chen, Z. Mechanistic Insights into the Anchorage of the Contractile Ring by Anillin and Mid1. Dev. Cell 2015, 33, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, A.; Tsurumura, T.; Yoshida, T.; Tsumori, Y.; Tsuge, H. Rho GTPase Recognition by C3 Exoenzyme Based on C3-RhoA Complex Structure. J. Biol. Chem. 2015, 290, 19423–19432. [Google Scholar] [CrossRef] [Green Version]
- Jank, T.; Eckerle, S.; Steinemann, M.; Trillhaase, C.; Schimpl, M.; Wiese, S.; van Aalten, D.M.; Driever, W.; Aktories, K. Tyrosine Glycosylation of Rho by Yersinia Toxin Impairs Blastomere Cell Behaviour in Zebrafish Embryos. Nat. Commun. 2015, 6, 7807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlmann, N.; Wroblowski, S.; Scislowski, L.; Lammers, M. RhoGDIα Acetylation at K127 and K141 Affects Binding toward Nonprenylated RhoA. Biochemistry 2016, 55, 304–312. [Google Scholar] [CrossRef]
- Yi, F.; Kong, R.; Ren, J.; Zhu, L.; Lou, J.; Wu, J.Y.; Feng, W. Noncanonical Myo9b-RhoGAP Accelerates RhoA GTP Hydrolysis by a Dual-Arginine-Finger Mechanism. J. Mol. Biol. 2016, 428, 3043–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, E.; Jaiswal, M.; Derewenda, U.; Reis, K.; Nouri, K.; Koessmeier, K.T.; Aspenstrom, P.; Somlyo, A.V.; Dvorsky, R.; Ahmadian, M.R. Deciphering the Molecular and Functional Basis of RHOGAP Family Proteins: A Systematic Approach toward Selective Inactivation of Rho Family Proteins. J. Biol. Chem. 2016, 291, 20353–20371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, H.; Li, F.; Wang, C.; Wang, N.; Jiang, Y.; Tang, Y.; Wu, J.; Shi, Y. Structural Basis for the Specific Recognition of RhoA by the Dual GTPase-Activating Protein ARAP3. J. Biol. Chem. 2016, 291, 16709–16719. [Google Scholar] [CrossRef] [Green Version]
- Dada, O.; Gutowski, S.; Brautigam, C.A.; Chen, Z.; Sternweis, P.C. Direct Regulation of P190RhoGEF by Activated Rho and Rac GTPases. J. Struct. Biol. 2018, 202, 13–24. [Google Scholar] [CrossRef]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular Localization of the Escherichia Coli Cytotoxic Necrotizing Factor CNF1 Cell-Binding and Catalytic Domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trajectory | Duration (ns) | Restrained Residues | Restraint Type | Force Constant (kcal/(mol.degrees)) |
|---|---|---|---|---|
| cis | 2 × 100 | P978, P768 | cis | 10 |
| free | 2 × 100 | - | - | - |
| tr_100 | 2 × 100 | P978, P768 | trans | 100 |
| tr768_100 | 2 × 100 | P768 | trans | 100 |
| tr968_100 | 2 × 100 | P978 | trans | 100 |
| tr_10 | 2 × 100 | P978, P768 | trans | 10 |
| tr768_10 | 2 × 100 | P768 | trans | 10 |
| tr968_10 | 2 × 100 | P978 | trans | 10 |
| RhoA 60–72 | Nter-T A G Q* E D Y D R L R P L |
| Ras 60–72 | Nter-T A G Q* E E Y S A M R D Q |
| System | Number of TIP3P Waters | Neutralizing Ion | Total Number of Atoms |
|---|---|---|---|
| CNF1 WT | 13,160 | One Na+ ion | 44,065 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paillares, E.; Marechal, M.; Swistak, L.; Tsoumtsa Meda, L.; Lemichez, E.; Malliavin, T.E. Conformational Insights into the Control of CNF1 Toxin Activity by Peptidyl-Prolyl Isomerization: A Molecular Dynamics Perspective. Int. J. Mol. Sci. 2021, 22, 10129. https://doi.org/10.3390/ijms221810129
Paillares E, Marechal M, Swistak L, Tsoumtsa Meda L, Lemichez E, Malliavin TE. Conformational Insights into the Control of CNF1 Toxin Activity by Peptidyl-Prolyl Isomerization: A Molecular Dynamics Perspective. International Journal of Molecular Sciences. 2021; 22(18):10129. https://doi.org/10.3390/ijms221810129
Chicago/Turabian StylePaillares, Eléa, Maud Marechal, Léa Swistak, Landry Tsoumtsa Meda, Emmanuel Lemichez, and Thérèse E. Malliavin. 2021. "Conformational Insights into the Control of CNF1 Toxin Activity by Peptidyl-Prolyl Isomerization: A Molecular Dynamics Perspective" International Journal of Molecular Sciences 22, no. 18: 10129. https://doi.org/10.3390/ijms221810129